
Abstract
The salinity stress tolerance in plants has been studied enormously, reflecting its agronomic relevance. Despite the extensive research, limited success has been achieved in relation to the plant tolerance mechanism. The beneficial interaction between Piriformospora indica and rice could essentially improve the performance of the plant during salt stress. In this study, the transcriptomic data between P. indica treated and untreated rice roots were compared under control and salt stress conditions. Overall, 661 salt-responsive differentially expressed genes (DEGs) were detected with 161 up- and 500 down-regulated genes in all comparison groups. Gene ontology analyses indicated the DEGs were mainly enriched in “auxin-activated signaling pathway”, “water channel activity”, “integral component of plasma membrane”, “stress responses”, and “metabolic processes”. Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that the DEGs were primarily related to “Zeatin biosynthesis”, “Fatty acid elongation”, “Carotenoid biosynthesis”, and “Biosynthesis of secondary metabolites”. Particularly, genes related to cell wall modifying enzymes (e.g. invertase/pectin methylesterase inhibitor protein and arabinogalactans), phytohormones (e.g. Auxin-responsive Aux/IAA gene family, ent-kaurene synthase, and 12-oxophytodienoate reductase) and receptor-like kinases (e.g. AGC kinase and receptor protein kinase) were induced in P. indica colonized rice under salt stress condition. The differential expression of these genes implies that the coordination between hormonal crosstalk, signaling, and cell wall dynamics contributes to the higher growth and tolerance in P. indica-inoculated rice. Our results offer a valuable resource for future functional studies on salt-responsive genes that should improve the resilience and adaptation of rice against salt stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A significant part of the cultivated and irrigated land in the world is affected by high salinity to the extents that pose a severe threat to agronomically important crops [1]. Soil salinity has a profound effect on crop productivity due to the toxic Na+ and Cl− ions accumulation and nutrient imbalance [2]. The salinity stress impaired the plant growth by reducing the soil osmotic potential that concomitantly causes physiological drought in the plant [3]. The disproportionate amount of Na+ and Cl− ions in the cell leads to structural damage of enzymes and other macroelements, disruption to the cell organelles as well as perturbation of plant metabolism and physiological functions such as photosynthesis, cellular respiration, and protein synthesis [2, 4].
Previous research provided information regarding cellular, metabolic, molecular, and physiological changes related to salt stress in various plants [5, 6]. Many genes involved in salt tolerance such as cyclophilin, Na+/H+ antiporter (NHX), salt overly sensitive (SOS), superoxide dismutase (SOD), pyrroline-5-carboxylate synthetase, have been identified and characterized in many plant varieties [7,8,9]. During the past decade, genetic manipulation of crops provided efficient ways to produce the salt-stress tolerant transgenic varieties [10, 11].
The constant demand to develop crops with higher salt tolerance has been an incentive for research. Studies have demonstrated that the plants associated with mycorrhizal fungi exhibit improved growth and yield under salt stress [12, 13]. For instance, the higher chlorophyll contents have been detected in the leaves of mycorrhizal plants, indicating that the symbiotic interaction plays a crucial role in regulating photosynthetic and other metabolic activities [12]. Further, mycorrhizal fungi have also been shown to influence stomatal conductance and transpiration in the host plants [14]. It has also been reported that the mycorrhizal symbiosis can improve the osmotic balance in the host plant by enhancing osmolytes production, such as proline, during salinity stress [15, 16].
P. indica, an axenically cultivable root endophyte of basidiomycota, has manifested growth-promotional activity in a wide range of plant species [17]. P. indica has shown enormous potential to ameliorate the plant vigor, nutrient acquisition, and provide tolerance against abiotic and biotic stresses [18]. Plant colonized with P. indica has shown higher antioxidants level during salt stress, which minimizes the salinity-induced lipid peroxidation and membrane fatty acid desaturation in leaves [19]. Further, a cyclophilin like A protein in P. indica (PiCyPA) has reported being involved in various abiotic stresses such as salinity [8]. Despite the profound research advances in the past years, progress is slow; therefore, it is imperative to develop salt-tolerant plants with higher yield and productivity by employing eco-friendly biological means. The in-depth analysis of stress tolerance mechanism in plant–microbe system will be effective in developing strategies to improve salt tolerance.
Taking advantage of RNA-seq technology, many studies were conducted to identify genes involved in plant development and stress responses [20, 21]. Here, we compared the transcriptome of rice root colonized with and without P. indica under control and salt stress condition by using RNA-seq analysis. The treated root samples were sequenced in order to identify the repertoires of salt-responsive differentially expressed genes (DEGs). Functional annotation of DEGs was performed to reveal biological processes and metabolic pathways involved in salt stress responses. The present work provides a further exploration into the molecular events involved in salt tolerance in rice during beneficial interaction.
Materials and methods
Plant, fungus co-cultivation, salt stress treatment and tissue sampling
Seeds of rice (Oryza sativa) var. Pusa basmati 1 were germinated aseptically on Murashige and Skoog [22] media for ten days. Rice seedlings were planted to the PNM agar medium [23] and inoculated with P. indica spores (~ 1 × 105 spores/ml) after 24 h. For salt stress, rice plants were placed on the PNM medium containing 0 M, 0.05 M, 0.1 M, 0.15 M, and 0.25 M NaCl with and without P. indica for 14 days. Rice seedlings were kept in a culture room equipped with 2000 lx fluorescent light, 28 ± 2 °C temperature. P. indica colonization in roots was checked by staining root tissues with 0.05% lactophenol blue and visualized under the light microscope at × 20 or × 40 (Additional Supplementary Fig. 1). Total four groups of treated and untreated root samples were collected (15 plants each) on 14 days after P. indica inoculation (dai) i.e. control roots (C), only salt-treated roots (Salt), only P. indica-treated roots (C + Pi), and salt- and P. indica- treated roots (Salt + Pi). Samples were immediately frozen in liquid nitrogen and stored at − 80 °C before the RNA extraction.
Determination of malondialdehyde (MDA) content
Rice leaves were collected after 14 dai with P. indica and salt treatment (0.25 M NaCl) to analyze the MDA content. Leaf tissues (0.1 g) were homogenized with 0.5 ml of 0.1% (w/v) trichloroacetic acid (TCA) and centrifuged at 10,000×g for 10 min. The resulting supernatant was separated and mixed with 1.5 ml of 20% (w/v) TCA and 0.5% (w/v) thiobarbituric acid (TBA). The mixer was incubated at 95 °C for 25 min. After cooling on the ice, mixer was centrifuged at 10,000×g for 10 min. The absorbance of the supernatant was recorded at 532 nm and 600 nm [24].
