
Abstract
The presence of a bacterial backbone in conventional eukaryotic expression plasmids may cause undesirable effects by triggering the immune responses in mammals and repression of episomal transgene expression. To avoid these problems, researchers have proposed the use of minicircle DNAs which are episomal vectors that have lost their bacterial backbone using a site-specific recombinase mediated recombination. In the present study, we have constructed a new minicircle DNA vector that carries an enhanced green florescent protein (EGFP) reporter gene using phage ΦC31 integrase-mediated recombination and homing endonuclease ISceI-mediated purification in E. coli. ΦC31 integrase expression was under the control of the araBAD promoter, whereas ISceI endonuclease was controlled by the tac promoter. This vector was transfected into CHO-K1 cells, which showed transient expression of EGFP up to 14 generations. Similar results were obtained upon transient transfection into HEK cells. In addition, PCR results on genomic DNA, demonstrated the EGFP-minicircle was episomal and did not integrate into the host genome. Our constructed parental plasmid expresses EGFP and could be used for the generation of episomal minicircle DNA with intent to carry out transient transfection of interested DNA fragments into the eukaryotic cells for various purposes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Conventional plasmids, used in gene therapy consist of two parts, a bacterial backbone and eukaryotic expression cassette [1]. The bacterial backbone that includes the origin of replication and antibiotic resistance gene may lead to immunity problems in vivo as their inflammatory unmethylated CpG motifs could interact with a variety of cytokines [2]. In addition, the bacterial backbone plays an important role in episomal transgene silencing [3]. The short duration of transgene expression is a major obstacle in the use of regular plasmids for gene therapy [4]. Chen et al. have shown that covalent linkage between the bacterial backbone and transgene expression cassette in conventional plasmids is responsible for silencing the transgene [3]. These researchers introduced a model for transgene silencing in regular plasmids. According to their study, a putative silencing complex was formed over the bacterial backbone after plasmid transfection. Silencing spreads to the transgene, leading to its inactivation [5]. Minicircle DNAs have been developed to avoid these problems. Gene expression levels from minicircle vectors have shown a significant increase in intensity and duration of gene expression in several in vitro and in vivo studies [4, 8, 9]. Minicircle DNA vectors lose their bacterial backbone by a site-specific recombinase-mediated recombination. These vectors are prepared in two steps. First, construction of a parental plasmid should be performed to carry a eukaryotic expression cassette flanked by two recognition sites of a site-specific recombinase. In the next step, ectopic expression of the recombinase results in recognition and recombination between two recognition sites. Therefore the minicircle is created, while it carries the eukaryotic expression cassette apart from the parental plasmid which encompasses the bacterial backbone [4, 6]. Thus far, a number of recombinases have been used for minicircle preparation, including tyrosine recombinases (λ, Cre, FLP) [7–9] and serine recombinases (ΦC31, ParA resolvase) [4, 6, 10]. The site-specific recombinase could be expressed either from the parental plasmid or the bacterial genome [11]. Effective control of the site-specific recombinase expression and activity for minicircle preparation is of utmost importance. Until now, several expression systems such as the temperature-sensitive λ cI857/p R promoter and PBAD/araC arabinose expression systems have been used to control such activities [4, 6, 8]. In the present study we have constructed a parental plasmid that encodes ΦC31 integrase under the control of the araBAD promoter which is regulated by induction with arabinose.
Efficient isolation of the minicircle from residual miniplasmid deduced from the backbone of the mother plasmid during recombination has yet to be accomplished. Although several methods have been used for minicircle purification [6], however the majority of studies emphasize ultracentrifugation of recombination products in cesium chloride [4]. Chen and coworkers have purified the minicircle DNA by degrading the remaining miniplasmid and parental plasmid in vivo via the expression of homing endonuclease ISceI under control of the araBAD promoter [12]. As seen with Southern blot analysis, the minicircle vectors do not integrate in the host genome and remain episomal [4, 13]. In this study we have constructed a cassette that encompasses homing endonuclease ISceI under the control of a tac promoter in a minicircle DNA vector. One of the main characteristics of minicircle vectors is that they are non-dividing and become diluted in dividing cells after several generations, which makes them suitable for transient gene expression [13]. In this study we have tested our minicircle vector by transient transfection into CHO cells in order to evaluate both its functionality and ability to remain episomally in these cells. Resultant data has shown evidence of EGFP expression up to 14 generations, after which the vector completely vanished. Gradual dilution of the minicircle vectors in HEK293 cells population was demonstrated too.
