
Abstract
Diabetes mellitus (DM) is a chronic disease that results in a variety of systemic complications. Recently, stem cell-based therapies have been proposed as potential modalities to manage DM related complications. Mesenchymal stem cell (MSC) based therapies are often considered as an ideal stem cell-based treatment for DM management due to their immunosuppressive characteristics, anti-inflammatory properties and differentiation potential. While MSCs show tremendous promise, the underlying functional deficits of MSCs in DM patients is not well understood. Using the MEDLINE database to define these functional deficits, our search yielded 1826 articles of which 33 met our inclusion criteria. This allowed us to review the topic and illuminate four major molecular categories by which MSCs are compromised in both Type 1 DM and Type II DM models which include: (1) changes in angiogenesis/vasculogenesis, (2) altered pro-inflammatory cytokine secretion, (3) increased oxidative stress markers and (4) impaired cellular differentiation and decreased proliferation. Knowledge of the deficits in MSC function will allow us to more clearly assess the efficacy of potential biologic therapies for reversing these dysfunctions when treating the complications of diabetic disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to the Centers for Disease Control and Prevention’s 2013–2015 National Health and Nutrition Examination Survey, 30.3 million Americans or nearly 10% of the total population of the United States, carry the diagnosis of diabetes mellitus [1]. The International Diabetes Foundation estimates that the total healthcare diabetes-related expenditure was over $850 billion dollars in 2017 [2]. Unfortunately, diabetics often suffer from additional pathologies such as retinopathy, cardiomyopathy, nephropathy, and neuropathy secondary to the effects of elevated blood glucose levels also known as hyperglycemia. The hyperglycemic state can be considered a physiological stressor that leads to the formation of a pathological microenvironment, which alters normal cellular processes [3]. While many of the cellular changes in diabetes are well established, little is known in regards to how diabetes alters the function of mesenchymal stem cells (MSCs) [4].
MSCs are multipotent, plastic-adherent cells with the potential to differentiate into a variety of cell types, such as chondroblasts, osteoblasts or adipocytes [5]. Research has demonstrated the tremendous therapeutic capabilities of MSCs, such as the use of autologous MSC transfer for ameliorating diabetic nephropathy [6, 7]. While normal MSCs may confer therapeutic benefits, their dysfunction in diabetes is thought to contribute to further complications. These complications include cardiomyopathy [8], nephropathy [9], retinopathy [10], and impaired bone and wound healing [11,12,13,14], the latter of which may result in lower extremity amputation, which is 10 times more common in diabetics [8, 15]. Additionally, MSCs isolated from diabetic states have shown a greater propensity to differentiate into adipocytes, in vitro [16].
MSCs can be isolated from a variety of tissues sources including adipose, bone marrow, amniotic fluid, umbilical cord tissue, and dental pulp [17]. MSCs harvested from bone marrow (BM-MSCs) have been studied most extensively but adipose derived MSCs, also known as adipose derived stem cells (ADSCs), are now of increasing interest. ADSCs are more abundant and more easily accessible than BM-MSCs, making them appealing as a therapeutic cell source [18]. Both cell types possess the same profile of surface markers (CD73+/CD90+/CD105+/CD31−/CD45−) and are able to differentiate into bone, cartilage or fat, although other functional differences have been reported [19]. The effects of MSCs in vivo are primarily mediated by paracrine and autocrine factors, the regulation of which differs between BM-MSCs and ADSCs [20]. The study of ADSCs is relatively recent and therefore the majority of the literature reviewed here examines the effect of diabetic pathology on BM-MSCs, although some reports of changes in ADSCs are included as well.
While diabetes is an extremely complicated disease process with many pathological consequences, some of which have been well established, little is known about how diabetes alters the function of MSCs. In this review, we aim to demonstrate how the physiologic stress of diabetes alters MSC function. In particular, we explore the properties of diabetic MSCs in terms of (1) angiogenesis/vasculogenesis, (2) cytokine secretion, (3) oxidative stress, and (4) proliferation/differentiation potential. By understanding the body of literature devoted to these topics, researchers and practitioners may gain valuable insight into prevention, treatment, and management of diabetes and its complications. Additionally, it is important to illuminate the changes in the diabetic stem cell microenvironment, which may facilitate the development and improvement of future MSC-related therapies.
Study design
A PubMed literature search was conducted on January 20th, 2018 for the following keywords: “Mesenchymal stem cell,” “Mesenchymal stem cells”, “MSC,” “MSCs,” “human mesenchymal stem cell,” “human mesenchymal stem cells,” “hMSC,” “hMSCs,” “bone marrow-derived mesenchymal stem cell,” “bone marrow-derived mesenchymal stem cells,” “bone marrow derived mesenchymal stem cell,” “bone marrow derived mesenchymal stem cells,” “BM-MSC,” “BM-MSCs,” “multipotent stromal cell,” “multipotent stromal cells,” “diabetes,” “diabetic,” “diabetes mellitus,” “insulin-resistant,” “insulin resistant,” “secretome,” “secreting,” “secrete,” “growth factors,” “yield,” “excrete,” “produce,” “dysfunction,” “dysfunctions,” “dysfunctional,” “malfunction,” “malfunctions,” “malfunctional,” “unfunctional,” “defect,” “defects,” “defective,” “ineffective,” “inhibit,” “inhibits,” “inhibitory,” “inhibition,” “impair,” “impairs,” “impaired.” A total of 1826 abstracts were reviewed for inclusion criteria. For additional information see PRISMA diagram of compiled search groups (Fig. 1).

Moher et al. [118]
1824 articles were obtained from search terms and two external sources were added. After reviewing all 1826 abstracts only 317 articles met the inclusion criteria for complete full-text assessment. After full-text assessment only 33 articles were found that met selection criteria
Study method
All 1826 abstracts were assessed and selected if they demonstrated the following inclusion criteria: (1) published within the past 20 years, (2) studied adipose derived or bone marrow derived MSCs acquired from a diabetic model (human or animal) or normal MSCs placed in a diabetic environment, (3) discussed changes in diabetic MSC gene expression. Changes in the function of MSCs were related to one of the four major categories discussed in the introduction: angiogenesis/vasculogenesis, cytokine secretion, oxidative stress, and proliferation/differentiation potential. Studies were excluded if they were literature reviews or used MSC transfer for treatment purposes without further investigation. Each selected article was then read and cross-referenced with our inclusion criteria once more, before being confirmed as part of the final cohort by a senior researcher (R.S.). In total, 33 articles met the inclusion criteria and were included in the review (Table 1).
Angiogenesis/vasculogenesis
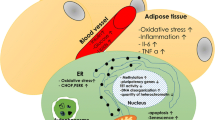
Some of the most severe complications in diabetics arise due to either macrovascular pathologies, such as myocardial infarction and cerebrovascular disease, or microvascular pathologies, such as diabetic nephropathy and retinopathy [21]. These vascular changes are attributed to the alteration of the body’s normal angiogenic and vasculogenic pathways, which to some degree involve MSC dysfunction. Vasculogenesis is defined as the de novo formation of blood vessels from precursor angioblasts, and angiogenesis is the process by which these newly formed vessels grow in conjunction with endothelial and smooth muscle cells [22]. In the diabetic state, certain tissues exhibit increased angiogenesis, while other tissue types display the opposite. This “angiogenic paradox,” as it is commonly referred to in the diabetic literature, renders medical management particularly complex. In this section, we explore the potential mechanisms for these changes, including alterations in MSC mobilization, extracellular vesicle (EV) secretion, and mediation of angiogenic signals.
Several studies have shown that diabetes alters the normal ability of MSCs to promote angiogenesis. In one such study, Rezaie et al. exposed human BM-MSCs in vitro, to serum isolated from either Type 2 diabetic (T2DM) patients, healthy donors, or to fetal bovine serum (FBS). After culturing the MSCs in these respective conditions, vascular endothelial growth factor (VEGF) RNA levels were quantified for each sample. The researchers found a two-fold decrease in VEGF expression from the MSCs in diabetic sera compared to both control groups (p < 0.05) [23]. The decrease in VEGF, a potent angiogenic factor, observed in MSCs cultured in a diabetic milieu has also been corroborated in other MSC studies. For example, Ribot et al. examined the supernatant of cultured tibial and femoral BM-MSCs from Zucker diabetic fatty rats and control lean rats. Proteomic analysis of the secretome demonstrated that VEGF concentrations were significantly decreased in the diabetic BM-MSCs (p < 0.001) and that the levels of αβ-crystallin, a chaperone for VEGF, were also significantly reduced (p < 0.05) [24]. Previous literature indicates that αβ-crystallin plays a major role in angiogenesis through the modulation of VEGF [25, 26]. Consequently, these findings indicate that the diabetic state alters MSC secretion of VEGF in vitro and suggest that this may be through attenuation of other angiogenic signals, such as αβ-crystallin.
The production of other angiogenic factors has also been shown to be altered as a consequence of diabetic MSC dysfunction. For example, Kim et al. measured the VEGF expression of BM-MSCs from streptozotocin induced diabetic and control rats by isolating RNA and performing RT-PCR experiments. The diabetic MSCs not only exhibited reduced expression of VEGF-A, but also a decrease in three other major angiogenic genes: angiopoietin 1, angiopoietin 2, and VEGF-C. Interestingly, this group also examined the therapeutic and regenerative potential of the MSCs in vivo by comparing the outcomes of injecting ischemic rat hind limbs with either normal or diabetic MSCs, or phosphate-buffered saline (PBS). Laser Doppler perfusion imaging (LDPI) was then performed to assess hind limb perfusion, which showed that injection of normal MSCs led to significantly greater perfusion of the ischemic hindlimb, compared to diabetic MSCs (p < 0.01). Furthermore, the lack of a therapeutic benefit observed after injection of diabetic MSCs relative to the negative control group suggests that diabetes may render MSCs essentially ineffective for improving ischemia if unaltered [16].