RNA extraction, library construction and transcriptome sequencing
Total RNA was extracted using the RNeasy plant mini kit (Qiagen) according to the manufacturer’s protocol. The RNA quality was assessed using 1% agarose gels and NanoDrop 1000 spectrophotometer (Thermo Scientific, USA). Total RNA was treated with DNaseI to remove DNA traces. The purity and quantity of RNA were analyzed by NanoDrop 1000 and 21,000 Bioanalyzer RNA Nanochip (Agilent Technologies GmbH, Germany). RIN (RNA integrity number) values were > 6 for all samples. In order to generate the cDNA library, total RNA was fragmented and amplified by illumina primer (oligonucleotide sequences© 2006–2010 Illumina Inc) and reverse transcriptase. After quality assessment with Agilent 2100, a total of four cDNA libraries were generated with the Illumina Hiseq 2500 platform following the manufacturer’s instructions (Illumina, USA).
Preprocessing, assembly, and data analysis
The paired-end raw reads were generated by Illumina Hiseq 2500 and analyzed by FastQC (v.0.11.3). The adapter sequences and low-quality bases were trimmed by AdaperRemoval-v2 (version 2.2.0) followed by rRNA removal by Sliva database using Bowtie2 (version 1.0.13). The clean reads were aligned to the Rice genome and gene model downloaded from MSU Rice Genome Annotation Project website. The read alignment was performed using the STAR program (version 2.5.3a). The aligned reads were then used for estimating the gene expression using the cufflinks (version 2.2.1). The normalized gene expression level was computed as fragments per kilobase (kb) pair of exon model per million fragments mapped (FPKM). Differential expression analysis between samples was analysed using the cuffdiff program of cufflinks package. Genes with log2 fold change ≥ 2 and/or p-value ≤ 0.05 were used as cutoff criteria for DEGs. The p-values were adjusted using Benjamini and Hotchberg’s False Discovery Rate method.
Functional categorization and annotation
The functional annotation of rice genes was performed with blastx search against the rice genome database. The GO-term distribution was generated using in-house scripts based on Uniprot annotations obtained for DEGs. The GO-enrichment analysis was executed using GOseq R package. GO terms with p-value ≤ 0.05 were depicted as significantly enriched. KEGG Orthology-Based Annotation System (KOBAS 3.0) was used to assess the enrichment of DEGs in KEGG pathways (Fisher’s exact test, p < 0.05) for metabolic pathway prediction. The expressed genes were used as the background set for GO and KEGG enrichment analyses.
Validation of RNA-Seq data by quantitative real-time PCR (qRT-PCR)
The RT-qPCR was performed on the Roche real-time PCR system (Roche, Switzerland) using LightCycler 480 SYBR Green I Master (Roche, Switzerland) according to the manufacturer’s instruction. The cycle threshold value (CT) was assessed and differential expression was calculated using the 2−ΔΔCT method [25] with the actin gene of rice as an endogenous reference [26]. All data are the means of three biological replicates and three technical replicates ± SD. Gene-specific primers used for qRT-PCR were listed in Supplementary Table S3.
Data availability
All the raw sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra) under the accession number PRJNA535402.
Results
Effect of salt treatment on the growth of rice seedlings in the presence and absence of P. indica
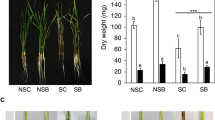
Rice seedlings were treated with different salt concentrations of 0 M, 0.05 M, 0.1 M, 0.15 M, and 0.25 M NaCl for 14 days with and without P. indica inoculation (Supplementary Fig. S1). Root length, shoot length, and fresh weight of treated and control plants, were measured and compared (Fig. 1). We observed that root and shoot length of rice plants were gradually decreased with increasing salt concentration in comparison to control. However, plants colonized with P. indica showed higher root and shoot length as compared to salt-treated plants without P. indica. A similar observation was recorded in the case of fresh weight of treated and control rice. The root length and fresh weight of P. indica-inoculated rice were shown slight increment at 0.25 M NaCl concentration in comparison to control; however, the shoot length of P. indica-inoculated rice was significantly higher as compared to uninoculated plant (Fig. 1). This result suggests that P. indica positively supports the plant growth during salt stress to a certain extent, however; the growth rate was slower with increasing salt concentration. Based on the above finding, we decided to explore the molecular events in rice affected by higher salinity stress and the molecular changes occurred in the presence of beneficial endophytic interaction during stress.

Measurement of root length (a), shoot length (b), and fresh weight (c) of rice under different concentration of salt (NaCl) in the presence and absence of P. indica interaction at 14 days after inoculation (dai). (d) Effects of P. indica interaction on MDA level under salt stress. The MDA content was measured in leaf samples collected after salt treatment (0.25 M NaCl) for 14 days with or without P. indica. Values are the mean ± SD (n = 15). Asterisks indicate statistical significance between P. indica treated and untreated plants (p-value < 0.05) based on t-test
Effect of P. indica colonization on lipid peroxidation in salt-treated rice
Salinity causes oxidative stress and lipid peroxidation in plant tissues which results in a higher level of MDA. Our result showed that the MDA content was increased upon salt treatment (0.25 M NaCl). However, P. indica colonized plant showed significantly lowered MDA level in both control and salt-treated plant (Fig. 1d).
RNA sequencing and mapping
We performed a comprehensive RNA-seq profile with rice root under the given high salt stress in the presence and absence of P. indica interaction to explore the global gene expression changes in root tissues. High-throughput sequencing of the four root RNA samples generated a total of 237.2 M reads of length 100 bp (paired-end), with over 11 Gbps of raw read data. An average 97.15% of total reads passed ≥ 30 Phred score. After trimming, removal of adapter and low-quality sequence, a total of 72,415,218, 52,503,258, 64,794,380, and 46,234,048 clean reads were obtained from RNA samples of C, C + Pi, Salt, and Salt + Pi respectively. The percentages of aligned reads were 97.28%, 94.86%, 98.27%, and 96.99% respectively. The summary of reads and mapping statistics is given in Tables 1 and 2. The total number of genes, unique genes, and common genes expressed in C, C + Pi, Salt, and Salt + Pi samples are provided in Supplementary Table S1.
Differential expression of rice genes in three comparison groups
A total of 1610 DEGs were identified in all sample combinations. The FPKM values of several DEGs were observed to be < 1; therefore genes with FPKM ≥ 1 at least in one sample were also taken into consideration. Out of 1610 DEGs only 661 genes were considered substantial between all the comparison groups (C vs Pi, C vs salt, C vs Salt + Pi, and Salt vs Salt + Pi), with 161 up- and 500 down-regulated genes (Fig. 2, Online Resource 1), indicating that the number of down-regulated genes was higher in the samples exposed to salinity. The total number of DEGs varied among the salt and P. indica-treated comparison groups. There were fewer DEGs in the C vs Pi group as compared to other treatment groups. The total number of up- and down-regulated DEGs in each group were shown in Figs. 2 and 3. FPKM plots and heatmap for expressed genes of all comparison groups were shown in Supplementary Fig. S2, Fig. S3, and Fig. S4. To explore the potential functions of these genes, all the transcripts were annotated using UniProt databases.