Materials and methods
Vector construction
We constructed the parental vector (pBAD.ΦC31.EGFP.ISceI) used to produce the minicircle DNA in the following three steps.
pBAD.EGFP construction
First, we amplified an EGFP expression cassette that encompassed the EGFP coding region in association with a CMV promoter and SV40 polyA by using the plasmid pEGFP-C1 (Clontech, USA) as a template with primers that introduced a Bst11071 restriction site at both the 5′ and 3′ ends. Furthermore, the primers introduced ΦC31 integrase recognition sites, attP at the head and attB at the tail of the amplified fragment (Table 1). An amplified fragment (1779 bp) was inserted into pTZ57R/T (Fermentas, Lithuania) to facilitate its digestion with Bst11071 (Fermentas). The treated EGFP expression cassette was purified and inserted into a pBAD.gIIIA vector (Invitrogen, USA) which generated a new recombinant vector, pBAD.EGFP (Fig. 1). The ligation process was performed using a TaKaRa ligation kit (TaKaRa, Japan). To ensure proper construction of the EGFP expression cassette, we sent the recombinant plasmid for sequencing (Takapozist, Iran).

Schematic representation of the EGFP expression cassette amplification and its insertion into the pBAD.gIIIA vector
pBAD.ΦC31.EGFP construction
The ΦC31 integrase coding DNA sequence was amplified using a plasmid, pCMV-INT (gifted from Professor M. Calos, Stanford University, CA, USA) as a template using a specific pair-primer with the following description. A mega forward primer introduced 80 bp from the backbone of pBAD.gIIIA vector encompassed the 3′ part of the araBAD promoter, which included a unique BamHI restriction site and bacterial backbone ribosome binding site. The reverse primer introduced and SacI restriction site (Table 1). We treated the amplified fragment (1904 bp) with the restriction enzymes BamHI and SacI (Fermentas) and inserted them into a recombinant vector pBAD.EGFP which generated a new plasmid, pBAD.ΦC31.EGFP (Fig. 2). Sequencing was performed by sending the new vector to Takapozist Company.

Schematic representation of the ΦC31 integrase coding region amplification and its insertion into the pBAD.EGFP vector
pBAD.ΦC31.EGFP.ISceI construction
In order to facilitate purification of the minicircle, the intron-encoded endonuclease ISceI coding sequences (US patent no.: 7,214,536 B2; Dujon et al.), which were synthesized (Takapozist, Iran) encompassed the NheI and PstI restriction sites at the 5′ and 3′ ends, respectively (Table 1). The PCR product was inserted into pTZ57R/T for proper digestion with the respected restriction enzymes (NheI and PstI, Fermentas). The NheI–PstI fragment was then inserted into the same sites in pCYB2 (Pasture Institute, Iran) under the control of a tac promoter (PTAC) created a new plasmid, pCYB2.ISceI (6001 bp; Fig. 3). Then, a DNA cassette from the pCYB2.ISceI that included the bacterial lacI gene, the tac promoter and ISceI coding sequence was amplified by using pCYB2.ISceI as the template with a forward primer that introduced NotI and ISceI restriction sites at the 5′ end and a reverse primer that introduced a NotI restriction site at the 3′ end (Table 1). The PCR product was inserted again into pTZ57R/T and digested with NotI (Fermentas). The NotI treated fragment was inserted into the NotI site of the recombinant plasmid pBAD.ΦC31.EGFP, which resulted in the generation of a parental plasmid, pBAD.ΦC31.EGFP.ISceI (Fig. 4).