The term “mobilopathy” is used to describe stem cells that fail to mobilize effectively from the bone marrow [27]. While BM-MSCs undergo a so-called “mobilopathy,” they also impact the mobilization of endothelial progenitor cells (EPCs) via paracrine signaling [24]. The movement of both MSCs and EPCs is essential for de novo blood vessel formation. To study this, Rezabakhsh et al. examined how incubating human MSCs in diabetic and non-diabetic serum influenced their angiogenic paracrine effects on endothelial cells (ECs), using a Matrigel plug assay in Wistar rats. Here, MSCs were pre-conditioned with either normal or diabetic serum for 7 days and then subsequently added to the Matrigel before being injected subcutaneously into the flanks of rats. After 7 days, immunohistochemistry was performed on the Matrigel plugs and showed that MSCs grown in diabetic serum had decreased levels of both vascular endothelial cadherin (VE-cadherin; p < 0.01) and alpha smooth muscle actin (α-SMA; p < 0.0001) [28]. These two proteins are essential for endothelial migration and vascular smooth muscle formation, respectively [29, 30]. Furthermore, since both are essential to de novo angiogenesis, they are often used together as a measure of blood vessel formation. The authors attributed the decreased blood vessel formation they observed, to changes in the secretome of MSCs, rendering them dysfunctional due to exposure to diabetic serum [28]. It is thought that the mobilization of both MSCs and EPCs from the bone marrow is essential in the initiation of vasculogenesis and angiogenesis. In the next section, we will also investigate the mechanism by which adiponectin modulates MSC mobilization.
To measure the initiation of angiogenesis and endothelial cell adhesion in vitro, researchers have also utilized blood vessel tube formation assays, often specifically studying the effect of diabetes on MSC-mediated angiogenesis [31]. For example, Rezabakhsh et al. collected human BM-MSCs from one healthy male donor and cultured them for 14 days in 10% solutions of either diabetic or normal sera. It was found that after seeding the BM-MSCs grown in diabetic serum conditions in growth factor depleted Matrigel substrate, they completely lost the ability to form blood vessel tubes within 8–12 h. In addition, the researchers went on to confirm that the diabetic sera decreased MSC to endothelial cell trans-differentiation by fourfold (p < 0.001). To assay for changes in MSC migration in the diabetic state, Rezabakhsh et al. cultured human MSCs in either diabetic or normal sera for 7 days and subsequently seeded the samples on a transwell migration assay containing stromal-cell derived factor-1 alpha (SDF-1α), an important regulator known to induce MSC migration [32, 33]. The researchers discovered that MSCs pre-treated with diabetic sera displayed a significantly decreased migration rate (p < 0.01) [28]. Taken together, these findings suggest that the normal pathways of MSC and endothelial cell communication, which are essential for angiogenesis and vasculogenesis, are dysfunctional in the diabetic state, in part due to changes in MSC migration.
In addition to the impaired angiogenic capacity of BM-MSCs as a result of diabetes, alterations to the angiogenic capabilities of ADSCs have been reported in diabetic states [34]. Using RT-qPCR, Rennert et al. studied the expression of angiogenic genes in ADSCs isolated from the inguinal fat pads of T2DM and wild-type (WT) mice [35]. The diabetic condition resulted in a dysfunctional signaling environment in situ, with a statistically significant decrease in the expression of essential angiogenic genes VEGF-A (p < 0.01) and SDF-1α (p < 0.05). Other studies have confirmed that ADSCs and BM-MSCs isolated from streptozotocin induced Type 1 diabetic (T1DM) mice also display significant decreases in VEGF-A expression (p < 0.005 and p = 0.02) [36, 37]. To investigate the ability of ADSCs to form vascular networks, Rennert et al. went on to isolate ADSCs from the stromal vascular fraction (SVF) of the inguinal fat pads of T2DM and WT mice and performed Matrigel tubule formation assays. The T2DM ADSCs displayed a near sixfold decrease in tubular structures (p < 0.001) versus WT controls, indicating that the pro-angiogenic effects of ADSCs are severely impaired by the diabetic state. The authors hypothesized that the decrease in angiogenesis may be an issue of proliferation, due to a decrease in the fraction of ADSCs in the SVF. This hypothesis was confirmed by flow cytometry analysis, in which the WT-SVF contained a significantly larger proportion of ADSCs (CD45−/CD31−/CD34+ cells) compared to T2DM-SVF and T1DM-SVF (p ≤ 0.01). The SVF refers to the aqueous fraction of lipoaspirate isolated following enzymatic digestion [38] and contains a heterogeneous group of cells—including subgroups with differing levels of angiogenic capacities [39]. In order to elucidate the changes that occur in diabetic adipose tissue, Rennert et al. employed single-cell transcriptional analyses and clustering of the gene profiles to determine that there were three major subpopulations of ADSCs. One subpopulation of ADSCs was specifically associated with an elevated angiogenic and vasculogenenic gene profile. This subpopulation of ADSCs, which was deemed essential for angiogenesis and vasculogenesis, was expressed at a lower proportion in the T2DM and T1DM cell populations compared to WT [35]. These findings suggest that the diabetic state impairs the overall angiogenic gene expression of ADSCs by selectively diminishing a subpopulation of ADSCs associated with vasculogenesis and angiogenesis.
Depending on the tissue type, the vascular complications that arise in diabetics are due to either increased or decreased vascular processes, a concept that has specifically been termed in this context, the “angiogenic paradox” [22, 40,41,42]. This paradox is due to the altered secretion of both pro-angiogenic and anti-angiogenic factors in diabetes, which makes the treatment of diabetic vascular complications problematic [40, 43, 44]. The modulation of VEGF secretion, by a number of upstream factors, results in what is described as an ‘angiogenic switch’ of VEGF and other associated angiogenic factors [40, 42, 43]. Although the MSC secretome has not been precisely characterized, it is thought to provide some of the upstream factors that mediate angiogenesis and vasculogenesis [41, 45]. Overall, the mechanisms that control normal angiogenic processes require a fine equilibrium between both stimulating and inhibiting factors. This delicate balance is often disturbed by a variety growth factors, cytokines, hematopoietic factors, and metabolic changes that arise differentially in specific tissues in the diabetic state [46].
While in the previous studies discussed, the levels of angiogenic factors and blood vessel formation were severely attenuated due to the dysfunction in diabetic MSCs, other tissues such as the retina and kidney display increased angiogenesis [47,48,49]. It is thought that diabetic retinopathy involves a loss of pericyte cells and the disruption of their interaction with ECs in the retina, which ultimately leads to increased EC proliferation and angiogenesis [50, 51]. It is hypothesized that the extracellular vesicles (EVs) of diabetic MSCs mediate this process in its earliest stages of pericyte detachment and in later stages of uncontrolled and excessive EC proliferation and angiogenesis. To demonstrate this, Beltramo et al. isolated EVs from human adipose tissue- and bone marrow-derived MSC cultures exposed to either high glucose (28.0 mmol/l, HG) or physiological conditions (5.6 mmol/l, NG). It was found that after adding the isolated EVs from the HG MSC cultures to sub-confluent human retinal pericyte cultures, an increased time-dependent detachment of pericytes was observed. This group further stressed the high-glucose and control MSC groups by exposing each to either hypoxic or normoxic conditions and discovered that EVs derived from MSC’s exposed to both high glucose and hypoxia together showed the highest pericyte detachment rate. These findings support the suggestion that EVs produced by MSCs mediate the early stages of diabetic retinopathy [10]. It is important to note that the micro- and macrovascular changes that occur in diabetes, in some cases, cause a state of physiologic hypoxia. Several authors, such as Kinnaird et al., have shown that MSC production of VEGF, as well as endothelial and smooth muscle cell migration, is increased several fold in response to hypoxia [51, 52]. In turn, it was also found that the EVs isolated from MSCs placed in the hyperglycemic/hypoxic conditions resulted in increased expression of VEGF from human retinal pericyte cultures [51]. This finding suggests that the MSC secretome may also mediate later stages of diabetic retinopathy where significant angiogenesis occurs. The changes described in the retina versus those seen in ischemic limbs are examples of the ‘angiogenic paradox’, which is shown to be at the very least, partially mediated by the secretome of MSCs. Although the diabetic retina displays increased MSC-mediated angiogenesis, most other diabetic tissue show the reverse [4]—as is seen in diabetic cardiomyopathy and diabetic wound healing processes [16, 53].
While much of the current literature describes the dysfunction in diabetic MSCs involved in angiogenesis, only a few investigators have studied mechanisms to recover or improve the lost MSC angiogenic potential. In one such study, Kim et al. hypothesized that administration of oxytocin could recover the angiogenic potential of diabetic MSCs [54]. This hypothesis was based on previous studies that successfully demonstrated the hormone’s ability to induce cardiomyogenesis [55] and endothelial cell migration [56], when administered to MSCs found in the myocardium. After isolating BM-MSCs from both non-diabetic and streptozotocin induced diabetic rats, Kim et al. quantified the angiogenic activity of MSCs by measuring tube length and tube area before and after treatment with oxytocin. It was found that with no treatment, the diabetic MSCs displayed a tenfold decrease in tube length and a sevenfold decrease in tube area. However, pre-treatment with 100 nM of oxytocin improved the diabetic MSC angiogenic potential, which showed a 2.6-fold increase in tube length and ten-fold increase in tube area compared to PBS treated diabetic BM-MSCs. The authors hypothesized that oxytocin treatment ameliorates impaired angiogenic function through induction of Krüppel-like factor 2 (KLF2), a critical angiogenic factor and master regulator of endothelial development. To confirm that KLF2 expression was induced by oxytocin, KLF2 mRNA levels were assessed in diabetic-MSCs treated with different concentrations of oxytocin. While KLF2 expression was impaired in diabetic MSCs compared to non-diabetic cells, its expression was recovered by oxytocin in a dose-dependent manner. Furthermore, to confirm whether KLF2 was a mediator of oxytocin’s ability to restore angiogenic potential, Kim et al. knocked down KLF2 in vitro using siRNA. The researchers found that oxytocin treatment was unable to restore KLF2 levels and induce tube formation in the siRNA transfected cells, which demonstrates that oxytocin’s ability to improve the angiogenic potential of diabetic MSCs can be attributed to its interaction with KLF2 [54].