Total number of up- and down-regulated DEGs of rice inresponse to salt stress and P. indica interaction. Total 90 (25 up/65 down), 139 (12 up/127 down), 308 (76 up/232 down), and 124 (48 up/76 down) DEGs were detected in C vs Pi, C vs Salt, C vs Salt + Pi, and Salt vs salt + Pi group combinations, respectively. DEGs were selected based on log2fold change (> two fold) and statistical significance of p ≤ 0.05

Venn diagrams show common, salt and P. indica specific DEGs in four comparison groups. a Up-regulated DEGs and b down-regulated DEGs, where A, B, C, and D indicate all DEGs in C vs Pi, C vs Salt, C vs Salt + Pi, and Salt vs salt + Pi groups, respectively. DEGs with log2fold change (> two fold) are shown in brackets
GO and KEGG enrichment analysis
We employed GO enrichment analysis to identify the major gene groups affected by salt stress and beneficial interaction with P. indica. We observed 13 up- and 30 down-regulated GO terms that were over-represented (p-value ≤ 0.01) in Salt vs Salt + Pi group. The most enriched up-regulated terms in biological processes (BP), cellular component (CC), and molecular function (MF) categories were ion transmembrane transport (2 DEGs), integral component of plasma membrane (3 DEGs), and syn-copalyl diphosphate synthase activity (1 DEG), respectively. Whereas the enriched down-regulated terms in BP, CC, and MF categories were response to stress (30 DEGs), extracellular region (10 DEGs), and catalytic activity (25 DEGs), respectively (Fig. 4a, b). Next, the most enriched GO term in the C vs Pi group were cytoplasmic, membrane-bounded vesicle (17 DEGs) and lipid metabolic process (18 DEGs) in up- and down-regulated term, respectively (Supplementary Fig. S5). In the C vs Salt sample, the most enriched up-regulated and down-regulated GO term were response to stress with 11 and 47 DEGs, respectively (Supplementary Fig. S6). Further, we found most enriched GO terms in the C vs Salt + Pi group with cytoplasmic, membrane-bounded vesicle (27 DEGs) and response to stress (90 DEGs) in the up- and down-regulated GO terms, respectively (Supplementary Fig. S7).

GO enrichment of DEGs in Salt vs salt + Pi group showing enriched BP, CC and MF categories. a, b indicate the up and down GO terms, respectively
Next, KEGG pathway analysis was performed to investigate the DEGs enriched in various metabolic pathways. In this analysis, 9 DEGs in C vs Pi, 4 DEGs in C vs Salt, 62 DEGs in C vs Salt + Pi group, and 9 DEGs in Salt vs Salt + Pi were classified into 4, 6, 7, and 7 functional categories, respectively. Carotenoid biosynthesis (9-cis-epoxycarotenoid dioxygenase 1, LOC_Os02g47510), cutin, suberin, and wax biosynthesis (WAX2, LOC_Os02g40784.1 and HOTHEAD, LOC_Os09g19930.1), and fatty acid biosynthesis pathways (acyl-desaturase, LOC_Os01g65830.1 and AMP-binding enzyme, LOC_Os11g35400.1) were significantly enriched among the up- and down-regulated DEGs in the C vs Pi group. In the C vs Salt group, the most enriched up- and down-regulated pathways were flavonoid biosynthesis (chalcone synthase, LOC_Os11g32650.1) and galactose metabolism (glycosyl hydrolases, LOC_Os04g56930.1 and LOC_Os04g45290.1), respectively. Metabolic pathways (alpha-amylase, LOC_Os09g28420.1; dehydrogenase, LOC_Os11g10510.1, and others) biosynthesis of secondary metabolites (lactate/malate dehydrogenase, LOC_Os06g01590.1; 3-ketoacyl-CoA synthase, LOC_Os02g49920.1, and others), and glycolysis/gluconeogenesis (dehydrogenase, LOC_Os11g10510.1; pyruvate decarboxylase, LOC_Os11g38910.1, and others) were the most enriched pathways among the down-regulated DEGs in the C vs Salt + Pi group. However, no significant pathway was detected in up-regulated DEGs in the C vs Salt + Pi group. On the other hand, in the Salt vs Salt + Pi group, pathways related to zeatin biosynthesis (cytokinin dehydrogenase, LOC_Os10g34230), carotenoid biosynthesis (9-cis-epoxycarotenoid dioxygenase 1, LOC_Os12g42280), and diterpenoid biosynthesis (ent-kaurene synthase, LOC_Os04g09900) were up-regulated, whereas protein processing in endoplasmic reticulum (hsp20/alpha crystallin family protein, LOC_Os01g04360 and LOC_Os03g16020), and phenylpropanoid biosynthesis (peroxidase, LOC_Os03g25370 and beta-glucosidase, LOC_Os09g31430) were among the down-regulated DEGs (Table 3). The enrichment analysis results for all GO enrichment and KEGG pathways are provided in Online Resource 3.
Identification of DEGs related to transcription factors and transporters
Transcription factors are the regulatory proteins which play an important role in stress responses. In our data, a total of 36 transcripts encoding for transcription factors were identified, among which 7 TFs were up-regulated whereas 29 TFs were found to be down-regulated. A total of 3, 6, 17, and 10 TF genes showed differential expression in the C vs Pi, C vs salt, C vs Salt + Pi, and Salt vs Salt + Pi groups, respectively. Among the 14 families represented in these DEGs, the bHLH (7 DEGs) and ERF family (6 DEGs) TFs were the most abundant followed by G2-like (5 DEGs) family. Besides, NAC (3 DEGs), WRKY- domain (3 DEGs), and MYB (2 DEGs) TFs were also significantly represented among DEGs (Supplementary Table S2).
In order to maintain Na+ homeostasis in plants, a variety of ion pumps and membrane proteins play a vital role. Here, we observed a total 33 DEGs encoding transporter proteins with 9 up-regulated and 24 down-regulated genes. Gene coding for amino acid transporter, aluminum-activated malate transporter, and auxin efflux carrier are up-regulated in the C vs Pi and C vs Salt + Pi groups. Further, nodulin MtN3 family protein, aquaporin protein, and peptide transporter PTR2 genes were up-regulated in the Salt vs Salt + Pi group. Surprisingly, gene encoding potassium channels KAT1, high-affinity potassium transporters (OsHKT1, OsHKT2), aquaporin protein, major facilitator superfamily antiporter and inorganic phosphate transporter were among the down-regulated genes (Table 4).
DEGs related to signaling and phytohormones
Signaling cascade mediated by protein kinases (PKs) play a critical role in response to environmental stresses as well as in plant growth. We observed that genes related to receptor protein kinases (RPKs) and AGC kinases were up-regulated in Salt vs Salt + Pi groups. On the other hand, gene encoding wall-associated kinases (OsWAK9), RPK, PK, wall-associated receptor kinase, and AGC kinases were up-regulated in C vs Salt + Pi group. However, other genes coding for receptor-like kinases (RLKs), lectin receptor-like kinases (LecRKs), mitogen-activated protein kinases (MAPKKK), ACG kinases, and other kinases were down-regulated in all groups (Table 4).