Schematic representation of the ISceI coding region and its insertion into the pCYB2 vector

Schematic representation of the amplified fragment that contains the lacI gene-ISceI-tac promoter in order to construct pBAD.ΦC31.EGFP.ISceI
Minicircle production
For minicircle production we cultured a single colony of bacterial transformed cells (TOP10 strain, Invitrogen, USA) with pBAD.ΦC31.EGFP.ISceI overnight. The next day, a portion of the culture was transferred to fresh LB broth (ratio: 4:1, v/v). A variety of conditions were tested to determine which condition provided optimal production of the minicircle. Appropriate production of the minicircle was attained by the following protocol. Simultaneous induction of an araBAD promoter and tac promoter was accomplished by the addition of 0.2 % (w/v) L-(+)-arabinose and 2 mM IPTG (w/v; Fermentas) to the cell culture at 32 °C with orbital shaking at 250 rpm for 120 min. This step was repeated by solitary addition of 2 mM IPTG (w/v) to ensure proper function of ISceI. Minicircle DNA preparation was performed by the QIAgen Plasmid Miniprep Kit (Qiagen). The extracted minicircle DNA was digested with a SacI restriction enzyme. This digestion was performed in order to discriminate the bands related to minicircle, miniplasmid and parental plasmid. Figure 5 illustrates the final step of minicircle DNA separation from the parental plasmid. In order to purify the minicircle DNA vector completely, a mixture of the SacI digested sample was electrophoresed. A respective band of minicircle DNA was extracted from agarose gel using QIAgen Gel Extraction Kit (Qiagen) and then was religated again using Mighty Mix DNA Ligation Kit (TaKaRa).

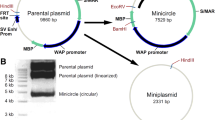
Maps of the parental plasmid, resultant minicircle and miniplasmid
Cell culture and transfection
CHO-K1 cells obtained from Royan Institute for Stem Cell Biology (Tehran) were cultured at a density of 5 × 103 cells/cm2 in DMEM/Ham’s F-12 (Sigma, USA) medium supplemented with 100 U/mL penicillin and streptomycin (Gibco, USA) and 10 % FCS (Gibco) at 5 % CO2. The day before transfection, CHO-K1 cells were trypsinized and counted. Approximately 2 × 105 cells were plated per well of 6-well plate (TPP, USA) in 2 mL of complete growth medium. Transfection of cells by EGFP containing minicircle DNA was carried out at 50–80 % of cell confluency. To this end, 2.5 μg of minicircle DNA was diluted into 500 μL of dilution medium (Medium without serum). After that 5 μL of Lipofectamine™ LTX (Invitrogen) was diluted into the above diluted DNA solution. 500 μL of the DNA-Lipofectamine™ LTX complexes were added to each well containing cells and complete growth medium according to the manufacturer’s instructions. One day post transfection, assessment of the green florescence protein for tracking the CHO cells expressing EGFP was performed using a fluorescence microscope (Olympus, Japan) and images were captured with an Olympus camera. The efficiency of transfection was assessed by flow cytometry. Next, transfected CHO cells were trypsinized, counted and diluted in a complete medium in order to achieve a single cell per well in a 96 well plate. Cells were tracked by a fluorescent microscope for 9 days. Finally, cells were harvested 5 days post-transfection for genomic DNA isolation (DNeasy Blood and Tissue Kit, Qiagen) and episomal vector extraction using the QIAprep Spin Miniprep Kit (Qiagen). Amplification of the EGFP expression cassette on the extracted episomal vector and isolated genomic DNA was performed by the primers mentioned in Table 1.
HEK293 cells were cultured in DMEM high glucose (Gibco) medium supplemented with 1 % penicillin and streptomycin (Gibco) and 10 % FCS (Gibco), 1 % l-glutamine (Gibco), 1 % non-essential amino acid (Gibco) at 5 % CO2. The day before transfection, 2.5 × 105 cells were plated well in 1 mL of complete medium in a 12-well dish. Transfection was performed using a transfection complex containing 1 μg of EGFP-minicircle DNA, 200 μL OPTI-MEM dilution media and 3 μL of Lipofectamine™ LTX (Invitrogen). The transfected cell population of one well in 12-well dish (TPP, USA) was monitored for EGFP expressing using fluorescence microscopy and flow cytometry every 4 days until day 21. The last day, genomic DNA was extracted from tracked cells and untransfected cells using (DNeasy Blood and Tissue Kit, Qiagen). Amplification of the EGFP expression cassette on the extracted genomic DNA was carried out by the primers mentioned in Table 1.