In summary, the diabetic state impairs the angiogenic functions of MSCs leading to pathological changes. While the modulation of angiogenic behavior is tissue dependent, in vitro studies have shown that diabetes alters MSC VEGF expression and decreases MSC migration necessary for vasculogenesis. Due to the limited number of studies, additional research is required to fully uncover the mechanisms by which diabetic MSCs are dysfunctional and play a role in mediating angiogenesis. With an improved understanding, novel therapeutics targeting the diabetic MSC can be developed to help ameliorate additional pathologies associated with diabetes.
Pro-inflammatory cytokines
Inflammatory cytokines are chronically elevated in diabetics and it is important to explore the effect of this altered state on the function and differentiation patterns of MSCs. MSC function is highly regulated by cytokines [57,58,59]. Changes in cytokine interactions can induce altered patterns in the MSC secretome [60], which mediates critical cell signaling and migratory pathways [61]. The disruption of these processes also has downstream effects on MSC differentiation. In this section, we will explore how the diabetic milieu affects MSCs and how these changes may contribute to furthering diabetic pathology.
In the diabetic milieu, several interleukins (IL) are elevated, most notably of which is IL-6, a pro-inflammatory cytokine [62]. Previous studies have shown that IL-6 has a role in modulating glucose uptake and insulin sensitivity [63]. In one experiment, Madhira et al. compared the secretomes of BM-MSCs isolated from obese (WNIN/Ob), insulin resistant (T2DM), lean control (lean) and parental control (control) rats, using RT-PCR. Their analysis showed that mRNA levels of inflammatory markers, specifically IL-6, were significantly elevated in the secretome of BM-MSCs from the diabetic mouse model (p < 0.05). They also found that the increase in IL-6 was proportional to the increase in thiobarbituric acid reacting species (TBARS), a marker of oxidative stress [59]. These results suggest that the diabetic milieu induces MSCs to contribute to the chronic inflammatory state. Altogether, the IL-6 signaling protein mediates the chronic low-grade inflammatory state of DM and is secreted at an increased level in diabetics by BM-MSCs.
TNF-α is another pro-inflammatory cytokine involved in the pathogenesis of insulin resistance [64]. A meta-analysis of Type 1 DM (T1DM) patients indicated consistently elevated levels of TNF-α [65]. During the immune response, TNF-α is primarily secreted by macrophages and to a lesser degree, by adipocytes [53]. On the molecular level, TNF-α signaling leads to an up-regulation of protein-tyrosine phosphatase, which dephosphorylates tyrosine residues on the insulin receptor leading to a down regulation of the insulin-signaling cascade [63]. This mechanism inhibits the effects of insulin and can further increase insulin resistance and decrease insulin sensitivity.
MSCs are important contributors to bone healing. Their differentiation into chondroblasts and osteoblasts mediates the process of endochondral ossification. The inflammatory process, which is equally important in bone healing, is altered in diabetics due to increased levels of inflammation in the diabetic state. To investigate the role of TNF-α on MSC-mediated fracture repair in diabetics, Ko et al. inflicted diabetic and non-diseased mice with closed-transverse femur fractures. Immunohistochemistry was performed on femoral calluses from both groups and it was observed that relative to normoglycemic mice, femoral calluses of diabetic mice had a 3.2-fold increase in TNF-α expression (p < 0.05). Next, bone formation for the callus site was quantified using haematoxylin and eosin (H&E) staining, which showed that the diabetic femoral calluses displayed a significant decrease in bone formation with a lower osteoblast count. To further delineated the role of TNF-α on MSC-mediated fracture repair in the diabetic state, Ko et al. also measured DNA fragmentation as an indicator of apoptosis and determined that the diabetic BM-MSCs demonstrated an 3.9 fold increase in apoptosis and reduced proliferation (p < 0.05). In addition, the intraperitoneal injection of pegsunercept (4 mg/kg), an inhibitor of TNF-α, was effective in reducing diabetic TNF-α levels to the same as normoglycemic femoral calluses and reducing MSC apoptosis to restore overall bone formation [66]. From these findings, the mechanisms of TNF-α damage to MSCs appear to include promoting MSC apoptosis, decreasing MSC osteoblast differentiation, and inhibiting overall MSC proliferation. Employing methods to attenuate the elevated levels of TNF-α observed in diabetics may be a promising strategy to improve bone formation post-fracture as a potential target for future therapies.
Chronic inflammation in diabetes is often worsened by obesity, a phenotype that has been shown to independently contribute to systemic inflammation [67, 68]. Adipose tissue is often forgotten as an endocrine tissue, nonetheless, its effect in metabolic regulation is crucial. Adiponectin is one important cytokine secreted by adipose tissues involved in both glucose regulation and free fatty acid oxidation [69]. Under normal conditions, adiponectin increases insulin sensitivity [70] and those with higher levels of adiponectin expression show a reduced incidence of diabetes [71]. In addition, in both diabetic and obese serum, adiponectin has shown markedly decreased levels when compared to serum from non-diseased individuals.
Researchers have shown that adiponectin is important for bone regeneration and is involved in mobilization of MSCs from the bone marrow to fracture sites, via mediating the interaction of SDF-1 with its receptor CXCR4 [72, 73]. Yu et al. isolated BM-MSCs from WT mice and treated them with adiponectin (10 g/ml), which they found to decrease SDF-1/CXCR-4 expression. In the non-diseased state, adiponectin creates a gradient of SDF-1 with high levels at the site of bone injury and low levels in the bone marrow [72]. The SDF-1 concentration gradient thereby regulates the migration of MSCs out of the bone marrow and towards the injured site [32]. However, when adiponectin levels are low, as is the case in diabetes, the concentration gradient or SDF-1 is altered and MSC migration is impaired. The impairment of this signaling pathway ultimately contributes to increased bone fracture risk and poor bone healing. These complications may potentially improve through therapies increasing adiponectin or modulating the SDF-1/CXCR-4 axis, allowing BM-MSCs to properly mobilize to sites of bone injury and differentiate accordingly.
Another important endocrine function of adipose tissue is its synthesis of leptin. Leptin is a key appetite regulating hormone released by MSCs, in addition to adipocytes, which binds to receptors in the hypothalamus to induce feelings of satiety [74]. Leptin also plays a major role in glycemic control by directly increasing insulin secretion, increasing insulin receptor sensitivity, and improving glucose uptake in peripheral tissues via an additional insulin-independent mechanism [75]. In animal models, leptin administration successfully treats T1DM and T2DM by inhibiting the hypothalamic–pituitary–adrenal axis, leading to increased lipolysis and decreased gluconeogenesis [76]. Leptin administration in both mice and humans has been utilized to decrease food intake, ultimately resulting in weight loss [77]. To investigate how diabetes alters BM-MSCs’ secretion of leptin in vitro, Madhira et al. analyzed BM-MSCs from diabetic rats and compared them to lean controls. It was observed that leptin secretion in diabetic BM-MSCs was lower than in their lean counterparts and the number of preadipocytes was significantly increased in the diabetic group (p < 0.05). The authors attributed the findings to BM-MSC functional impairment due to the long-standing exposure to the diabetic environment within the rats. These terminal changes due to diabetic pathology, also described as the “disease memory” of the BM-MSCs, contribute to the altered differentiation and may be due to leptin resistance or a defective leptin receptor [59]. In summary, leptin is a crucial regulator of insulin and hyperglycemia systemically and its expression is decreased in BM-MSCs due to extended exposure within the diabetic environment.
In conclusion, the diabetic state alters MSC expression of genes involved in the inflammatory process. The metabolic changes seen in diabetes, such as the impairment of glucose metabolism, results in increased fatty acid oxidation and metabolic inflammation. Furthermore, altered metabolism seen in diabetic states also activates pathways that lead to the dysregulation of inflammatory cytokines, such as TNF-α, adiponectin, and leptin, all of which can directly or indirectly stimulate additional insulin resistance. Ultimately, a positive feedback of insulin resistance and inflammation occurs in diabetes, with MSCs significantly contributing to the changes in the microenvironment that mediate these processes.
Oxidative stress markers
The pathogenesis of diabetes involves oxidative stressors [78] that contribute to complications of the disease, such as diabetic nephropathy and cardiomyopathy [71, 72, 79]. While the presence of oxidative stress in diabetics is well established, the effects on MSCs has not been clearly delineated. Under normal non-disease conditions, MSC cell populations are sensitive to oxidative stress and display some antioxidant properties of their own [80]. Chronic hyperglycemia alters normal mitochondrial metabolic processes and protein kinase C function, which then leads to increased formation of reactive oxygen species (ROS), as well as reduced levels of glutathione, an antioxidant necessary for ROS scavenging [81,82,83]. As a result, oxidative stressors are present at much higher levels in diabetes and cannot be effectively managed by the body’s normal antioxidant mechanisms. In this section we will review the effects of oxidative stress on diabetic MSC function.
