
Abstract
Autosomal recessive osteopetrosis is a severe fatal disorder with an average incidence of around 1:250,000. It is diagnosed soon after birth or within the 1st year of life with severe symptoms of abnormal bone remodelling. This study was aimed to identify the underlying genetic cause of the disease in a Pakistani family segregating infantile malignant osteopetrosis in autosomal recessive pattern. Whole exome sequencing of the proband was performed using the 51 Mb SureSelect V4 library kit and sequenced using the Illumina HiSeq2500 sequencing system. The reads were analysed using standard bioinformatic data analysis pipeline. The genotype of candidate variants was confirmed in the proband and his normal parents by Sanger sequencing. Two novel homozygous missense variants were found in the same codon 204 of CLCN7 NM_001287.5:c.[610A>T;612C>G] predicting p.(Ser204Trp) variant in the protein. Sanger sequencing and RFLP assay verified that both these variants were heterozygous in the unaffected parents. Moreover, these variants were not detected in the unrelated healthy Pakistani subjects (200 chromosomes), ExAC, dbSNP, or the 1000 Genomes Project data. Multiple bioinformatics tools unanimously predicted the p.(Ser204Trp) variant as deleterious. CLCN7 mutation p.(Ser204Trp) is the likely cause of the osteopetrosis disease in the Pakistani family. This study expands the restricted spectrum of CLCN7 mutations associated with infantile malignant osteopetrosis and indicates clinical significance of whole exome sequencing in the diagnosis of clinically and genetically heterogenous osteopetrosis phenotype. These data should be helpful in the improved genetic counselling, carrier identification and prenatal diagnosis of the affected family.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteopetrosis comprises a clinically and genetically heterogeneous group of conditions that share the hallmark of increased bone density on radiographs. The increase in bone density results from abnormalities in osteoclast differentiation or function [1]. Symptoms and severity can vary from asymptomatic to fatal in infancy. Several major types of osteopetrosis have been described, which are usually distinguished by their pattern of inheritance: autosomal dominant, autosomal recessive, or X-linked. An intermediate group also exists in which the defect can be inherited as either a recessive or a dominant trait. The prevalence of autosomal dominant osteopetrosis has been estimated at up to 1:20,000. Autosomal recessive osteopetrosis is less common, with a prevalence around 1:250,000 [2]. Mutations in at least ten genes have been linked to different forms of osteopetrosis accounting for 70% of all cases and therefore the search continues for the genes responsible for the remainder [3].
Autosomal recessive infantile malignant osteopetrosis is the most severe form of the disease. It is diagnosed soon after birth or within the 1st year of life with severe symptoms of abnormal bone remodelling, including significant hematologic abnormalities with bone marrow failure and extramedullary hematopoiesis, resulting in hepatosplenomegaly, a characteristic macrocephaly with frontal bossing, exophthalmos, bone fractures and failure to thrive [4]. Mutations in the TCIRG1, CLCN7, OSTM1 and SNX10 genes have been reported as the underlying cause of this fatal disease. However, a milder form of the autosomal recessive osteopetrosis is caused by TNFSF11 mutations and an intermediate form by PLEKHM1 mutations. Autosomal recessive osteopetrosis may also be associated with hypogammaglobulinemia or renal tubular acidosis caused by mutations in the TNFRSF11A and CA2 genes, respectively [5]. In the current study we ascertained a Pakistani family affected with autosomal recessive infantile malignant osteopetrosis through whole exome sequencing and identified two novel homozygous missense variants in CLCN7 as the likely cause of the disease.
Materials and methods
Family and pedigree information
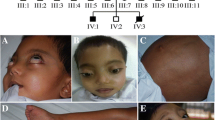
The family affected with autosomal recessive malignant infantile osteopetrosis was recruited from Lady Reading Hospital Peshawar. The family originated from a remote village of Landi Kotal, a town in the Federally Administered Tribal Areas of Pakistan. There were two affected individuals (IV-1, IV-2) in the family born to consanguineous parents (Fig. 1). Informed consent was sought from the head of the family (III-3) for the study presented here and for the publication of the identifiable figures and pedigree information. The study was approved by the institutional review board of Quaid-i-Azam University.