Results
As described in “Materials and methods” section, we constructed the parental plasmid in three steps.
pBAD.EGFP construction
PCR analysis of pEGFP-C1 as a template generated a 1779 bp band that was related to the EGFP expression cassette (Fig. 6a). Insertion of this cassette into the pBAD.gIIIA vector generated a recombinant vector, pBAD.EGFP. One cut digestion with ScaI confirmed the length of this recombinant vector to be 5925 bp (Fig. 6b).

Steps for the construction of pBAD.EGFP. a Amplification of EGFP expression cassette (1779 bp). b Digestion of pBAD.EGFP with ScaI. M is a DNA marker
pBAD.ΦC31.EGFP construction
ΦC31 integrase coding region (ΦC31 integrase) that had a length of 1904 bp was successfully amplified (Fig. 7a) and inserted into the pBAD.EGFP recombinant vector, which generated a new plasmid termed pBAD.ΦC31.EGFP. The linearized recombinant plasmid with BamHI digestion was 7690 bp in length (Fig. 7b).

Steps for construction of pBAD.ΦC31.EGFP. a Amplification of the ΦC31 integrase coding region. b Digestion of pBAD.ΦC31.EGFP with BamHI. M is a DNA marker
pBAD.ΦC31.EGFP.ISceI construction
A DNA fragment encoding ISceI, with a size of 726 bp (Fig. 8a) was properly inserted into pCYB2 which led to the generation of a new vector, pCYB2.ISceI. The recombinant vector was digested with NheI and produced a linear band of 6001 bp (Fig. 8b). As described in “Materials and methods” section, a DNA cassette from pCYB2.ISceI encompassing the bacterial lacI gene, tac promoter and ISceI coding sequence was amplified from pCYB2.ISceI (2654 bp). We inserted the PCR product, lacI-PTAC-ISceI (Fig. 8c) into the NotI site of the recombinant plasmid pBAD.ΦC31.EGFP, which resulted in generation of a parental plasmid, pBAD.ΦC31.EGFP.ISceI as confirmed via digestion with ScaI (10,336 bp, Fig. 8d).

Steps for construction of pBAD.ΦC31.EGFP.ISceI.LacI. a ISceI coding region. b Digestion of pCYB2.ISceI with NheI. c LacI-PTAC-ISceI amplification (2654 bp). d Digestion of pBAD.ΦC31.EGFP.ISceI.LacI with ScaI. M is a DNA marker
Minicircle preparation
Escherichia coli TOP10 cells were transformed with the parental plasmid. Production of ΦC31 integrase was stimulated after induction with 0.2 % L-(+)-arabinose at 32 °C for 120 min, thereby recombination between attB and attP sites caused generation of a minicircle DNA that carried EGFP. Two concentrations of 0.2 and 0.5 % of L-(+)-arabinose at 0, 60, 120, 180 and 240 min were tested for induction of araBAD promoter activity. The results of these two concentrations were similar in terms of integrase production. Therefore, we chose concentration of 0.2 % and optimal time for induction was 120 min (data not shown). To obtain better resolution for agarose gel electrophoresis, the resultant DNA from the induction was digested with ScaI prior to loading. A product band (1733 bp) related to the linearized minicircle was observed along with three additional bands of lengths 8603, 6638 and 3698 bp. The 6638 and 3698 bp bands represented unrecombined parental plasmid, whereas the 8603 bp band was derived from the miniplasmid (Fig. 9a). In order to eliminate any residual parental bacterial plasmid and miniplasmid, the inducible ISceI gene in conjunction with its recognition site was included in the structure of the parental plasmid. Therefore we applied a variety of IPTG concentrations (1, 2, 3 mM) for induction of ISceI endonuclease production as described in “Materials and methods” section. ISceI produces linear forms of miniplasmid and parental plasmid. However, those circular of parental plasmid which were remain untreated could be extracted by plasmid extraction kit. Interestingly, at concentration of 2 mM, the yield of the undesirable product (ISceI-untreated miniplasmid) were lower compared to the other concentration of IPTG (Fig. 9b). In this condition, we obtained approximately 2.5 mg semi-purified minicircle DNA vector in a 250 mL of bacterial culture. This semi-purified minicircle was extracted using QIAgen Plasmid Maxiprep Kit. This yield of production could be improved by further experiments to optimize the conditions. As shown in Fig. 9c, Minicircle DNA vector was purified completely after SacI digestion of the mixture followed by electroporation. Using gel extraction, minicircle related band was isolated and re-ligated. Electroporation of the re-ligated minicircle DNA confirmed a sole DNA band free from undesired contamination with residual backbones and parental miniplasmid.