In order delineate if diabetic MSCs contribute to oxidative stress, Ali et al. isolated Wharton jelly-derived MSCs and treated them with serum from either Type 2 diabetics or non-diabetic patients. They observed that MSCs exposed to diabetic serum possessed increased malondialdehyde levels, an indicator for oxidative stress (p < 0.001). In addition, MSCs treated with diabetic serum had decreased activity of antioxidants enzymes, superoxide dismutase and glutathione reductase (p < 0.01 and p < 0.001, respectively) [84]. Another group, Yan et al., investigated the source and impact of oxidative stress in T2DM mouse MSCs [85]. They cultured BM-MSCs from control and diabetic mice and subsequently assayed for 2,7-dichlorofluorescein diacetate (DCF), a marker for oxidative stress, and Nox4, a gene hypothesized to be a cause of oxidative stress in diabetics [85, 86]. As expected, diabetic MSCs demonstrated a greater than fivefold increase in DCF and a 1.5-fold increase in Nox4 when compared to controls (p < 0.05). To confirm the effect of Nox4 expression on the increased oxidative stress in diabetic MSCs, Yan et al. pretreated diabetic MSCs with Nox4 siRNA and discovered that the knockdown of Nox4 restored DCF levels to that of the WT and a Cy3 siRNA control condition (p < 0.05) [85]. Taken together, these findings indicate that diabetic MSCs, unlike non-diseased MSCs, contribute to oxidative stress by both production of oxidative stressors and reduction of key antioxidant enzymes.
The production of ROS, in part by diabetic MSCs and other dysfunctional processes in diabetes, results in the formation of advanced glycation end products (AGEs) [87]. AGEs are formed when glucose is added to cytosolic proteins through a process known as glycation [88]. As a consequence of AGE accumulation, vascular compromise occurs in diabetic tissue including the retina and kidney glomeruli [5]. AGEs also interact with specific surface receptor proteins, such as scavenger receptors and the receptor for advanced glycation end products (RAGEs) [89]. By binding to their respective receptors, AGEs can alter intracellular signaling pathways and interfere with normal MSC functions [90, 91]. Researchers have found that both AGEs and ROS production were significantly increased in diabetic BM-MSCs and in diabetic tissue (p < 0.01 and p < 0.05, respectively) [92, 93]. Bone marrow-derived MSCs isolated from streptozotocin-induced diabetic rats, for example, have been shown to have an almost 90% increase in RAGE expression when compared to MSCs from control rats (p < 0.01) [92]. When AGEs interact with RAGEs, pro-inflammatory processes are initiated stimulating further increases in ROS and nitric oxide production, exacerbating oxidative stress in a positive feedback loop. More importantly, AGEs have been shown to affect cell surface receptors on MSCs influencing them to differentiate into adipocytes [16, 83, 85].
In conclusion, altered mitochondrial metabolism in diabetes plays a major role in the formation of ROS and oxidative stress. The hyperglycemic environment of diabetes creates AGEs from increased systemic ROS, and the RAGE surface receptors on MSCs interact with AGEs to trigger additional oxidative stress and nitric oxide formation. Normal MSC antioxidant properties are impaired, but oxidative stress can be managed via antioxidant supplementation [85]. The increased oxidative stress present in the diabetic state alters the function of MSCs. These changes ultimately result in altered differentiation, decreased proliferation and increased apoptosis, all of which will be described in greater detail in the following section.
Impaired differentiation and decreased proliferation
Many of the complications that arise in diabetes as a result of MSC dysfunction are due to the impaired differentiation and proliferation of MSCs. Normal MSCs have the potential to differentiate into a variety of cell types, most notably osteoblasts, chondroblasts, and adipocytes [94]. However, disruptions in the balance of MSC differentiation patterns can lead to various pathologies and downstream effects seen in diabetes [95].
To investigate the proliferation rates of diabetic MSCs, Kim et al. isolated diabetic BM-MSCs and compared them to BM-MSCs derived from healthy rats. Cell counts were obtained on a daily basis for 12 days and a significant decrease in MSC proliferation was observed beginning on day 7 in the diabetic MSC group (p < 0.05). The difference in proliferation continued until day 12, resulting in a twofold decrease in cell number in diabetic MSCs compared to normal controls (p < 0.001) by the last day of the study [16]. In addition to exhibiting reduced proliferation rates, the diabetic MSCs also showed an increased preference to differentiate into adipocytes. Additionally, ADSCs have also been shown to display an increased tendency to differentiate into adipocytes in diabetic states [34]. Altogether, this data suggests that diabetes impairs the overall proliferative capacity of MSCs and promotes their disproportionate differentiation into adipocytes, which may contribute to disease burden [16, 35].
Several in vitro studies have investigated the process by which diabetic MSCs undergo adipogenesis to a greater degree than normal MSCs [16, 85, 96]. To elucidate the mechanisms behind reduced multipotency in diabetic MSCs, Yan et al. compared the differentiation patterns of MSCs from diabetic mice and healthy controls in adipogenic media for 72 h [71]. The diabetic MSCs demonstrated a fourfold increase in adipocyte differentiation (p < 0.05), strongly suggesting that the diabetic state alone influences MSC differentiation patterns towards adipogenesis. This group suggested that the increased tendency toward adipocyte differentiation of MSCs was related to oxidative stress in the diabetic environment. They tested this hypothesis by pre-treating diabetic MSCs with the antioxidant N-acetylcysteine (NAC), before culturing the cells [85]. For the diabetic MSCs, NAC pre-treatment successfully reversed the adipogenic influence on differentiation observed in the diabetes group [71]. While the exact cellular mechanisms involved in the process require further investigation, these results provide preliminary evidence supporting the idea that increased oxidative stress from the diabetic environment plays an important role in adipogenic differentiation of diabetic MSCs.
Although an increase in adipogenic differentiation of diabetic MSCs is consistently evident throughout the literature, Barbagallo et al. went one step further to show that once matured, the resulting adipogenic cells were dysfunctional and did not possess the full gene profile of typical adipocytes [97]. These results are consistent with the adipose tissue dysfunction that is commonly seen in diabetic states. Diabetic MSCs show increased adipogenesis, but it is possible that the mature cells that result, consistently show disrupted cellular pathways resulting in the dysfunction of the adipose tissue.
As mentioned previously, the presence of oxidative stress in a high glucose environment leads to the formation of AGEs, which can bind to the scavenger receptor on the surface of MSCs [98]. It is believed that AGEs may also have a role in stimulating MSC differentiation into adipocytes in diabetes. In an experiment performed by Lee et al., MSCs and preadipocytes were isolated from the SVF of high-fat diet (HFD) fed mice, a model for early Type 2 diabetes and impaired glucose tolerance [99], to measure gene expression of the scavenger receptor and other associated proteins. Researchers found a greater expression of both scavenger receptor class A member 5 (SCARA5), and preadipocyte factor 1 (pref-1) in both the MSCs and preadipocytes of the HFD mice (p < 0.05) relative to healthy controls. While the increase in preadipocyte commitment induced by the HFD condition was expected, the concurrent increase in SCARA5 expression alluded to an association between SCARA5 and adipocyte differentiation. To test this hypothesis, Lee et al. knocked down SCARA5 via RNA interference in preadipocyte cells from the HFD mice. This approach successfully decreased adipocyte differentiation, while also significantly reducing the expression of key adipogenic markers PPARg and C/EPBa measured at 48 h (p < 0.05). To further evaluate the role of the SCARA5 receptor in the induction of adipogenesis, Lee et al. created a SCARA5-overexpressing murine stem cell virus (MSCV) and transfected the plasmid into normal MSC cells. MSC cells transfected with the MSCV containing SCARA5 showed enhanced adipocyte differentiation and significantly elevated levels of triglyceride content compared to MSCs transfected with a control MSCV plasmid (p < 0.05) [100]. These results indicate that SCARA5 expression and activity play critical roles in both early MSC differentiation into preadipocytes and the terminal differentiation of preadipocytes into functional adipocytes, which appears to be dysfunctional in the setting of diabetes.
The increased tendency of diabetic MSCs to differentiate into adipocytes is often coupled with a simultaneous impairment in their ability to differentiation into osteoblasts. It is well known that diabetics are at an increased risk for bone fractures and osteoporosis, which some researchers attribute to impaired osteogenic differentiation of diabetic MSCs [8, 101]. In one study performed by Silva et al., BM-MSCs from both streptozotocin induced diabetic and control rats were cultured for 12 days in osteogenic media. Subsequently, qPCR gene analysis was performed at days 5 and 12, which showed that the diabetic BM-MSC cultures demonstrated significantly reduced expression of two osteogenic markers, alkaline phosphate and runt-related transcription factor-2 (RUNX2, p ≤ 0.05), indicating that osteogenic differentiation potential was compromised in the diabetic state. Furthermore, Silva et al. cultured diabetic and healthy control MSCs and assayed DNA content to estimate cell proliferation on days 1, 5, 8, and 12. The diabetic MSC cultures displayed significantly reduced cell numbers on days 8 and 12 when compared to controls, suggesting that overall MSC proliferation is impaired in the diabetic state (p < 0.05). To further elucidate the mechanism behind this finding, an assay measuring the activity of the pro-apoptotic factor, caspase-3, was performed that showed increased apoptosis in the diabetic MSCs on days 8 and 12 (p < 0.05) [102]. Similar findings have been observed in human MSCs cultured from adipose tissue derived from diabetic patients [103]. When cultured in osteogenic media, BM-MSCs exhibit reduced differentiation and increased apoptosis, which may in part explain the increased risk of bone fracture and impaired bone repair seen in diabetes [104, 105].
Attention has recently shifted to elucidate the mechanism by which reduced osteogenic potential of diabetic MSCs occurs. First hyperglycemia, which is the hallmark of diabetes, has been shown to significantly decrease the osteogenic differentiation potential of MSC in vitro (p < 0.05) [98]. This reduced potential may also be explained by decreased levels of proteins such as RUNX2, which has been shown to effect activation of downstream osteogenic factors such as osteopontin, osteocalcin, and osteoprotegerin, in the diabetic state [102]. In addition, Wang et al. measured bone morphogenetic protein-2 (BMP-2), an upstream activator of RUNX2, and determined that its expression by MSCs was decreased in the high glucose state. This group also found that the addition of exogenous BMP-2 to diabetic MSCs resulted in normalization of RUNX2 levels and their osteogenic differentiation [106]. These findings suggest that BMP-2 may be a potential target to improve the impaired osteogenic differentiation potential of MSCs in diabetes.