The plant hormone signaling pathway is one of the crucial pathways which activate the required physiological changes during adverse conditions to ensure the plant growth and development [27]. Genes related to plant hormones biosynthesis and signaling, were also differentially expressed in response to salt and P. indica. We found that genes related to ent-kaurene synthase, 12-oxophytodienoate reductase, cytokinin dehydrogenase, and 9-cis-epoxycarotenoid dioxygenase were up-regulated in Salt vs Salt + Pi groups. Genes such as OsIAA26, AIR12, auxin efflux carrier, OsSAUR50, jasmonate-induced protein, phytosulfokines precursor were up-regulated in the C vs Salt + Pi group. Further, in the C vs Pi group, gibberellin receptor GID1L2 and 9-cis-epoxycarotenoid dioxygenase were among the up-regulated DEGs. On the contrary, genes encoding OsSAUR10, cytokinin-O-glucosyltransferase 2, 1-aminocyclopropane-1-carboxylate oxidase, gibberellin receptor GID1L2, and 12-oxophytodienoate reductase were down-regulated in C vs Salt group (Table 4).
Identification of genes related to antioxidants and other stresses
Salt exposure can cause oxidative stress and induces various anti-oxidative enzymes along with the other stress-responsive genes. We observed that two genes related to peroxidase and glutaredoxin (OsGrx_C8) were up-regulated in C vs Salt + Pi group, whereas one gene related to peroxidase was up-regulated in Salt vs Salt + Pi groups (Table 4).
Among other stress and disease-related genes, SCP-like extracellular protein, chitinase (CHIT4), laccase and pathogenesis-related Bet v I family protein were induced exclusively in the C vs Pi group. Gene encoding OsSub12, legume lectins beta domain-containing protein, and RALFL28 were up-regulated in C vs Salt + Pi group, whereas OsSub12, antifreeze glycoprotein, and thaumatin related genes were up-regulated specifically in Salt vs Salt + Pi groups. However, many stress-related genes encoding salt stress root protein RS1, disease resistance protein, hypersensitive-induced response protein, chitinases (CHIT2, CHIT3), dehydration stress-induced protein, wound-induced protein (WIP3, WIP4, WIP5), and metallothionein were found to be down-regulated in all salt-treated group (Table 4).
Identification of cell wall-related DEGs
Cell wall dynamics play an important role in providing resistance to plants against biotic and abiotic stresses. Evidently, structural alteration in the cell wall during salt stress has been reported in many plants [28, 29]. A significant number of genes associated with cell wall modification and metabolism have been observed to be differentially expressed in our study. Two genes encoding beta-expansion and one arabinogalactan peptide were up-regulated in C vs Pi. Genes involved in cell wall modification such as pectinesterase, pectin methylesterase inhibitor, polygalacturonase inhibitor, arabinogalactan peptide 23, glycin-rich cell wall structural protein 2, CSLF3, glyoxal oxidase, exostosin family domain-containing protein, elastin precursor, and membrane-associated DUF588 domain-containing protein were up-regulated in the C vs Salt + Pi and Salt vs Salt + Pi groups. On the contrary, several genes related to glycosyl hydrolase, endoglucanase, and beta-glucosidase were down-regulated in the salt-treated groups (Table 4).
Genes involved in secondary metabolism and other processes
We, further, observed that genes related to diterpenoid metabolisms such as terpene synthase and ent-kaurene synthase were overexpressed in the C vs Pi and Salt vs Salt + Pi groups respectively. Several genes related to lipid transfer proteins or lipid transfer protein-like (LTPs or LTPL), metallothionein, thaumatin, cytochrome P450 and others were responded to salt stress and P. indica in this data. We noticed that genes belong to LTPL121 and LTPL101 protease inhibitor/seed storage/LTP family protein were up-regulated in C vs Pi and C vs Salt + Pi groups, respectively. Apart from that, genes related to ubiquitin, Pollen Ole e I allergen and extensin family protein, and thaumatin were induced in the C vs Salt + Pi and Salt vs Salt + Pi groups (Online Resource 2).
RNA-Seq data validation
To validate the gene expression patterns obtained by RNA-Seq, we conducted qRT-PCR on 11 selected salt-responsive DEGs correspond to each condition. The genes were selected from DEGs that were expressed in at least one of the comparison group including both up- and down-regulated genes. The results of qRT-PCR experiment for the selected genes are shown in Fig. 5 and Online Resource 4. We observed that the relative expression patterns of the qRT-PCR results were consistent with the RNA-Seq data.

Validation of RNA-Seq results by using qRT-PCR analysis. We selected 11 genes from all the treated rice groups for real-time PCR and used rice actin2 gene as an endogenous reference. Values are means of three replicates and the bar represents the ± SD
Discussion
Salinity stress is destructive to the plant growth and affects crop production worldwide [2]. Recent studies have provided significant evidence regarding the mechanism of stress regulation in plants emphasizing morphological, physiological, and molecular responses [30, 31]; however, exploitable results on plant resilience are still relatively limited.
In our study, rice colonized with P. indica has shown higher growth in comparison to the plants without P. indica during high salt stress (0.25 M NaCl). We also observed that the significant reduction in MDA content in P. indica inoculated rice during salt stress suggesting that P. indica helps the plant to overcome the harmful effect of salinity induced lipid peroxidation. We, further, employed an RNA-Seq based transcriptomic approach to investigate the gene expression in rice in response to salt stress and beneficial interaction with P. indica. In our transcriptome analysis, overall, 235.94 million clean reads were generated from four samples and a total of 1610 DEGs were identified in the four comparison groups. GO enrichment analysis of DEGs revealed that ion membrane transport, response to stress, endogenous stimulus, flavonoid biosynthetic process, secondary metabolic process, auxin-activated signaling pathway, and oxidoreductase activity were activated in response to P. indica during salt stress. KEGG analysis revealed the pathways that were mainly involved in zeatin biosynthesis, carotenoid biosynthesis, diterpenoid biosynthesis, flavonoid biosynthesis, fatty acid elongation, biosynthesis of secondary metabolites and cutin, suberine, and wax biosynthesis in response to P. indica and salt stress. These observations imply that the coordinated response of gene networks involved in various metabolic pathways related to transport, plant hormone signaling, stress responses, secondary metabolites biosynthesis etc. is crucial for the plant adaptation to environmental stresses as reported previously [32, 33].