Improvement in minicircle production. a Induction of ΦC31 integrase production by addition of 0.2 % arabinose after 120 min. The minicircle was produced according to the protocol described in “Materials and Methods” section. b Strategy for induction of ISceI expression performed using serial concentrations of IPTG (1–3 mM) as described in “Materials and Methods” section. Note that the efficient concentration of IPTG was 2 mM. Also please note that in each lane 500 ng of SacI digested DNA was loaded with. Hence, upon digestion with SacI, parental plasmid with the length of 10,336 bp, divides and yields into two bands with size of 6638 and 3698 bp. This is due to the presence of two SacI sites in parental plasmid. However, minicircle and miniplasmid have a unique SacI site. c Minicircle DNA vector was purified completely using electroporation of SacI digested mixture and gel extraction of respective band and re-ligation. Final EGFP-minicircle vector was digested again with SacI restriction enzyme prior to electrophoresis. M is a DNA marker
Monitoring of minicircle in CHO cells
The EGFP containing minicircle DNA was transfected into CHO-K1 cells. One day post-transfection, a green fluorescence color was observed in 70 % of the cells as determined by flow cytometry (Supplementary Fig. 1). Solitary isolated cells that expressed EGFP were plated in a 96 micro well plate and tracked by fluorescence microscopy. The data showed a gradual dilution of the minicircle vectors in all grown colonies as evidenced by a decrease in fluorescence intensity overtime. This intensity had mostly diminished by day nine which was assigned for 14th generation of cells (Fig. 10a). To examine whether the minicircle had integrated into the genome, amplification of the EGFP expression cassette was performed both on extracted episomal vector and isolated genomic DNA (Fig. 10b). We observed a 1671 bp product in the extracted episomal vector that was derived from transfected cells (CHO+); this band was absent in the CHO− (control) cells (Fig. 10c). Interestingly, this product was not detectable in extracted genomic DNA from both CHO+ and CHO− cells, demonstrating the episomal state of transfected minicircle DNA (Fig. 10d).