Cramer et al. showed that under increasing glucose concentrations, ADSCs cultured from both diabetic and control patients, demonstrated progressively lower levels of mitosis as well as increased levels of apoptosis. The decreased proliferation was confirmed by violet staining followed by cell counting. Increased apoptosis was also confirmed via an apoptosis assay, where flow cytometry was performed to detect 7-amino-actinomycin D (7-AAD), which was observed in high glucose concentrations and exaggerated in diabetic MSCs versus their control counterparts. The results discussed support the idea that these cells display “disease memory”—a notion theorizing that cells are terminally altered by their exposure to a disease state. The high glucose environment also induces senescence, visualized through B-galactosidase staining, in both healthy and diseased cells, but diabetic MSCs underwent cellular senescence at a significantly higher rate (p < 0.05) [107]. In another study, Gu et al. compared the proliferation and apoptotic rates of BM-MSCs from non-obese diabetic (NOD) mice and WT controls and determined that BM-MSCs from NOD mice displayed significantly decreased proliferation (p < 0.01) and increased apoptosis (p < 0.05), consistent with previous literature [108, 109]. Furthermore, Gu et al. went on to demonstrate that the changes in proliferative and apoptotic rates were associated with p21 expression, which led them to believe that these changes were due alterations in the NF-κB-p53/p21 pathway. To test this hypothesis, the researchers transfected the BM-MSCs from NOD mice with p21 siRNA. The knockdown of p21 expression significantly increased proliferation rates (p < 0.05) and decreased apoptotic rates (p < 0.05), suggesting that p21 expression plays a role in the altered proliferation and apoptosis seen in NOD mice [108].
Overall, MSCs in high glucose environments show signs of increased apoptosis, and impairment in proliferation and differentiation, which is most notable in that it predetermines the preference of MSCs to differentiate into adipocytes over chondroblasts or osteoblasts.
Discussion
Perturbations in MSC-mediated angiogenesis lead to several complications, most notably diabetic nephropathy and chronic foot ulcers as a result of delayed and impaired wound healing, which affect 40% and 15% of the diabetic population respectively [86, 100]. The imbalance of VEGF seen in different organ systems poses a complex challenge in the treatment of diabetic complications. For example, a therapy that increases levels of VEGF systemically could ameliorate diabetic cardiomyopathy, while exacerbating diabetic retinopathy [40, 110]. Targeted therapy, such as altering local levels of VEGF in the setting of diabetic retinopathy is feasible [111], but also impractical to control the numerous systemic changes in VEGF at each site individually. The levels of VEGF and changes in angiogenesis are both directly proportional to AKT1 [44] and eNOS levels [16, 85], two downstream targets of VEGF, supporting the notion that angiogenic changes are most likely specific to the VEGF-AKT1–eNOS pathway. Future studies should aim to identify the pathways and genes involved with alterations in angiogenesis at each individual tissue affected.
A systemic increase in pro-inflammatory cytokines mediates the phenotype of chronic inflammation seen in diabetics. There has been much promise with direct inhibition of both TNF-α and the IL class of cytokines, in reducing inflammation in diabetics [66, 112]. Decreases in MSC secretion of adiponectin ultimately lead to reduced MSC migration, which could explain why diabetics have impaired healing at sites of tissue damage. While adiponectin-based therapy has shown some promise in improving MSC migration, wound healing, and bone formation, additional research is needed to determine if adiponectin treatment is specifically appropriate for diabetic patients [72]. Systemic levels of leptin, another major adipokine, are increased in diabetic patients. The increase is mostly due to the preferred adipocyte differentiation of diabetic MSCs and not to changes in the MSC secretome. By normalizing MSC adipocyte differentiation in diabetics, we may see normalization of leptin levels as well.
Elevated levels of oxidative stress in the diabetic state lead to formation of AGEs, which can be detrimental to surrounding cellular function. Increased AGE formation is coupled with increased formation of AGE receptors, including the RAGE and the scavenger receptors, both of which are found on the surface of MSCs. Lee et al. determined that knocking down the scavenger receptor, SCARA5, could effectively impair differentiation of preadipocytes into adipocytes [100], however, future research is necessary to understand the ultimate fate of these cells. It is possible that these cells can de-differentiate into MSCs, and therefore be used to ameliorate diabetic complications. At the moment, oxidative stress can be effectively ameliorated through dietary supplementation of cysteine and glycine [113].
Decreased bone density is another serious sequela of diabetes and a thorough understanding of the osteogenic processes is necessary to prevent complications, such as hip fractures. It appears that the compounding changes in the microenvironment of diabetes foster cellular pathways that decrease osteogenesis and increase adipogenesis, limiting the multipotency of diabetic MSCs. The classic T2DM phenotype includes obesity and altered bone metabolism; both of these characteristics can be explained by an increased tendency of MSCs to undergo adipogenesis over osteogenesis under diabetic conditions. Moreover, the changes to the aforementioned adipocytokines such as leptin and adiponectin compound the effects of the impaired multipotency seen in diabetic MSCs and result in an increased total fat mass [80, 85].
The era of big data and personalized medicine will likely uncover treatment strategies and approaches not previously possible. One such approach was demonstrated by a group at Stanford University, who employed single-cell transcriptional analysis to uncover an MSC phenotype critical to diabetic wound healing. With the assistance of high-throughput microfluidics and advanced mathematical modeling, the group tested 386 surface markers and uncovered that a specific MSC subtype, DPP4+/CD55+, was critical to wound healing in a murine model of diabetes, and that this subtype is greatly reduced in the adipose stores of the human diabetic population [114].
One potential therapeutic approach currently being explored by The Center for Tissue Engineering at UC Irvine involves the use of mechanical shear forces to modulate desired stem cell phenotypes in diseased tissue. For example, liposuction aspirate was collected from diabetic individuals undergoing routine procedures at the Veterans Affairs Long Beach Health Care System (n = 3) using a 3 mm cannula. Each adipose sample was then split into two groups. The control group was adipose tissue that was left unprocessed. The experimental group was then subjected to intersyringe processing that exerts a maximal shear force of 3326 dyne/cm2 as previously described [115]. After isolation of the SVF using standard collagenase techniques [94] and flow cytometric analysis, we have indeed observed an increase in the critical DPP4+/CD55 + MSC subpopulation described by Rennert et al. (Fig. 2, p = 0.001). We will soon begin testing the therapeutic potential of this methodology in a murine model of diabetes.

Histogram representing phenotypic change in DPP4+/CD55 + expression on human diabetic MSCs after application of mechanical shear stress
One limitation of our review worth noting is the absence of a section on the paracrine mechanisms of MSCs and ADSCs. It is now understood that stem cells exert a majority of their effects through paracrine and autocrine mechanisms. Previous studies have focused on the capacity for stem cells to differentiate into diverse cell types, however recent evidence suggests that the therapeutic effects of stem cells are a result of their secreted factors [116]. These processes have not been extensively investigated in the literature and therefore were not thoroughly discussed in an individual section. Additionally, much of the articles reviewed focused on the changes to the angiogenic potential of D-MSCs, with less of an emphasis on the other topics we reviewed. We believe that future studies that include exosome analysis of secreted factors will help elucidate the effects of MSCs in both diabetic and non-diabetic states.
Conclusion
Complications related to diabetic MSC dysfunction contribute to the major pathological changes seen in the growing diabetic population. Regenerative medicine utilizing MSC transplantation has shown great promise in ameliorating multiple diabetic complications, including chronic wound healing and impaired bone healing, primarily by the improved differentiation patterning and secretory capacity brought on by added MSCs [117]. These therapies are especially promising because MSCs are easily accessible and can effectively evade immune responses, which is especially appealing for diabetic patients due to the lack of effective treatment options available when complications arise. Further investigation of the changes in cytokine secretion, oxidative stress competence, and differentiation potential in the diabetic stem cell niche is still necessary to improve future MSC based therapies.
Changes in the MSC microenvironment mediate many of the common diabetic complications. Previous literature has highlighted how altered angiogenesis, cytokine secretion, oxidative stress, and proliferation and differentiation impairment recapitulate these changes. To further understand the secretome of MSCs, additional research needs to elucidate the cellular mechanisms in play. We believe that additional experimental analyses, including novel tools in exosome analysis could help answer some of the questions and advance future endeavors in regenerative and MSC based therapies for diabetics.