Transcription factors control the expression of numerous target genes during stress. Here, in P. indica colonized rice, genes related to ERF, bZIP and WRKY TF family are induced during salt stress which are involved in the regulation of many hormones and stress-responsive genes [33]. High salt stress results in an intracellular ionic imbalance due to excess of sodium ion (Na+). The synchronized action of various ion pumps help in the exclusion of excess Na+ ions and vacuolar compartmentalization thereby preventing ionic toxicity in the cytosol. In this study, the activation of the transporter genes such as aquaporin, peptide transporter PTR2, and nodulin MtN3 family protein in Salt vs Salt + Pi group suggests their role in the maintenance of water flow and plant growth in the presence of beneficial interaction during salt stress [34, 35]. An ALMT gene (LOC_Os04g34010) is up-regulated in C vs Salt + Pi group which has considered important for plant tolerance to abiotic stress as well as in stomatal opening and GABA signaling [36]. Genes related to OsHKT1;1, OsHKT2;1, potassium channel KAT1, aquaporin protein, major facilitator superfamily antiporter were down-regulated in our data in contrast to previous studies [37, 38]. However, in some reports, genes for OsHKT1 and OsAKT1 were found to be down-regulated after the osmotic shock and salt stress [39, 40].
Protein kinases participate in many plant signal transduction pathways and play a crucial role in response to environmental stresses as well as plant development [41]. Here, the up-regulation of genes related to wall-associated kinases (OsWAK9), AGC kinases, RPKs, and PKs in the C vs Salt + Pi and Salt vs Salt + Pi groups implicating their role in cell wall expansion and activation of stress responses [42]. Gene related to AGC kinases (LOC_Os12g42020) or barren inflorescence 2 was reported to be involved in salt tolerance in rice seedlings [43]. Our result suggests that these protein kinases might regulate fungal invasion inside the root cortex by regulating cell wall-cytoskeleton interface, priming defense/immune system of the plant thereby conferring resistance against stresses.
The interplay between plant hormone signal transduction and stress signaling pathways confers plant acclimatization to adverse conditions. Studies revealed that symbiotic interaction with P. indica affects the plant hormone regulation and homeostasis and is required for plant growth and immunity [18, 44, 45]. We observed the overexpression of a jasmonate-induced protein (LOC_Os12g12720) in the C vs Salt + Pi group which indicates the increase in JA activity. Further, a gene encoding 12-oxophytodienoate reductase which is involved in jasmonic acid (JA) biosynthesis was found to be induced in Salt vs Salt + Pi. An increase in JA has been observed in tomato, rice, and other plants during salt stress and might be responsible for reduced photosynthesis activity [46, 47]. Abscisic acid (ABA) is a phytohormone that contributes to plant survival under hostile conditions [48]. Two genes related to 9-cis-epoxycarotenoid dioxygenase 1 involved in ABA biosynthesis were found to be up-regulated in C vs Pi and Salt vs Salt + Pi groups. Two genes encoding gibberellin receptor GID1L2 which are involved in the gibberellins signaling pathway, and gene encoding ent-kaurene synthase (LOC_Os04g09900), a key enzyme in gibberellic acid (GA) synthesis, were up-regulated in the C vs Pi and Salt vs Salt + Pi groups, respectively. It was reported that the gibberellin receptor is required for P. indica host colonization and GA induced defense and metabolism [18]. Further, genes involved in ethylene biosynthesis and cytokinin signaling (1-aminocyclopropane-1-carboxylate oxidase and cytokinin-O-glucosyltransferase 2) were down-regulated. Also, a cytokinin degrading enzyme, cytokinin dehydrogenase, was up-regulated in response to P. indica and salt stress indicating that the ethylene and cytokinin signaling might be compromised in the presence of high salt and endophytic colonization which is essentially required for stress tolerance [18, 27]. Furthermore, several auxin gene family members were induced in C vs Salt + Pi group such as OsIAA26 (LOC_Os09g35870), putative auxin-induced protein (LOC_Os08g41290), auxin efflux carrier (LOC_Os11g04190) and OsSAUR50 (LOC_Os09g37452). A gene code for OsIAA6 was reported to be induced in rice under high salt and drought conditions [49]. Similarly, the Arabidopsis SAUR41 subfamily genes were shown to regulate cell expansion during salinity stress [50] which further supports our results. The differential expression pattern of phytohormones might explain that P. indica actively involved in the modulation of hormonal crosstalk in rice enabling plant growth as well as immunity to cope with salt stress.
Further, we observed that many stress and disease-related genes such as laccase, chitinase, and pathogenesis-related Bet v I family proteins were induced in C vs Pi group. Moreover, genes related to OsSub12 (LOC_Os02g10520), thaumatin (LOC_Os12g43450), legume lectins beta domain-containing protein (LOC_Os04g01950), and RALFL28 (LOC_Os06g29730) were induced in the C vs Salt + Pi and Salt vs Salt + Pi groups. These genes play essential roles in plant immunity, pathogen recognition, defense, abiotic stress, and flower development which suggests that P. indica influences these genes to provide tolerance against salt stress [51,52,53]. On the other hand, several genes related to WIP, chitinase, hypersensitive-induced response protein, salt stress root protein RS1, and disease-related proteins were shown down-regulation upon salt treatment insinuating that P. indica might be regulating the expression of these genes in order to lower the defense response to prevent further damage by salt stress. Apart from this, genes such as peroxidase and glutaredoxin (OsGrx) were among the up-regulated DEGs in the C vs Salt + Pi and Salt vs Salt + Pi groups. This result suggests that these antioxidants help in alleviating the toxic effects of oxidative stress in salt-stressed rice during symbiotic interaction.
Cell wall architecture is a crucial factor in stress-responsive signaling pathways. Cell wall proteins and cell wall-modifying enzymes such as xyloglucan endo-β-transglucosylases/hydrolases, expansins, and pectin acetylesterase are involved in modulating cell wall extensibility and plasticity [54,55,56]. In this transcriptome profile, we observed that genes encoding pectinesterase (LOC_Os01g20980, LOC_Os07g41650), pectin methylesterase inhibitor (PMEIs, LOC_Os04g01570), polygalacturonase inhibitor (PGIPs, LOC_Os05g01380), alpha-amylase (LOC_Os02g52700), and arabinogalactan proteins (OsAGP, LOC_Os06g21410 and LOC_Os08g37630) were up-regulated in C vs Pi, C vs Salt + Pi and Salt vs Salt + Pi group. The activities of PGIPs and PMEIs are important to maintain the pectin level for cell wall rigidity during disease and salt stress as evident by earlier studies [28, 57]. Arabinogalactan protein possesses adhesive and water-holding properties, reported to be induced upon salt exposure [58, 59]. Further, the activation of genes related to cellulose synthase-like protein (LOC_Os07g36750) glycin-rich cell wall structural protein 2 (LOC_Os10g31330), exostosin family domain-containing protein (LOC_Os12g12290), and membrane-associated DUF588 domain-containing protein (LOC_Os12g38100) indicates their involvement in the regulation of ROS, cell differentiation and development, and cell wall integrity during stress condition [60,61,62]. In the C vs Pi group, genes related to beta-expansin (LOC_Os06g47360 and LOC_Os09g29710), acanthoscurrin-1 (LOC_Os07g09620) and arabinogalactan (LOC_Os06g21410) were found to be induced. The P. indica colonized rice seems to undergo a major cell wall alteration under high salinity by influencing the expression of cell wall-modifying proteins and thereby maintaining the wall-plasma membrane continuum to provide the necessary protection against stress.