Tracking of the minicircle in CHO cells. a Minicircle vectors were diluted in tracked colonies as shown by decreased fluorescence intensity in the cells. Note that expression of EGFP terminated after 9 days post-transfection. Upper panels from left to right indicate fluorescent cells on days 2, 3, 5, 6, 7, 8, and 9, respectively. Below panels are phase contrast pictures of their upper counterparts. Bar is 50 µm for figures of day 2, 3, 4, and 100 µm for figures of day 6 and 7 and 200 µm for figures of day 8 and 9. b Isolated genomes derived from CHO cells. Both were transfected with minicircle DNA (CHO+) and control sample (CHO−). c PCR of the vector solution derived from CHO cells. Note that the respective band (1671 bp) for minicircle DNA was detected in CHO+ cells. d PCR analysis of CHO cell-derived genome. No product was identified in the samples which showed the episomal state of minicircle DNA in transfected cells. M is a DNA marker
Monitoring of minicircle in HEK293 cells
Tracking of EGFP-minicircle in HEK293 cells was performed with a total population of transfected cells. Transfected HEK293 cells were monitored for expressing of EGFP using both fluorescence microscopy and flow cytometry every 4 days, starting day 2 until day 22 post-transfection (Fig. 11 a, b). Gradual dilution of minicircle vectors in HEK293 cells population was monitored by intensity of EGFP. These data were confirmed again by flow cytometry (Supplementary Fig. 2). Meanwhile, amplification of the EGFP expression cassette on the extracted genomic DNA yielded no product due to integrated minicircle DNA (data not shown). In order to compare the intensity of EGFP expression of semi-purified minicircle product with completely purified minicircle DNA, both of them were transfected into the mycoplasma-free HEK293 cells in the same conditions and FACS analysis was performed next day. The GFP signal intensity and percent of transfected cells were similar; interpreting that presence of contaminants (parental miniplasmids and residual backbones) in semi-purified sample did not interfere with expression of EGFP and transfection efficiency (Supplementary Fig. 3).

Monitoring of EGFP-minicircle in HEK293 cells. A population of transfected cells was monitored for cells expressing EGFP using fluorescence microscopy (a) and flow cytometry (b) every 4 days, starting day 2 until day 22 post transfection. Bar is 200 µm
Discussion
Previous studies have demonstrated that minicircle DNA vectors lack a bacterial backbone and thus express high levels of transgenes. These vectors are safe and useful for gene therapy [4]. In the current study we have successfully produced and partially purified a minicircle DNA vector that carried an EGFP reporter gene by using ΦC31 integrase mediated recombination and homing endonuclease ISceI mediated purification in E. coli. The ΦC31 integrase belongs to the resolvase/invertases family and is derived from a Streptomyces temperate phage. Different studies have used a variety of recombinases to produce a minicircle, including serine-recombinases including ΦC31 [4] and ParA resolvase [10] in addition to tyrosine recombinases such as λ [8], Cre [9], and FLP [7].These studies have shown recombination-mediated ΦC31 integrase to be more efficient than the other recombinases [4]. In this study we used the ΦC31 integrase which was constructed under the control of an araBAD inducible promoter. Expression by the araBAD promoter was induced using L-(+)-arabinose at a concentration of 0.2 % for 120 min. This optimal condition was also reported in previous studies [4]. We used attB (length: 34 bp) and attP (length: 39 bp) which have been considered as the minimal functional sizes recognizable for ΦC31 integrase [12]. ΦC31 integrase functions through aforementioned sites and creates a minicircle. However, there are several unrecombined parental plasmids in the final product which are the result of a lack/low expression of an arabinose transporter (AreE) in a subset of bacterial cells. Thus ΦC31 integrase could not be induced, which results in the failure of minicircle production [14]. In order to prevent the presence of unrecombined parental and minicircle plasmids, Chen and coworkers have used an ISceI endonuclease, which is an intron-encoded endonuclease which recognizes a region of 18 bp at target DNA, resulting in efficient purification of the minicircle DNA [12]. The main reason for selection of this endonuclease was its absence in the E. coli genome [12]. Chen et al. have co-expressed ISceI and ΦC31 integrase coding sequences under control of araBAD promoter. However, in this study we expressed ΦC31 integrase and ISceI endonuclease under control of araBAD and tac promoter, respectively. Induction of ISceI endonuclease expression was performed in several concentrations of IPTG, where we determined that 2 mM IPTG was more efficient.