Pedigree of the family affected with autosomal recessive malignant infantile osteopetrosis. Both affected individuals died at the age of 12 months. The blood samples were available for the proband (IV-2) and parents (III-1 and III-2)
Whole exome sequencing
Genomic DNA was extracted from peripheral blood of the affected individual (IV-2) and parents (III-2, III-3) using a QIAamp Blood DNA Mini Kit (Qiagen, Hilden, Germany). The affected individual (IV-1) was not available as she had died before the start of the study. Genomic DNA of the affected individual (IV-2) was subjected to whole exome paired-end sequencing analysis with 100X coverage by generating 51 Mb SureSelect V4 libraries (Agilent Technologies, USA). Shearing, hybridization using RNA-based Library Baits, Target capture and bridged amplification were subsequently carried out. The imaging and extension was achieved in automated cycles by mounting the clusters-bearing flow cell onto the Illumina HiSeq 2000/2500 sequencer (Illumina, San Diego, CA, USA). The raw reads were processed to measure the base quality and recalibration. It was achieved using Genome Analysis Toolkit. In addition, variant score call, small deletions and insertion evaluation was also attained using GATK3.v4. Further evaluation included marking duplicates using PicardTool.
Variant filtering
Because the family presented with an autosomal recessive mode of inheritance, we filtered candidate variants using the following criteria: (1) homozygous or compound heterozygous in the affected individual, and heterozygous in obligate carriers for the mutant allele; (2) present in coding exons or splice junctions; (3) non-synonymous, frame-shift, gain/loss of stop codons; (4) having a functional effect, e.g., predicted to be pathogenic or deleterious by in silico prediction software including Polyphen-2 and SIFT; (5) minor allele frequency < 0.01 in the 1000 Genomes Project data (http://www.1000genomes.org/); and (6) not present in our in-house exome sequence data obtained from 30 unrelated healthy individuals from local Pakistani population.
Sanger sequencing and RFLP assay
Sanger sequencing was used to validate the potentially causative variants (NM_001287.5: c.610A>T and c.612C>G) in the affected individual (IV-2), his parents and 200 ethnically matched healthy control chromosomes. The reference sequence of CLCN7 (ENSG00000103249) was obtained from the Ensembl genome browser (http://www.ensembl.org/). The primer sequences (upstream 5′-TCCCAGGGCTCTGACTGTGT-3′ and downstream 5′-CGTAGGTAGGGACACCCGCC-3′) for PCR amplification covering c.610A>T and c.612C>G in exon 7 of CLCN7 were designed manually. The amplicon was amplified in each sample and screened by DNA cycle sequencing using a Big Dye Terminator v3.1 Cycle Sequencing Kit in an ABI 3730 Genetic Analyzer (Applied Biosytems, Foster City, CA). Sequence variants were identified via BioEdit sequence alignment editor version 6.0.7 (http://www.mbio.ncsu.edu/bioedit/bioedit.html).
The identified variants obliterated an MspA1I site, so its presence was assayed by PCR amplification of CLCN7 exon 7 from genomic DNA, digestion of the product with MspA1I, and separation of the resulting fragments by agarose gel electrophoresis with direct visualization using ethidium bromide.
In silico analysis
PredictProtein was used for protein structure prediction and sequence analysis (http://www.predictprotein.org). The 3-D CLCN7 structure (P51798) was superimposed with mutated (p.204S > W) CLCN7 generated through I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). The visualization and analysis of molecular structures was performed using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/). The PROVEAN (http://provean.jcvi.org), SIFT (http://sift.jcvi.org/), and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) were used to predict the deleterious effects of the variant p.(Ser204Trp) on protein function.
Results
Clinical history and phenotype
The affected children IV-1 and IV-2 were born to a consanguineous couple in Peshawar city of Khyber Pakhtunkhwa Province of Pakistan (Fig. 1). The proband IV-2 (Fig. 2) was delivered preterm by C-section at maternity home and was hospitalized for 7 days for incubator care. At the age of 4 months, he was presented in the Lady Reading Hospital Peshawar with episodes of body stiffness. His physical examination revealed dysmorphic facies and protruded eye balls, normal precordium, normal heart sounds, slightly enlarged liver and spleen and clear chest. The femoral pulse was palpable (90/min) and regular with normal volume. The child was bottle-fed and vaccinated at the time of presentation. The chest X-rays revealed normal size cardiac shadow and increased density of bones (sclerosis) (Fig. 2). The computed tomography scan of the brain showed dilated external and internal CSF spaces indicating brain atrophy. Eye examination revealed bilateral optic atrophy. The echocardiography (ECG) showed normal structure and function of the heart. The echoencephalography (EEG) was also normal. The findings of biochemical investigations were as follows [reference ranges in parenthesis]: random blood glucose 97 mg/dL [65–165], urea 14 mg/dL [18–45], creatinine 0.4 mg/dL [0.42–1.06], uric acid 1.7 mg/dL [1.7–5.1], CK-Nac 309 U/L [41–330], CK-MB 110 U/L [5–20], total calcium 8.7 mg/dL [8.5–10.5], phosphorous 3.7 mg/dL [2.5–7.5], total bilirubin 0.4 mg/dL [up to 1.0], ALT 29 U/L [up to 40], total proteins 5.9 mg/dL [5.7–8.0], albumin 4.6 mg/dL [3.5–5.2], C3 level 106 mg/dL [70–196], C4 level 15 mg/dL [13–38], CRP level 3.8 mg/dL [< 6.0], ferritin level 150 mg/dL [10–160], CA 125-II 4.5 U/mL [< 35], total IgE 01 IU/mL [< 150], haemoglobin 10.4 g/dL [11.5–13.5], and alkaline phosphatase, 1295 U/L [up to 645]. He died of pneumonia at the age of 12 months after repeated hospitalization like her elder sister (IV-1) who also died with osteopetrosis at the age of 12 months. Both parents (III-2 and III-3) of the patients were healthy and had no symptoms of osteopetrosis or other diseases.