Furthermore, many genes related to cytochrome P450 were up-regulated in P. indica treated roots which regulate the plant growth and defense mechanism during salt stress by catalyzing secondary metabolites, regulation of plant hormone metabolism, and other metabolic processes as evident by previous research [45, 63]. Apart from this, many genes related to ubiquitin, LTPLs, and pollen Ole e I allergen and extensin family protein, were also showed altered expression in salt and P. indica treated rice. These genes involved in plant development, defense and stress regulation, indicating rice-P. indica symbiotic system employed the unique coping mechanism against higher salinity by modulating the functions of these genes [64, 65]. Also, a gene related to lipase (LOC_Os01g51360) that involved in lipid peroxidation, was downregulated in Salt vs Salt + Pi group, which further supports our result that P. indica can alleviate the process of lipid peroxidation. Due to low transcript expression, many previously studied salt-responsive genes could not be included in our study except one gene related to osmolyte (TPP, LOC_Os02g44235).
Based on the above results, we proposed a schematic model of the signaling cascades involving important rice DEGs in response to high salt stress and P. indica interaction (Fig. 6). Since most of the genes showed down-regulation, the common protection mechanism of rice against salt stress seems to be controlled in a specific manner at the higher salinity condition. However, P. indica colonized rice seem to follow the pathways that rely on maintaining cell wall integrity and plant growth which is consistent with the improved growth performance in the Salt + Pi treated rice.

A schematic representation of cell signaling cascade based on differential gene expression pattern in P. indica colonized roots in response to salt stress. (Genes related to SOS1 (Na+/H+ antiporter), NHX (Na+/H+ exchanger), and Ca+2 sensor were not detected in the present study and only included in the model for easier explanation)
Conclusion
We conclude that the transcriptome of P. indica colonized rice root under salt stress revealed the expression variations among several genes with emphasis on the genes related to cell wall remodeling, phytohormones, and receptor-like kinases, suggesting P. indica positively influences the plant growth during salt stress. Functional annotation of salt-responsive DEGs revealed the intricate networks of biological processes and metabolic pathways that might be crucial for growth and stress tolerance in rice. Several DEGs were identified in this study such as RALFL28 (LOC_Os06g29730), membrane-associated DUF588 domain-containing protein (LOC_Os12g38100), and pollen Ole e I allergen and extensin family protein (LOC_Os12g28770) which were not clearly described earlier for their role in symbiotic interaction during stress. Further functional validation of these genes will establish their role in fungal mediated stress response in plants. The results demonstrated here, offer a fresh perspective into the activation and regulation of salt tolerance mechanisms during plant–microbe beneficial interaction and provide a valuable addition to the genetic improvement of plant development and salt tolerance.
References
Zhu JK (2001) Over expression of a delta-pyrroline-5-carboxylate synthetase gene and analysis of tolerance to water and salt stress in transgenic rice. Trends Plant Sci 6:66–72. https://doi.org/10.1104/pp.108.4.1387
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 59:651–681. https://doi.org/10.1146/annurev.arplant.59.032607.092911
Jahromi F, Aroca R, Porcel R, Ruiz-Lozano JM (2008) Influence of salinity on the in vitro development of Glomus intraradices and on the in vivo physiological and molecular responses of mycorrhizal lettuce plants. Microb Ecol 55:45–53. https://doi.org/10.1007/s00248-007-9249-7
Juniper S, Abbott LK (1993) Vesicular-arbuscular mycorrhizas and soil salinity. Mycorrhiza 4:45–57. https://doi.org/10.1007/BF00204058
Hasegawa PM, Bressan RA, Zhu JK, Bohnert HJ (2000) Plant cellular and molecular responses to high salinity. Annu Rev Plant Physiol Plant Mol Biol 51:463–499. https://doi.org/10.1146/annurev.arplant.51.1.463
Roy SJ, Negrao S, Tester M (2014) Salt resistant crop plants. Curr Opin Biotechnol 26:115–124. https://doi.org/10.1016/j.copbio.2013.12.004
Apse MP, Aharon GS, Snedden WA, Blumwald E (1999) Salt tolerance conferred by overexpression of a vacuolar Na+/H+ antiport in Arabidopsis. Science 285(5431):1256–1258. https://doi.org/10.1126/science.285.5431.1256
Trivedi DK, Bhatt H, Pal RK, Tuteja R, Garg B, Johri AK, Bhavesh NS, Tuteja N (2013) Structure of RNA-interacting Cyclophilin A-like protein from Piriformospora indica that provides salinity-stress tolerance in plants. Sci Rep 3:300. https://doi.org/10.1038/srep03001
Maach M, Baghour M, Akodad M et al (2020) Overexpression of LeNHX4 improved yield, fruit quality and salt tolerance in tomato plants (Solanum lycopersicum L.). Mol Biol Rep 47(6):4145–4153. https://doi.org/10.1007/s11033-020-05499-z
Grover A, Aggarwal PK, Kapoor A, Katiyar-Agarwal S, Agarwal M, Chandramouli A (2003) Addressing abiotic stresses in agriculture through transgenic technology. Curr Sci 84:355–367
Zafar SA, Zaidi SS, Gaba Y, Singla-Pareek SL, Dhankher OP, Li X, Mansoor S, Pareek A (2020) Engineering abiotic stress tolerance via CRISPR/Cas-mediated genome editing. J Exp Bot 71(2):470–479. https://doi.org/10.1093/jxb/erz476
Sheng M, Tang M, Chen H, Yang B, Zhang F, Huang Y (2008) Influence of arbuscular mycorrhizae on photosynthesis and water status of maize plants under salt stress. Mycorrhiza 18:287–296. https://doi.org/10.1007/s00572-008-0180-7
Tisarum R, Theerawitaya C, Samphumphuang T, Polispitak K, Thongpoem P, Singh HP, Cha-um S (2020) Alleviation of salt stress in upland rice (Oryza sativa L. ssp. indica cv. Leum Pua) using arbuscular mycorrhizal fungi inoculation. Front Plant Sci 11:348. https://doi.org/10.3389/fpls.2020.00348
Augé RM, Toler HD, Sams CE, Nasim G (2008) Hydraulic conductance and water potential gradients in squash leaves showing mycorrhiza-induced increases in stomatal conductance. Mycorrhiza 18(3):115–121. https://doi.org/10.1007/s00572-008-0162-9