The tac promoter is a strong, inducible promoter. This promoter is a hybrid promoter that consists of the -35 region of the trp promoter and the -10 region of the lacUV5 promoter/operator. The tac promoter could be activated by addition of the respective inducer, IPTG. Expression of the tac promoter is repressed by the lacI protein. The mutated lacI q allele used in parental plasmid increases the intracellular concentration of lacI repressor, resulting in strong repression of the tac promoter in the ground state [15]. Using this and previous in vivo procedures, it is impossible to purify the minicircle DNA completely. Small amounts of un-recombinant parental plasmid and miniplasmid remain anyway. In a similar study, Chen and coworkers produced minicircle DNA and purified it. As they reported, the yield of final minicircle DNA was 1 mg/L using CsCl equilibrium gradient centrifugation while 3–15 % parental plasmid and miniplasmid were reported as contaminants [4]. Kay et al., have indicated that production and purification of minicircle using genetically modified E. coli, yielded 3.4–4.8 mg/L with 0.4–1.5 % contamination [11]. In the present study, the yield of semi-purified minicircle DNA vector was 2.5 mg per 250 mL of bacterial culture. To get rid of such contaminants, researchers have used several in vitro approaches including affinity chromatography [17] and digestion of parental plasmid with restriction enzymes, followed by dephosphorylating/re-exposing process, and a selective ligation of minicircle [18]. In the structure of our parental plasmid the existence of multiple cloning sites after the EGFP coding region facilitated insertion of interest DNAs, either in the form of a chimeric DNA or separately in parental plasmid. Therefore, we also performed similar experiment using digestion of parental plasmid, residual backbone and minicircle DNA in the mixture sample followed by electroporation and extraction of minicircle respective band.
The presence of a multi cloning site downstream of EGFP coding sequences in our miniplasmid facilitates the insertion of interest DNA in shape of either chimeric structures with EGFP or a separated translating unit flanking by intervening sequences like IRES and 2A peptide encoding sequences. In case of 2A peptide, the downstream gene is often translated at lower levels (~10 %), although it has not effect on the expression upstream gene, EGFP [19].
As minicircles belong to episomal vectors their numbers could be diluted after several generations of transfected cells [4, 13]. Different cell lines such as CHO [7], NIH 3T3 (murine fibroblast), H460 (human nonsmall cell lung carcinoma), 3LL (mouse Lewis lung carcinoma), human aortic smooth muscle (HSM), and rabbit aortic smooth muscle (RSM) [8], HeLa cells [9] have been used to study the potency of minicircles for gene expression and functionality. In this study, to track the produced minicircle and assessment of its functionality, the produced minicircle was transfected into CHO-K1 cells. The molecular properties, transfection and genetic manipulation methods of CHO-K1 cell line has been greatly characterized [16]. Our EGFP expressing minicircle gradually became diluted in all tracked CHO colonies as EGFP fluorescence intensity was terminated on day 9. According to PCR results on genomic DNA, EGFP-minicircle remained at episomal state and did not integrate into the CHO genome. Monitoring of EGFP-minicircle in HEK293 cells was also performed. In this experiment a population of transfected cells was tracked for GFP+ cells using flow cytometry and fluorescence microscopy every 4 days. Similarly, 22 days after transfection GFP+ ware disappeared completely. This procedure of cell tracking has already been used by Jia et al. to indicate the dilution of the minicircle vector during cell proliferation [13].
This study such as previous studies on minicircle DNA vectors [4, 7–12] was performed in the small scale. However, large scale production of our minicircle vector is possible, though we did not test it in the present study. It is supposed that reduction of IPTG concentration by using 32 copies of ISceI sites instead of one restriction site could improve the yield of mini circle DNA production and thus should be used for further scale up strategies. Implementation of 32 copies of ISceI sites was already shown to improve the efficiency of ISceI digestion [11].
Conclusion
We have constructed a parental plasmid expressing EGFP which could be used for the generation of episomal minicircle DNA with intent to carry out transient transfection of interested DNA fragments into the eukaryotic cells for various purposes.