References
Centers for Disease Control and Prevention (2017) National Diabetes Statistics Report (2017): estimate of diabetes and its burden in the United States. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf
Federation ID (2017) IDF Diabetes Atlas. International Diabetes Federation, Brussels
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414(6865):813–820. https://doi.org/10.1038/414813a
van de Vyver M (2017) Intrinsic mesenchymal stem cell dysfunction in diabetes mellitus: implications for autologous cell therapy. Stem Cells Dev 26(14):1042–1053. https://doi.org/10.1089/scd.2017.0025
Williams AR, Hare JM (2011) Mesenchymal stem cells: biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res 109(8):923–940. https://doi.org/10.1161/circresaha.111.243147
Mizuno H, Tobita M, Uysal AC (2012) Concise review: adipose-derived stem cells as a novel tool for future regenerative medicine. Stem Cells 30(5):804–810. https://doi.org/10.1002/stem.1076
Nagaishi K, Mizue Y, Chikenji T, Otani M, Nakano M, Konari N, Fujimiya M (2016) Mesenchymal stem cell therapy ameliorates diabetic nephropathy via the paracrine effect of renal trophic factors including exosomes. Sci Rep 6:34842. https://doi.org/10.1038/srep34842
de Paula DRM, Capuano V, Filho DM, Carneiro A, de Oliveira Crema V, de Oliveira LF, Rodrigues ARA, Montano N, da Silva VJD (2017) Biological properties of cardiac mesenchymal stem cells in rats with diabetic cardiomyopathy. Life Sci 188:45–52. https://doi.org/10.1016/j.lfs.2017.08.034
Baban B, Liu JY, Payne S, Abebe W, Yu JC, Mozaffari MS (2016) Status of stem cells in diabetic nephropathy: predictive and preventive potentials. EPMA J 7:21. https://doi.org/10.1186/s13167-016-0070-6
Beltramo E, Lopatina T, Berrone E, Mazzeo A, Iavello A, Camussi G, Porta M (2014) Extracellular vesicles derived from mesenchymal stem cells induce features of diabetic retinopathy in vitro. Acta Diabetol 51(6):1055–1064. https://doi.org/10.1007/s00592-014-0672-1
Parajuli A, Liu C, Li W, Gu X, Lai X, Pei S, Price C, You L, Lu XL, Wang L (2015) Bone’s responses to mechanical loading are impaired in type 1 diabetes. Bone 81:152–160. https://doi.org/10.1016/j.bone.2015.07.012
Thomas SWAD, Lakey JRT, Krishnan R, Banyard DA, Salibian AA, Wirth GA, Toranto J, Paydar K, Evans GRD (2014) Adipose derived stem cells and diabetic wound healing: a promising therapeutic modality. CellR4 2(6):e1309
Kuo YR, Wang CT, Cheng JT, Kao GS, Chiang YC, Wang CJ (2016) Adipose-derived stem cells accelerate diabetic wound healing through the induction of autocrine and paracrine effects. Cell Transpl 25(1):71–81. https://doi.org/10.3727/096368915x687921
Gao D, Gu C, Wu Y, Xie J, Yao B, Li J, Feng C, Wang J, Wu X, Huang S, Fu X (2014) Mesenchymal stromal cells enhance wound healing by ameliorating impaired metabolism in diabetic mice. Cytotherapy 16(11):1467–1475. https://doi.org/10.1016/j.jcyt.2014.05.014
Hoffstad O, Mitra N, Walsh J, Margolis DJ (2015) Diabetes, lower-extremity amputation, and death. Diab Care 38(10):1852–1857. https://doi.org/10.2337/dc15-0536
Kim H, Han JW, Lee JY, Choi YJ, Sohn YD, Song M, Yoon YS (2015) Diabetic mesenchymal stem cells are ineffective for improving limb ischemia due to their impaired angiogenic capability. Cell Transpl 24(8):1571–1584. https://doi.org/10.3727/096368914x682792
Arutyunyan I, Elchaninov A, Makarov A, Fatkhudinov T (2016) Umbilical cord as prospective source for mesenchymal stem cell-based therapy. Stem Cells Int 2016:6901286. https://doi.org/10.1155/2016/6901286
Fernandes M, Valente SG, Sabongi RG, Gomes Dos Santos JB, Leite VM, Ulrich H, Nery AA, da Silva Fernandes MJ (2018) Bone marrow-derived mesenchymal stem cells versus adipose-derived mesenchymal stem cells for peripheral nerve regeneration. Neural Regen Res 13(1):100–104. https://doi.org/10.4103/1673-5374.224378
Lin CS, Xin ZC, Dai J, Lue TF (2013) Commonly used mesenchymal stem cell markers and tracking labels: limitations and challenges. Histol Histopathol 28(9):1109–1116. https://doi.org/10.14670/hh-28.1109
Li CY, Wu XY, Tong JB, Yang XX, Zhao JL, Zheng QF, Zhao GB, Ma ZJ (2015) Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res Ther 6:55. https://doi.org/10.1186/s13287-015-0066-5
Cheng R, Ma JX (2015) Angiogenesis in diabetes and obesity. Rev Endocr Metab Disord 16(1):67–75. https://doi.org/10.1007/s11154-015-9310-7
Risau W, Sariola H, Zerwes HG, Sasse J, Ekblom P, Kemler R, Doetschman T (1988) Vasculogenesis and angiogenesis in embryonic-stem-cell-derived embryoid bodies. Development 102(3):471–478
Rezaie J, Mehranjani MS, Rahbarghazi R, Shariatzadeh MA (2018) Angiogenic and restorative abilities of human mesenchymal stem cells were reduced following treatment with serum from diabetes mellitus type 2 patients. J Cell Biochem 119(1):524–535. https://doi.org/10.1002/jcb.26211
Ribot J, Caliaperoumal G, Paquet J, Boisson-Vidal C, Petite H, Anagnostou F (2017) Type 2 diabetes alters mesenchymal stem cell secretome composition and angiogenic properties. J Cell Mol Med 21(2):349–363. https://doi.org/10.1111/jcmm.12969
Kase S, He S, Sonoda S, Kitamura M, Spee C, Wawrousek E, Ryan SJ, Kannan R, Hinton DR (2010) AlphaB-crystallin regulation of angiogenesis by modulation of VEGF. Blood 115(16):3398–3406. https://doi.org/10.1182/blood-2009-01-197095
Shi Y, Su C, Wang JT, Du B, Dong LJ, Liu AH, Li XR (2015) Temporal and spatial changes in VEGF, alphaA- and alphaB-crystallin expression in a mouse model of oxygen-induced retinopathy. Int J Clin Exp Med 8(3):3349–3359
DiPersio JF (2011) Diabetic stem-cell “mobilopathy”. N Engl J Med 365(26):2536–2538. https://doi.org/10.1056/NEJMcibr1112347
Rezabakhsh A, Cheraghi O, Nourazarian A, Hassanpour M, Kazemi M, Ghaderi S, Faraji E, Rahbarghazi R, Avci CB, Bagca BG, Garjani A (2017) Type 2 diabetes inhibited human mesenchymal stem cells angiogenic response by over-activity of the autophagic pathway. J Cell Biochem 118(6):1518–1530. https://doi.org/10.1002/jcb.25814
Vestweber D (2008) VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol 28(2):223–232. https://doi.org/10.1161/atvbaha.107.158014
Yuan SM (2015) Alpha-smooth muscle actin and ACTA2 gene expressions in vasculopathies. Braz J Cardiovasc Surg 30(6):644–649. https://doi.org/10.5935/1678-9741.20150081
Arnaoutova I, Kleinman HK (2010) In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc 5(4):628–635. https://doi.org/10.1038/nprot.2010.6
Gong J, Meng HB, Hua J, Song ZS, He ZG, Zhou B, Qian MP (2014) The SDF-1/CXCR4 axis regulates migration of transplanted bone marrow mesenchymal stem cells towards the pancreas in rats with acute pancreatitis. Mol Med Rep 9(5):1575–1582. https://doi.org/10.3892/mmr.2014.2053
De Becker A, Riet IV (2016) Homing and migration of mesenchymal stromal cells: how to improve the efficacy of cell therapy? World J Stem Cells 8(3):73–87. https://doi.org/10.4252/wjsc.v8.i3.73
Jumabay M, Moon JH, Yeerna H, Bostrom KI (2015) Effect of diabetes mellitus on adipocyte-derived stem cells in rat. J Cell Physiol 230(11):2821–2828. https://doi.org/10.1002/jcp.25012
Rennert RC, Sorkin M, Januszyk M, Duscher D, Kosaraju R, Chung MT, Lennon J, Radiya-Dixit A, Raghvendra S, Maan ZN, Hu MS, Rajadas J, Rodrigues M, Gurtner GC (2014) Diabetes impairs the angiogenic potential of adipose-derived stem cells by selectively depleting cellular subpopulations. Stem Cell Res Ther 5(3):79. https://doi.org/10.1186/scrt468
Cianfarani F, Toietta G, Di Rocco G, Cesareo E, Zambruno G, Odorisio T (2013) Diabetes impairs adipose tissue-derived stem cell function and efficiency in promoting wound healing. Wound Repair Regen 21(4):545–553. https://doi.org/10.1111/wrr.12051
Jin P, Zhang X, Wu Y, Li L, Yin Q, Zheng L, Zhang H, Sun C (2010) Streptozotocin-induced diabetic rat-derived bone marrow mesenchymal stem cells have impaired abilities in proliferation, paracrine, antiapoptosis, and myogenic differentiation. Transpl Proceed 42(7):2745–2752. https://doi.org/10.1016/j.transproceed.2010.05.145
Nguyen A, Guo J, Banyard DA, Fadavi D, Toranto JD, Wirth GA, Paydar KZ, Evans GR, Widgerow AD (2016) Stromal vascular fraction: a regenerative reality? Part 1: current concepts and review of the literature. J Plast Reconstr Aesthet Surg 69(2):170–179. https://doi.org/10.1016/j.bjps.2015.10.015
Guo J, Nguyen A, Banyard DA, Fadavi D, Toranto JD, Wirth GA, Paydar KZ, Evans GR, Widgerow AD (2016) Stromal vascular fraction: a regenerative reality? Part 2: mechanisms of regenerative action. J Plast Reconstr Aesthet Surg 69 (2):180–188. https://doi.org/10.1016/j.bjps.2015.10.014
Duh E, Aiello LP (1999) Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes 48(10):1899–1906