Kumar A, Sharma S, Mishra S (2010) Influence of arbuscular mycorrhizal (AM) fungi and salinity on seedlings growth, solute accumulation, and mycorrhizal dependency of Jatropha curcas L. J Plant Growth Regul 29:297–306. https://doi.org/10.1007/s00344-009-9136-1
Ruiz-Lozano JM (2003) Arbuscular mycorrhizal symbiosis and alleviation of osmotic stress: new perspectives for molecular studies. Mycorrhiza 13:309–317. https://doi.org/10.1007/s00572-003-0237-6
Verma S, Varma A, Rexer K, Hassel A, Kost G, Sarbhoy A, Bisen P, Bütehorn B, Franken P (1998) Piriformospora indica, gen. et sp. nov, a new root-colonizing fungus. Mycologia 90:896–903. https://doi.org/10.1080/00275514.1998.12026983
Schäfer P, Pfiffi S, Voll LM, Zajic D, Chandler PM, Waller F et al (2009) Manipulation of plant innate immunity and gibberellin as factor of compatibility in the mutualistic association of barley roots with Piriformospora indica. Plant J 59(3):461–474. https://doi.org/10.1111/j.1365-313X.2009.03887.x
Waller F, Achatz B, Baltruschat H (2005) The endophytic fungus Piriformospora indica reprograms barley to salt-stress tolerance, disease resistance, and higher yield. Proc Natl Acad Sci USA 102:13386–13391. https://doi.org/10.1073/pnas.0504423102
Gazara RK, de Oliveira EAG, Rodrigues BC et al (2019) Transcriptional landscape of soybean (Glycine max) embryonic axes during germination in the presence of paclobutrazol, a gibberellin biosynthesis inhibitor. Sci Rep 9:9601. https://doi.org/10.1038/s41598-019-45898-2
Cadavid IC, Guzman F, de Oliveira-Busatto L, de Almeida RMC, Margis R (2020) Transcriptional analyses of two soybean cultivars under salt stress. Mol Biol Rep 47:2871–2888. https://doi.org/10.1007/s11033-020-05398-3
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Johnson JM, Sherameti I, Ludwig A, Nongbri PL, Sun C, Lou B, Varma A, Oelmüller R (2011) Protocols for Arabidopsis thaliana and Piriformospora indicacocultivation: a model system to study plant beneficial traits. Endocyt Cell Res 21:101–113
Heath RL, Packer L (1968) Photoperoxidation in isolated chloroplasts. I. Kinetics and stoichiometry of fatty acid peroxidation. Arch Biochem Biophys 125:189–198. https://doi.org/10.1016/0003-9861(68)90654-1
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Zafar SA, Hameed A, Ashraf M et al (2020) Agronomic, physiological and molecular characterisation of rice mutants revealed the key role of reactive oxygen species and catalase in high-temperature stress tolerance. Funct Plant Biol 47(5):440–453. https://doi.org/10.1071/fp19246
Bielach A, Hrtyan M, Tognetti V (2017) Plants under stress: involvement of auxin and cytokinin. Int J Mol Sci 18:1427
An P, Li X, Zheng Y, Matsuura A, Abe J, Eneji AE, Tanimoto E, Inanaga S (2014) Effects of NaCl on root growth and cell wall composition of two soya bean cultivars with contrasting salt tolerance. J Agro Crop Sci 200:212–218. https://doi.org/10.1111/jac.12060
Fan P, Nie L, Jiang P, Feng J, Lv S, Chen X, Bao H, Guo J, Tai F, Wang J et al (2013) Transcriptome analysis of Salicornia europaea under saline conditions revealed the adaptive primary metabolic pathways as early events to facilitate salt adaptation. PLoS ONE 8:e80595. https://doi.org/10.1371/journal.pone.0080595
Formentin E, Sudiro C, Perin G, Riccadonna S, Barizza E, Baldoni E, Lavezzo E, Stevanato P, Sacchi GA, Fontana P, Toppo S, Morosinotto T, Zottini M, Lo Schiavo F (2018) Transcriptome and cell physiological analyses in different rice cultivars provide new insights into adaptive and salinity stress responses. Front Plant Sci 9:204. https://doi.org/10.3389/Ffpls.2018.00204
Hanin M, Ebel C, Ngom M, Laplaze L, Masmoudi K (2016) New insights on plant salt tolerance mechanisms and their potential use for breeding. Front Plant Sci 7:1787. https://doi.org/10.3389/Ffpls.2016.01787
Li N, Liu H, Sun J et al (2018) Transcriptome analysis of two contrasting rice cultivars during alkaline stress. Sci Rep 8(1):9586. https://doi.org/10.1038/s41598-018-27940-x
Rahman H, Jagadeeshselvam N, Valarmathi R et al (2014) Transcriptome analysis of salinity responsiveness in contrasting genotypes of finger millet (Eleusine coracana L.) through RNA sequencing. Plant Mol Biol 85:485. https://doi.org/10.1007/s11103-014-0199-4
Denancé N, Szurek B, Noël LD (2014) Emerging functions of nodulin-like proteins in nonnodulating plant species. Plant Cell Physiol 55(3):469–474. https://doi.org/10.1093/pcp/pct198
Kapilan R, Vaziri M, Zwiazek JJ (2018) Regulation of aquaporins in plants under stress. Biol Res 51:4. https://doi.org/10.1186/s40659-018-0152-0
Palmer AJ, Baker A, Muench SP (2016) The varied functions of aluminium-activated malate transporters–much more than aluminium resistance. Biochem Soc Trans 44(3):856–862. https://doi.org/10.1042/BST20160027
Golldack D, Quigley F, Michalowski CB et al (2003) Salinity stress tolerant and sensitive rice (Oryza sativa L.) regulate AKT1-type potassium channel transcripts differently. Plant Mol Biol 51:71–81. https://doi.org/10.1023/A:1020763218045
Wang R, Jing W, Xiao L, Jin Y, Shen L, Zhang W (2015) The rice high-affinity potassium transporter1;1 is involved in salt tolerance and regulated by an MYB-type transcription factor. Plant Physiol 168(3):1076–1090. https://doi.org/10.1104/pp.15.00298
Fuchs I, Stölzle S, Ivashikina N et al (2005) Rice K+ uptake channel OsAKT1 is sensitive to salt stress. Planta 221:212–221. https://doi.org/10.1007/s00425-004-1437-9
Laurie S, Feeney KA, Maathuis FJM, Heard PJ, Brown SJ, Leigh RA (2002) A role for HKT1 in sodium uptake by wheat roots. Plant J 32:139–149. https://doi.org/10.1046/j.1365-313X.2002.01410.x
Stone JM, Walker JC (1995) Plant protein kinase families and signal transduction. Plant Physiol 108(2):451–457. https://doi.org/10.1104/pp.108.2.451
Wagner A, Kohorn BD (2001) Wall-associated kinases are expressed throughout plant development and are required for cell expansion. Plant Cell 13(303):318. https://doi.org/10.1105/tpc.13.2.303
Yu J, Zao W, He Q, Kim TS, Park YJ (2017) Genome-wide association study and gene set analysis for understanding candidate genes involved in salt tolerance at the rice seedling stage. Mol Genet Genomics 292:1391–1403. https://doi.org/10.1007/s00438-017-1354-9