References
Gill D, Pringle I, Hyde S (2009) Progress and prospects: the design and production of plasmid vectors. Gene Ther 16:165–171
Klinman DM (2004) Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol 4:249–259
Chen Z, He C, Meuse L, Kay M (2004) Silencing of episomal transgene expression by plasmid bacterial DNA elements in vivo. Gene Ther 11:856–864
Chen ZY, He CY, Ehrhardt A, Kay MA (2003) Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther 8:495–500
Chen ZY, Riu E, He CY, Xu H, Kay MA (2008) Silencing of episomal transgene expression in liver by plasmid bacterial backbone DNA is independent of CpG methylation. Mol Ther 16:548–556
Mayrhofer P, Schleef M, Jechlinger W (2009) Use of minicircle plasmids for gene therapy. Methods Mol Biol 542:87–104
Nehlsen K, Broll S, Bode J (2006) Replicating minicircles: generation of nonviral episomes for the efficient modification of dividing cells. Gene Ther Mol Biol 10:233–244
Darquet A, Cameron B, Wils P (1997) A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene Ther 4:1341–1349
Bigger BW, Tolmachov O, Collombet JM, Fragkos M, Palaszewski I, Coutelle C (2001) An araC-controlled bacterial Cre expression system to produce DNA minicircle vectors for nuclear and mitochondrial gene therapy. J Biol Chem 276:23018–23027
Jechlinger W, Azimpour Tabrizi C, Lubitz W, Mayrhofer P (2004) Minicircle DNA immobilized in bacterial ghosts: in vivo production of safe non-viral DNA delivery vehicles. J Mol Microbiol Biotechnol 8:222–231
Kay MA, He CY, Chen ZY (2010) A robust system for production of minicircle DNA vectors. Nat Biotechnol 28:1287–1289
Chen ZY, He CY, Kay MA (2005) Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum Gene Ther 16:126–131
Jia F, Wilson KD, Sun N, Gupta DM, Huang M, Li Z, Panetta NJ, Chen ZY, Robbins RC, Kay MA, Longaker MT, Wu JC (2010) A nonviral minicircle vector for deriving human iPS cells. Nat Methods 7:197–199
Siegele DA, Hu JC (1997) Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci 94:8168–8172
Baneyx F (1999) Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 10:411–421
Xu X, Nagarajan H, Lewis NE et al (2011) The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol 29:735–741
Kobelt D, Schleef M, Schmeer M, Aumann J, Schlag PM, Walther W (2013) Performance of high quality minicircle DNA for in vitro and in vivo gene transfer. Mol Biotechnol 53:80–89
Dong Y, Aied A, Li J, Wang Q, Hu X, Wang W (2013) An in vitro approach for production of non-scar minicircle DNA vectors. J Biotechnol 166:84–87
Trichas G, Begbie J, Srinivas S (2008) Use of the viral 2A peptide for bicistronic expression in transgenic mice. BMC Biol 6:40
Acknowledgments
This study was funded by a Grant-in-aid of research from Royan Institute for Biotechnology in support of Nafiseh Sanei Ata-Abadi for obtaining her M.Sc. degree from the University of Isfahan.
Conflict of interest
None of the authors has any conflicts of interest to disclose and all authors support submission to this journal.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2015_3864_MOESM1_ESM.doc
Supplementary Fig. 3 Dot blot of flow cytometry to measure the EGFP intensity.Flow cytometry was carried out to compare the rate of green fluorescence in transfected mycoplasma free HEK293 cells with semi-purified minicircle product (a) and in vitro completely purified minicircle (b).Supplementary material 1 (DOC 29 kb)
11033_2015_3864_MOESM2_ESM.tif
Supplementary Fig. 1 Dot blot of flow cytometry to measure the EGFP intensity.Flow cytometry was carried out to determine the rate of transfected CHO cells expressing EGFP. Please see materials and methods section for further elucidationSupplementary material 2 (TIFF 52 kb)
11033_2015_3864_MOESM3_ESM.tif
Supplementary Fig. 2 Dot blot of flow cytometry to measure the EGFP intensity.Flow cytometry was carried out to determine the rate of green fluorescence in transfected HEK cells different days post transfection as described in Materials and MethodsSupplementary material 3 (TIFF 588 kb)
Rights and permissions
About this article

Cite this article
Sanei Ata-Abadi, N., Dormiani, K., Khazaie, Y. et al. Construction of a new minicircle DNA carrying an enhanced green florescent protein reporter gene for efficient expression into mammalian cell lines. Mol Biol Rep 42, 1175–1185 (2015). https://doi.org/10.1007/s11033-015-3864-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-015-3864-z