Gangadaran P, Rajendran RL, Lee HW, Kalimuthu S, Hong CM, Jeong SY, Lee SW, Lee J, Ahn BC (2017) Extracellular vesicles from mesenchymal stem cells activates VEGF receptors and accelerates recovery of hindlimb ischemia. J Controll Release 264:112–126. https://doi.org/10.1016/j.jconrel.2017.08.022
Tahergorabi Z, Khazaei M (2012) Imbalance of angiogenesis in diabetic complications: the mechanisms. Int J Prev Med 3(12):827–838
Xu L, Kanasaki K, Kitada M, Koya D (2012) Diabetic angiopathy and angiogenic defects. Fibrogenes Tissue Repair 5(1):13. https://doi.org/10.1186/1755-1536-5-13
Wirostko B, Wong TY, Simo R (2008) Vascular endothelial growth factor and diabetic complications. Prog Retin Eye Res 27(6):608–621. https://doi.org/10.1016/j.preteyeres.2008.09.002
Merino-Gonzalez C, Zuniga FA, Escudero C, Ormazabal V, Reyes C, Nova-Lamperti E, Salomon C, Aguayo C (2016) Mesenchymal stem cell-derived extracellular vesicles promote angiogenesis: potencial clinical application. Front Physiol 7:24. https://doi.org/10.3389/fphys.2016.00024
Kota SK, Meher LK, Jammula S, Kota SK, Krishna SV, Modi KD (2012) Aberrant angiogenesis: the gateway to diabetic complications. Indian J Endocrinol Metab 16(6):918–930. https://doi.org/10.4103/2230-8210.102992
Endo M, Yanagisawa K, Tsuchida K, Okamoto T, Matsushita T, Higuchi M, Matsuda A, Takeuchi M, Makita Z, Koike T (2001) Increased levels of vascular endothelial growth factor and advanced glycation end products in aqueous humor of patients with diabetic retinopathy. Horm Metab Res 33 (5):317–322. https://doi.org/10.1055/s-2001-15122
Selim KM, Sahan D, Muhittin T, Osman C, Mustafa O (2010) Increased levels of vascular endothelial growth factor in the aqueous humor of patients with diabetic retinopathy. Ind J Ophthalmol 58(5):375–379. https://doi.org/10.4103/0301-4738.67042
Mironidou-Tzouveleki M, Tsartsalis S, Tomos C (2011) Vascular endothelial growth factor (VEGF) in the pathogenesis of diabetic nephropathy of type 1 diabetes mellitus. Curr Drug Targets 12(1):107–114
Tarr JM, Kaul K, Chopra M, Kohner EM, Chibber R (2013) Pathophysiology of diabetic retinopathy. ISRN Ophthalmol. https://doi.org/10.1155/2013/343560
Mazzeo A, Beltramo E, Iavello A, Carpanetto A, Porta M (2015) Molecular mechanisms of extracellular vesicle-induced vessel destabilization in diabetic retinopathy. Acta Diabetol 52(6):1113–1119. https://doi.org/10.1007/s00592-015-0798-9
Kinnaird T, Stabile E, Burnett MS, Lee CW, Barr S, Fuchs S, Epstein SE (2004) Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res 94(5):678–685. https://doi.org/10.1161/01.Res.0000118601.37875.Ac
Lai J, Chen F, Chen J, Ruan G, He M, Chen C, Tang J, Wang DW (2017) Overexpression of decorin promoted angiogenesis in diabetic cardiomyopathy via IGF1R-AKT-VEGF signaling. Sci Rep 7:44473. https://doi.org/10.1038/srep44473
Kim YS, Kwon JS, Hong MH, Kang WS, Jeong HY, Kang HJ, Jeong M, Ahn Y (2013) Restoration of angiogenic capacity of diabetes-insulted mesenchymal stem cells by oxytocin. BMC Cell Biol 14:38. https://doi.org/10.1186/1471-2121-14-38
Danalache BA, Paquin J, Donghao W, Grygorczyk R, Moore JC, Mummery CL, Gutkowska J, Jankowski M (2007) Nitric oxide signaling in oxytocin-mediated cardiomyogenesis. Stem Cells 25(3):679–688. https://doi.org/10.1634/stemcells.2005-0610
Cattaneo MG, Chini B, Vicentini LM (2008) Oxytocin stimulates migration and invasion in human endothelial cells. Br J Pharmacol 153(4):728–736. https://doi.org/10.1038/sj.bjp.0707609
Yuan Y, Shi M, Li L, Liu J, Chen B, Chen Y, An X, Liu S, Luo R, Long D, Zhang W, Newsholme P, Cheng J, Lu Y (2016) Mesenchymal stem cell-conditioned media ameliorate diabetic endothelial dysfunction by improving mitochondrial bioenergetics via the Sirt1/AMPK/PGC-1alpha pathway. Clin Sci 130(23):2181–2198. https://doi.org/10.1042/cs20160235
Khan M, Ali F, Mohsin S, Akhtar S, Mehmood A, Choudhery MS, Khan SN, Riazuddin S (2013) Preconditioning diabetic mesenchymal stem cells with myogenic medium increases their ability to repair diabetic heart. Stem Cell Res Ther 4(3):58. https://doi.org/10.1186/scrt207
Madhira SL, Challa SS, Chalasani M, Nappanveethl G, Bhonde RR, Ajumeera R, Venkatesan V (2012) Promise(s) of mesenchymal stem cells as an in vitro model system to depict pre-diabetic/diabetic milieu in WNIN/GR-Ob mutant rats. PloS ONE 7(10):e48061. https://doi.org/10.1371/journal.pone.0048061
Ranganath SH, Levy O, Inamdar MS, Karp JM (2012) Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 10(3):244–258. https://doi.org/10.1016/j.stem.2012.02.005
Kyurkchiev D, Bochev I, Ivanova-Todorova E, Mourdjeva M, Oreshkova T, Belemezova K, Kyurkchiev S (2014) Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells 6(5):552–570. https://doi.org/10.4252/wjsc.v6.i5.552
van de Vyver M, Niesler C, Myburgh KH, Ferris WF (2016) Delayed wound healing and dysregulation of IL6/STAT3 signalling in MSCs derived from pre-diabetic obese mice. Mol Cell Endocrinol 426:1–10. https://doi.org/10.1016/j.mce.2016.02.003
Nieto-Vazquez I, Fernandez-Veledo S, de Alvaro C, Lorenzo M (2008) Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes 57(12):3211–3221. https://doi.org/10.2337/db07-1062
Moller DE (2000) Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab 11(6):212–217
Qiao YC, Chen YL, Pan YH, Tian F, Xu Y, Zhang XX, Zhao HL (2017) The change of serum tumor necrosis factor alpha in patients with type 1 diabetes mellitus: a systematic review and meta-analysis. PloS ONE 12(4):e0176157. https://doi.org/10.1371/journal.pone.0176157
Ko KI, Coimbra LS, Tian C, Alblowi J, Kayal RA, Einhorn TA, Gerstenfeld LC, Pignolo RJ, Graves DT (2015) Diabetes reduces mesenchymal stem cells in fracture healing through a TNFalpha-mediated mechanism. Diabetologia 58(3):633–642. https://doi.org/10.1007/s00125-014-3470-y
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Investig 112(12):1821–1830. https://doi.org/10.1172/jci19451
Qatanani M, Lazar MA (2007) Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev 21(12):1443–1455. https://doi.org/10.1101/gad.1550907
Oda N, Imamura S, Fujita T, Uchida Y, Inagaki K, Kakizawa H, Hayakawa N, Suzuki A, Takeda J, Horikawa Y, Itoh M (2008) The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism 57(2):268–273. https://doi.org/10.1016/j.metabol.2007.09.011
Aleidi S, Issa A, Bustanji H, Khalil M, Bustanji Y (2015) Adiponectin serum levels correlate with insulin resistance in type 2 diabetic patients. Saudi Pharm J 23(3):250–256. https://doi.org/10.1016/j.jsps.2014.11.011
Duncan BB, Schmidt MI, Pankow JS, Bang H, Couper D, Ballantyne CM, Hoogeveen RC, Heiss G (2004) Adiponectin and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes 53(9):2473–2478
Yu L, Tu Q, Han Q, Zhang L, Sui L, Zheng L, Meng S, Tang Y, Xuan D, Zhang J, Murray D, Shen Q, Cheng J, Kim SH, Dong LQ, Valverde P, Cao X, Chen J (2015) Adiponectin regulates bone marrow mesenchymal stem cell niche through a unique signal transduction pathway: an approach for treating bone disease in diabetes. Stem Cells 33(1):240–252. https://doi.org/10.1002/stem.1844
Zhang B, Liu N, Shi H, Wu H, Gao Y, He H, Gu B, Liu H (2016) High glucose microenvironments inhibit the proliferation and migration of bone mesenchymal stem cells by activating GSK3beta. J Bone Miner Metab 34(2):140–150. https://doi.org/10.1007/s00774-015-0662-6
Huang L, Li C (2000) Leptin: a multifunctional hormone. Cell Res 10(2):81–92. https://doi.org/10.1038/sj.cr.7290038
Meek TH, Morton GJ (2012) Leptin, diabetes, and the brain. Indian J Endocrinol Metab 16(Suppl 3):S534–S542. https://doi.org/10.4103/2230-8210.105568
Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, Rothman DL, Shulman GI (2014) Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Med 20(7):759–763. https://doi.org/10.1038/nm.3579
Flier JS (2012) Hormone resistance in diabetes and obesity: insulin, leptin, and FGF21. Yale J Biol Med 85(3):405–414
Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107(9):1058–1070. https://doi.org/10.1161/circresaha.110.223545
Nigro E, Scudiero O, Monaco ML, Palmieri A, Mazzarella G, Costagliola C, Bianco A, Daniele A (2014) New insight into adiponectin role in obesity and obesity-related diseases. BioMed Res Int 2014:658913. https://doi.org/10.1155/2014/658913
Denu RA, Hematti P (2016) Effects of oxidative stress on mesenchymal stem cell biology. Oxid Med Cell Longev 2016:2989076. https://doi.org/10.1155/2016/2989076
Ha H, Hwang IA, Park JH, Lee HB (2008) Role of reactive oxygen species in the pathogenesis of diabetic nephropathy. Diab Res Clin Pract 82(Suppl 1):S42–S45. https://doi.org/10.1016/j.diabres.2008.09.017