Michal-Johnson J, Lee YC, Camehl I, Sun C, Yeh KW, Oelmuller R (2013) Piriformospora indica promotes growth of chinese cabbage by manipulating auxin homeostasis-role of auxin in symbiosis. In: Varma A, Kost G, Oelmüller R (eds) Piriformospora indica: sebacinales and their biotechnological applications. Springer, Berlin
Vadassery J, Ritter C, Venus Y, Camehl I, Varma A, Shahollari B, Novák O, Strnad M, LudwigMüller J, Oelmüller R (2008) The role of auxins and cytokinins in the mutualistic interaction between Arabidopsis and Piriformospora indica. Molecular Plant 21:1371–1383. https://doi.org/10.1094/MPMI-21-10-1371
Gardner H (1995) Biological roles and biochemistry of the lipoxygenase pathway. HortScience 30:197. https://doi.org/10.21273/HORTSCI.30.2.197
Kang D, Seo Y, Lee J, Ishii R, Kim K, Shin D, Park S, Jang S, Lee I (2005) Jasmonic acid differentially affects growth, ion uptake and abscisic acid concentration in salt-tolerant and salt sensitive rice cultivars. J Agron Crop Sci 191:273–282. https://doi.org/10.1111/j.1439-037X.2005.00153.x
Keskin BC, Sarikaya AT, Yüksel B, Memon AR (2010) Abscisic acid regulated gene expression in bread wheat (Triticum aestivum L.). Aust J Crop Sci 4:617–625
Jung H, Lee DK, Choi YD, Kim JK (2015) OsIAA6, a member of the rice Aux/IAA gene family, is involved in drought tolerance and tiller outgrowth. Plant Sci 236:304–312. https://doi.org/10.1016/j.plantsci.2015.04.018
Qiu T, Qi M, Ding X et al (2020) The SAUR41 subfamily of SMALL AUXIN UP RNA genes is abscisic acid inducible to modulate cell expansion and salt tolerance in Arabidopsis thaliana seedlings. Ann Bot 125(5):805–819. https://doi.org/10.1093/aob/mcz160
Carvalho AO, Gomes VM (2011) Plant defensins and defensin-like peptides- biological activities and biotechnological applications. Curr Pharm Design 17(38):4270–4293. https://doi.org/10.2174/138161211798999447
Figueiredo A, Monteiro F, Sebastiana M (2014) Subtilisin-like proteases in plant-pathogen recognition and immune priming: a perspective. Front Plant Sci 5:739. https://doi.org/10.3389/fpls.2014.00739
Sharma A, Hussain A, Mun BG, Imran QM, Falak N, Lee SU, Yun BW (2016) Comprehensive analysis of plant rapid alkalization factor (RALF) genes. Plant Physiol Biochem 106:82–90. https://doi.org/10.1016/j.plaphy.2016.03.037
Cosgrove D (2005) Growth of the plant cell wall. Nat Rev Mol Cell Biol 6:850–861. https://doi.org/10.1038/nrm1746
Eklöf JM, Brumer H (2010) The XTH gene family: an update on enzyme structure, function, and phylogeny in xyloglucan remodeling. Plant Physiol 153:456–466. https://doi.org/10.1104/pp.110.156844
Sénéchal F, Wattier C, Rustérucci C, Pelloux J (2014) Homogalacturonan-modifying enzymes: structure, expression, and roles in plants. J Exp Bot 65:5125–5160. https://doi.org/10.1093/jxb/eru272
Kalunke RM, Tundo S, Benedetti M, Cervone F, De Lorenzo G, D'Ovidio R (2015) An update on polygalacturonase-inhibiting protein (PGIP), a leucine-rich repeat protein that protects crop plants against pathogens. Front Plant Sci 6:146. https://doi.org/10.3389/fpls.2015.00146
Lamport DTA, Kieliszewski MJ, Showalter AM (2006) Salt stress upregulates periplasmic arabinogalactan proteins: using salt stress to analyse AGP function. New Phytol 169:479–492. https://doi.org/10.1111/j.1469-8137.2005.01591.x
Zhao C, Zayed O, Zeng F et al (2019) Arabinose biosynthesis is critical for salt stress tolerance in Arabidopsis. New Phytol 224(1):274–290. https://doi.org/10.1111/nph.15867
Liang J, Guo S, Sun B, Liu Q, Chen X, Peng H, Zhang Z, Xie Q (2018) Constitutive expression of REL1 confers the rice response to drought stress and abscisic acid. Rice (New York, NY) 11(1):59. https://doi.org/10.1186/s12284-018-0251-0
Madson M, Dunand C, Li X, Verma R, Vanzin GF, Caplan J, Shoue DA, Carpita NC, Reiter WD (2003) The MUR3 gene of arabidopsis encodes a xyloglucan galactosyltransferase that is evolutionarily related to animal exostosins. Plant Cell 15(7):1662–1670. https://doi.org/10.1105/tpc.009837
Zhu J, Lee BH, Dellinger M, Cui X, Zhang C, Wu S, Nothnagel EA, Zhu JK (2010) A cellulose synthase-like protein is required for osmotic stress tolerance in Arabidopsis. Plant J 63(1):128–140. https://doi.org/10.1111/j.1365-313X.2010.04227.x
Li W, Shao M, Yang J, Zhong W, Okada K, Yamane H, Qian G, Liu F (2013) Oscyp71Z2 involves diterpenoid phytoalexin biosynthesis that contributes to bacterial blight resistance in rice. Plant Sci 207:98–107. https://doi.org/10.1016/j.plantsci.2013.02.005
Chen M, Xu J, Devis D, Shi J, Ren K, Searle I, Zhang D (2016) Origin and functional prediction of pollen allergens in plants. Plant physiol 172(1):341–357. https://doi.org/10.1104/pp.16.00625
Pii Y, Astegno A, Peroni E, Zaccardelli M, Pandolfini T, Crimi M (2009) The Medicago truncatula N5 gene encoding a root-specific lipid transfer protein is required for the symbiotic interaction with Sinorhizobium meliloti. Mol Plant Microbe Interact 22(12):1577–1587. https://doi.org/10.1094/MPMI-22-12-1577
Acknowledgements
The financial support of Science and Engineering Research Board (SERB), Department of Science and Technology, Government of India, New Delhi (Grant Sanction Number SB/YS/LS-111/2014 2014) is greatly acknowledged. The authors are also grateful to AgriGenome Labs Pvt Ltd team for transcriptome sequencing and data analysis.
Author information
Authors and Affiliations
Contributions
N conducted the experiments and wrote the initial drafts of manuscript. RKG performed the data analysis, and helped in the manuscript editing. SK contributed in performing quantitative RT-PCR for data validation. SI and KA contributed in sample collection and RNA extraction. N and MZA planned and designed the research, evaluated the scientific implications of the transcriptome data, and prepared the final maunscript. All author approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
The present study did not involve any Human participants and/or Animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nivedita, Gazara, R.K., Khan, S. et al. Comparative transcriptome profiling of rice colonized with beneficial endophyte, Piriformospora indica, under high salinity environment. Mol Biol Rep 47, 7655–7673 (2020). https://doi.org/10.1007/s11033-020-05839-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05839-z