a Phenotypic appearance and b chest X-rays of the proband (IV-2) showing normal size cardiac shadow and increased bone density
Whole exome sequencing
Whole exome sequencing data were analysed for the identification of a causative variant. On average, 99% of bases had a phred score higher than 20; total number of bases in the reads were 5.9 Gb; and average depth of the target region was 117. Out of 73,202 variants 18,948 were missense, nonsense, frameshift, indels or splice site variants.
The list of identified variants was narrowed down after excluding those that did not show a recessive mode of inheritance, those with a minor allele frequency > 0.01 based on the 1000 Genomes Project data, those causing neutral or no functional effects based on in silico analyses to predict pathogenic effect, and those that were not present in our in-house exome sequence data of an unrelated healthy Pakistani population (n = 30). As a result, four homozygous missense variants remained as potential candidates: NM_001287.5:c.[610A>T;612C>G], NM_CFHR3:c.[542C>A] and NM_ HERC1:c.14491C>T. Sanger sequencing and RFLP assay verified two variants NM_001287.5:c.[610A>T;612C>G] in the CLCN7 as homozygous in the affected individual and heterozygous in the unaffected parents (Fig. 3a). The candidate variants in CFHR3 and HERC1 did not co-segregate with the disease phenotype in autosomal recessive pattern of inheritance.

a Sequencing chromatograms of exon 7 of CLCN7 (NM_001287.5) amplified from the gDNA of the proband (IV-2) (top), the heterozygous parents (middle) and unrelated healthy controls (bottom). Arrows indicate positions of two missense variants (c.610A>T and c.612C>G) predicting p.(Ser204Trp) variation in the protein sequence. b Protein sequence alignment of CLCN7 from different vertebrate species indicating highly conserved residue p.204Ser (boxed). c The superimposition of 3D structure of CLCN7 (P51798) with mutated p.(Ser204Trp) protein generated through I-TASSER showed remarkable difference. The visualization and analysis of molecular structures was performed using UCSF Chimera
The CLCN7 variants c.610A>T and c.612C>G lie in the same codon (1st and 3rd position, respectively) in exon 7 predicting the substitution of serine with tryptophan at amino acid position 204 p.(Ser204Trp). We screened ethnically matched healthy control chromosomes (n = 200) to ensure that the variants did not represent neutral polymorphisms in this population, and verified that these were not present outside the family. These variants were also not listed in Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/), dbSNP (https://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genomes Project data (http://www.1000genomes.org/), Human Gene Mutation Database (HGMD, http://www.hgmd.org/) or ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/).
In silico analysis
In multiple bioinformatics tools including PROVEAN, SIFT and PolyPhen-2, the CLCN7 variant p.(Ser204Trp) was predicted to be deleterious. The residue p.204Ser is highly conserved among vertebrates (Fig. 3b) and represents one of the three chloride binding sites in the predicted CLCN7 protein structure based on similarity [6]. Moreover, it lies in a region of highly conserved residues of CLCN7 with ConSurf conservation score 9 on a scale of 0–9. The secondary structure composition predicted through PredictProtein showed that alpha helix content in mutant CLCN7 was reduced from 47.45% (normal) to 45.34% while beta strand percentage increased from 6.71 (normal) to 8.70. The superimposition of wild and mutant CLCN7 protein structures predicted through ITASSER indicated remarkable structural difference (Fig. 3c).
Discussion
We ascertained a Pakistani family affected with autosomal recessive infantile malignant osteopetrosis and identified two novel homozygous missense variants in CLCN7 through whole exome sequencing. The two nucleotide variants lie in the same codon encoding serine at amino acid position 204. Each of the two single nucleotide variants c.610A>T [p.(Ser204Cys)] and c.612C>G [p.(Ser204Arg)] is predicted to be deleterious through SIFT and PolyPhen-2. However, both these variants are homozygous in the patient and heterozygous in the parents indicating their segregation in cis leading to a single amino acid substitution p.(Ser204Trp) in the protein. We suggest p.(Ser204Trp) substitution is the likely cause of the osteopetrosis disease in the family.