Lee HB, Yu MR, Yang Y, Jiang Z, Ha H (2003) Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol 14(8 Suppl 3):S241–S245
Hakki Kalkan I, Suher M (2013) The relationship between the level of glutathione, impairment of glucose metabolism and complications of diabetes mellitus. Pak J Med Sci 29(4):938–942
Ali F, Aziz F, Wajid N (2017) Effect of type 2 diabetic serum on the behavior of Wharton’s jelly-derived mesenchymal stem cells in vitro. Chronic Dis Transl Med 3(2):105–111. https://doi.org/10.1016/j.cdtm.2017.02.006
Yan J, Tie G, Wang S, Messina KE, DiDato S, Guo S, Messina LM (2012) Type 2 diabetes restricts multipotency of mesenchymal stem cells and impairs their capacity to augment postischemic neovascularization in db/db mice. J Am Heart Assoc 1(6):e002238. https://doi.org/10.1161/jaha.112.002238
Schroder K, Wandzioch K, Helmcke I, Brandes RP (2009) Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29 (2):239–245. https://doi.org/10.1161/atvbaha.108.174219
Nowotny K, Jung T, Hohn A, Weber D, Grune T (2015) Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 5(1):194–222. https://doi.org/10.3390/biom5010194
Singh VP, Bali A, Singh N, Jaggi AS (2014) Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol 18(1):1–14. https://doi.org/10.4196/kjpp.2014.18.1.1
Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A (2003) Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J 375(Pt 3):581–592. https://doi.org/10.1042/bj20030763
Xue J, Rai V, Singer D, Chabierski S, Xie J, Reverdatto S, Burz DS, Schmidt AM, Hoffmann R, Shekhtman A (2011) Advanced glycation end product recognition by the receptor for AGEs. Structure 19(5):722–732. https://doi.org/10.1016/j.str.2011.02.013
Aikawa E, Fujita R, Asai M, Kaneda Y, Tamai K (2016) Receptor for advanced glycation end products-mediated signaling impairs the maintenance of bone marrow mesenchymal stromal cells in diabetic model mice. Stem Cells Dev 25(22):1721–1732. https://doi.org/10.1089/scd.2016.0067
Stolzing A, Sellers D, Llewelyn O, Scutt A (2010) Diabetes induced changes in rat mesenchymal stem cells. Cells Tissues Organs 191(6):453–465. https://doi.org/10.1159/000281826
Ogawa N, Yamaguchi T, Yano S, Yamauchi M, Yamamoto M, Sugimoto T (2007) The combination of high glucose and advanced glycation end-products (AGEs) inhibits the mineralization of osteoblastic MC3T3-E1 cells through glucose-induced increase in the receptor for AGEs. Horm Metab Res 39(12):871–875. https://doi.org/10.1055/s-2007-991157
Banyard DA, Salibian AA, Widgerow AD, Evans GR (2015) Implications for human adipose-derived stem cells in plastic surgery. J Cell Mol Med 19(1):21–30. https://doi.org/10.1111/jcmm.12425
Deng X, Xu M, Shen M, Cheng J (2018) Effects of type 2 diabetic serum on proliferation and osteogenic differentiation of mesenchymal stem cells. J Diab Res 2018:5765478. https://doi.org/10.1155/2018/5765478
Moseley KF, Doyle ME, Jan De Beur SM (2018) Diabetic serum from older women increases adipogenic differentiation in mesenchymal stem cells. Endocr Res. https://doi.org/10.1080/07435800.2018.1441868
Barbagallo I, Li Volti G, Galvano F, Tettamanti G, Pluchinotta FR, Bergante S, Vanella L (2017) Diabetic human adipose tissue-derived mesenchymal stem cells fail to differentiate in functional adipocytes. Exp Biol Med 242(10):1079–1085. https://doi.org/10.1177/1535370216681552
Thornalley PJ (1998) Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol Biol 44(7):1013–1023
Winzell MS, Ahren B (2004) The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 53(Suppl 3):S215–S219
Lee H, Lee YJ, Choi H, Seok JW, Yoon BK, Kim D, Han JY, Lee Y, Kim HJ, Kim JW (2017) SCARA5 plays a critical role in the commitment of mesenchymal stem cells to adipogenesis. Sci Rep 7(1):14833. https://doi.org/10.1038/s41598-017-12512-2
Brown ML, Yukata K, Farnsworth CW, Chen DG, Awad H, Hilton MJ, O’Keefe RJ, Xing L, Mooney RA, Zuscik MJ (2014) Delayed fracture healing and increased callus adiposity in a C57BL/6J murine model of obesity-associated type 2 diabetes mellitus. PloS ONE 9(6):e99656. https://doi.org/10.1371/journal.pone.0099656
Silva JC, Sampaio P, Fernandes MH, Gomes PS (2015) The osteogenic priming of mesenchymal stem cells is impaired in experimental diabetes. J Cell Biochem 116(8):1658–1667. https://doi.org/10.1002/jcb.25126
Kornicka K, Houston J, Marycz K (2018) Dysfunction of mesenchymal stem cells isolated from metabolic syndrome and type 2 diabetic patients as result of oxidative stress and autophagy may limit their potential therapeutic use. Stem Cell Rev 14(3):337–345. https://doi.org/10.1007/s12015-018-9809-x
Sundararaghavan V, Mazur MM, Evans B, Liu J, Ebraheim NA (2017) Diabetes and bone health: latest evidence and clinical implications. Ther Adv Musculoskelet Dis 9(3):67–74. https://doi.org/10.1177/1759720x16687480
Wang W, Zhang X, Zheng J, Yang J (2010) High glucose stimulates adipogenic and inhibits osteogenic differentiation in MG-63 cells through cAMP/protein kinase A/extracellular signal-regulated kinase pathway. Mol Cell Biochem 338(1–2):115–122. https://doi.org/10.1007/s11010-009-0344-6
Wang J, Wang B, Li Y, Wang D, Lingling E, Bai Y, Liu H (2013) High glucose inhibits osteogenic differentiation through the BMP signaling pathway in bone mesenchymal stem cells in mice. EXCLI J 12:584–597
Cramer C, Freisinger E, Jones RK, Slakey DP, Dupin CL, Newsome ER, Alt EU, Izadpanah R (2010) Persistent high glucose concentrations alter the regenerative potential of mesenchymal stem cells. Stem Cells Dev 19(12):1875–1884. https://doi.org/10.1089/scd.2010.0009
Gu Z, Jiang J, Xia Y, Yue X, Yan M, Tao T, Cao X, Da Z, Liu H, Liu H, Miao Y, Li L, Wang Z (2013) p21 is associated with the proliferation and apoptosis of bone marrow-derived mesenchymal stem cells from non-obese diabetic mice. Exp Clin Endocrinol Diab 121(10):607–613. https://doi.org/10.1055/s-0033-1354380
Meng Y, Ji J, Tan W, Guo G, Xia Y, Cheng C, Gu Z, Wang Z (2016) Involvement of autophagy in the procedure of endoplasmic reticulum stress introduced apoptosis in bone marrow mesenchymal stem cells from nonobese diabetic mice. Cell Biochem Funct 34(1):25–33. https://doi.org/10.1002/cbf.3161
Chou E, Suzuma I, Way KJ, Opland D, Clermont AC, Naruse K, Suzuma K, Bowling NL, Vlahos CJ, Aiello LP, King GL (2002) Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: a possible explanation for impaired collateral formation in cardiac tissue. Circulation 105(3):373–379
Gupta N, Mansoor S, Sharma A, Sapkal A, Sheth J, Falatoonzadeh P, Kuppermann B, Kenney M (2013) Diabetic retinopathy and VEGF. Open Ophthalmol J 7:4–10. https://doi.org/10.2174/1874364101307010004
Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T (2009) Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diab Care 32(9):1663–1668. https://doi.org/10.2337/dc09-0533
Sekhar RV, McKay SV, Patel SG, Guthikonda AP, Reddy VT, Balasubramanyam A, Jahoor F (2011) Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diab Care 34(1):162–167. https://doi.org/10.2337/dc10-1006
Rennert RC, Januszyk M, Sorkin M, Rodrigues M, Maan ZN, Duscher D, Whittam AJ, Kosaraju R, Chung MT, Paik K, Li AY, Findlay M, Glotzbach JP, Butte AJ, Gurtner GC (2016) Microfluidic single-cell transcriptional analysis rationally identifies novel surface marker profiles to enhance cell-based therapies. Nat Commun 7:11945. https://doi.org/10.1038/ncomms11945
Banyard DA, Sarantopoulos CN, Borovikova AA, Qiu X, Wirth GA, Paydar KZ, Haun JB, Evans GR, Widgerow AD (2016) Phenotypic analysis of stromal vascular fraction after mechanical shear reveals stress-induced progenitor populations. Plast Reconstr Surg 138(2):237e–247e. https://doi.org/10.1097/prs.0000000000002356
Vizoso FJ, Eiro N, Cid S, Schneider J, Perez-Fernandez R (2017) Mesenchymal stem cell secretome: toward cell-free therapeutic strategies in regenerative medicine. Int J Mol Sci 18 (9). https://doi.org/10.3390/ijms18091852
Mulder GD, Lee DK, Jeppesen NS (2012) Comprehensive review of the clinical application of autologous mesenchymal stem cells in the treatment of chronic wounds and diabetic bone healing. Int Wound J 9(6):595–600. https://doi.org/10.1111/j.1742-481X.2011.00922.x
Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6(7):e1000097. https://doi.org/10.1371/journal.pmed1000097
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors whose names are listed immediately above certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Rights and permissions
About this article

Cite this article
Fijany, A., Sayadi, L.R., Khoshab, N. et al. Mesenchymal stem cell dysfunction in diabetes. Mol Biol Rep 46, 1459–1475 (2019). https://doi.org/10.1007/s11033-018-4516-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-4516-x