CLCN7 encodes the Cl−/H+ exchanger CLC-7 chloride channel of the osteoclast membrane, which is required for acidification of the resorption lacunae. It is primarily localized to the lysosomal compartment, where it interacts with OSTM1 to mediate Cl−/H+ exchange [7]. Mutations in the CLCN7 affect the function of osteoclast-mediated extracellular acidification, resulting in the disturbed dissolution of the bone inorganic matrix and a series of clinical features [3]. Mutations in the CLCN7 are responsible for about 75% of cases of autosomal dominant osteopetrosis, 13% of cases of autosomal recessive osteopetrosis, and all known cases of intermediate autosomal osteopetrosis [8,9,10]. Therefore, CLCN7 mutations database related to infantile malignant osteopetrosis is restricted to only six missense mutations (HGMD). This study expands the mutation spectrum of the CLCN7 underlying infantile malignant osteopetrosis.
Conclusion
The study indicates the clinical significance of molecular diagnosis of clinically and heterogenous osteopetrosis disease through whole exome sequencing. It should help in prenatal diagnosis and improved genetic counselling of the affected family.
References
Stark Z, Savarirayan R (2009) Osteopetrosis. Orphanet J Rare Dis 4:5
Sobacchi C, Villa A, Schulz A, Kornak U (2007) CLCN7-related osteopetrosis. In: Adam MP, Ardinger HH, Pagon RA et al (eds) GeneReviews. University of Washington, Seattle, Seattle (WA), pp 1993–2018
Okamoto N, Kohmoto T, Naruto T, Masuda K, Komori T, Imoto I (2017) Novel CLCN7 compound heterozygous mutations in intermediate autosomal recessive osteopetrosis. Hum Genome Var 4:17036
Kumar KJ, Bandaru K, Prashanth SN, Mamatha S (2013) Malignant infantile osteopetrosis. Indian J Hum Genet 19:90–92
Phadke SR, Fischer B, Gupta N, Ranganath P, Kabra M, Kornak U (2010) Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis. Indian J Med Res 131:508–514
Gaudet P, Michel PA, Zahn-Zabal M, Britan A, Cusin I, Domagalski M, Duek PD, Gateau A, Gleizes A, Hinard V, Rech de Laval V, Lin JJ, Nikitin F, Schaeffer M, Teixeira D, Lane L, Bairoch A (2017) The neXtProt knowledgebase on human proteins: 2017 update. Nucl Acids Res 45:D177–D182
Barvencik F, Kurth I, Koehne T, Stauber T, Zustin J, Tsiakas K, Ludwig CF, Beil FT, Pestka JM, Hahn M, Santer R, Supanchart C, Kornak U, Fattore AD, Jentsch TJ, Teti A, Schulz A, Schinke T, Amling M (2014) CLCN7 and TCIRG1 mutations differentially affect bone matrix mineralization in osteopetrotic individuals. J Bone Miner Res 29:982–991
Campos-Xavier AB, Saraiva JM, Ribeiro LM, Munnich A, Cormier-Daire V (2003) Chloride channel 7 (CLCN7) gene mutations in intermediate autosomal recessive osteopetrosis. Hum Genet 112:186–189
Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M, Andolina M, Flanagan A, Horwitz EM, Mihci E, Notarangelo LD, Ramenghi U, Teti A, Van Hove J, Vujic D, Young T, Albertini A, Orchard PJ, Vezzoni P, Villa A (2003) Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res 18:1740–1747
Pangrazio A, Pusch M, Caldana E, Frattini A, Lanino E, Tamhankar PM, Phadke S, Lopez AG, Orchard P, Mihci E, Abinun M, Wright M, Vettenranta K, Bariae I, Melis D, Tezcan I, Baumann C, Locatelli F, Zecca M, Horwitz E, Mansour LS, Van Roij M, Vezzoni P, Villa A, Sobacchi C (2010) Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: report of 20 novel mutations. Hum Mutat 31:E1071–E1080
Acknowledgements
Muhammad Aman Khan is supported by Higher Education Commission of Pakistan through Indigenous Ph.D. Fellowship Scheme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Khan, M.A., Ullah, A. & Naeem, M. Whole exome sequencing identified two novel homozygous missense variants in the same codon of CLCN7 underlying autosomal recessive infantile malignant osteopetrosis in a Pakistani family. Mol Biol Rep 45, 565–570 (2018). https://doi.org/10.1007/s11033-018-4194-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-4194-8