
Abstract
Rice grain amylose contents (ACs) is a key quantitative trait influencing eating and cooking quality. Regulating the expression level of Waxy, a key gene controlling ACs, and in turn fine-tuning the grain ACs, is an ideal approach to improve grain quality of rice varieties. Based on CRISPR/Cas9 genome editing technology, we designed eight targets in the cis-regulatory region of Wxa background, screened phenotypic changes of the transgenic lines and generated eight novel Waxy alleles with altered grain ACs. Among the eight alleles, we found that a 407-bp non-homologous substitution (NHS) in the 5′UTR-intron caused by genome editing regulated Waxy expression and decreased grain ACs by 2.9%. Moreover, embedding the 407-bp NHS into the cis-regulatory region of Wxb allele can also affect gene activity. Our work suggested the effect of 5'UTR-intron on Waxy gene expression regulation, and provided a potentially useful allele in breeding that can finely adjust rice grain ACs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Genome editing technology could be applied in crop breeding through generating advantageous alleles (Hua et al. 2019; Chen et al. 2019; Zhang et al. 2020). In previous works, it has been proven that knocking out one or several undesirable trait-associated genes via genome editing has the potential to improve crop corresponding traits (Chen et al. 2019; Zhang et al. 2020). For example, knocking out bread wheat (Triticum aestivum) MLOs gained powdery mildew-resistance (Wang et al. 2014), and knocking out rice (Oryza sativa) PYL1, PYL4, and PYL6 improved grain yield (Miao et al. 2018). However, for most genes, only a few specific alleles could be used for crop improvements, while defective functions of the genes usually produce few beneficial effects, even decrease crop quality and yield severely (Shen et al. 2018; Yano et al. 2019). Screening for fine-tuned alleles of target genes, especially quantitative trait genes, is one of the best ways to solve the problem. Recently, multiple precise genome editing tools for generating targeted variations efficiently (e.g., base-editors and prime editing system) have been developed (Hua et al. 2019; Chen et al. 2019; Zhang et al. 2020), which facilitated the fine-tuned allele screening.
In crop genomes, abundant functional alleles for crop breeding come from not only coding regions but also non-coding regions. Recent studies found that genome editing in non-coding regions, especially the cis-regulatory region (including 5′UTR, promoter, and distal regulatory elements), could improve crops efficiently (Oliva et al. 2019; Liu et al. 2021; Song et al. 2022). For example, a new rice line with broad-spectrum resistance to bacterial blight was developed by genome editing in the promoter of three gene encoding sugar transporters (SWEET11, SWEET13, and SWEET14) (Oliva et al. 2019). Furthermore, editing cis-regulatory regions showed a high potential for creating diverse quantitative variation in a highly efficient way (Rodríguez-Leal et al. 2017; Mohammadi et al. 2017). As a pioneer work in tomato, cis-regulatory alleles of three tomato genes (SlCLV3, COMPOUND INFLORESCENCE, and SELF PRUNING) were generated rapidly and efficiently, by designing a CRISPR/Cas9 construct with multiple sgRNAs targets in the promoter. Then, a series of alleles with continuum of traits were obtained through phenotype screening (Rodríguez-Leal et al. 2017). However, most genome editing lines in non-coding regions do not have phenotypic changes. It was reported that non-coding region, even very close to coding region, could have high tolerance to genetic perturbations (Wang et al. 2021).
In rice, Waxy encoding a granule-bound starch synthase is the major quantitative trait gene of amylose content (AC), which is an important trait influencing grain quality (Tian et al. 2009). The natural allelic variations of Waxy decide diverse ACs (Zhang et al. 2019), and too high grain ACs are generally related to poor eating and cooking quality. There are two major natural Waxy alleles (Wang et al. 1995; Isshiki et al. 1998): Wxa (usually exists in indica cultivars) and Wxb (mainly distributed in japonica cultivars). The main difference between Wxa and Wxb is a G/T polymorphism in the first splicing site of Waxy in 5′UTR. The Wxb allele, which undergoes an abnormal splicing process, has tenfold lower protein level than Wxa, resulting in a low grain AC (Larkin and Park 2003).
Creating waxy varieties by genome editing of Waxy gene showed higher-efficiency than trait introgression in conventional breeding that usually needs multi-generation backcrossing (Gao et al. 2020). CRISPR/Cas9 technology was firstly applied to target Waxy coding sequences to create null alleles which resulted in glutinous rice (Zhang et al. 2018), whereas moderate grain ACs (neither too glutinous nor too non-adhesive) are preferred in rice grain quality improvement, which relies on alleles with fine-tuned Waxy activity. Recently, fine-tuning rice grain ACs was implemented by genome editing in cis-regulatory region of Waxy (Zeng et al. 2020; Huang et al. 2020). More variations which can regulate Waxy activity are needed to fine-tune grain ACs for diverse rice breeding goals. There have been evidences in plants for the cis-regulatory elements in 5′UTR-introns with effects on gene expression (Kim et al. 2006; Zeng et al. 2017; Vetrici et al. 2021). Zeng et al. constructed Waxy alleles with altered splicing patterns and fine-tuned AC, by targeting the splicing site in 5′UTR intronic (Zeng et al. 2020). However, cis-regulatory elements inside the 5′UTR-intron region (more than 1 kb) of Waxy are still unrevealed, and whether editing inside the 5′UTR-intron region can fine-tune the Waxy level is still unknown.
To explore the cis-regulatory region, especially the 5′UTR-intron, to generate more appropriate target sites for adjusting grain ACs, in this study, we used 8 sgRNA in one CRISPR/Cas9 transgene system to rapidly create dozens of random alleles with variations in the cis-regulatory region of Waxy. Coupled with phenotype screening, new Waxy alleles with fine-tuned ACs were generated.
Results
Strategy for constructing novel Waxy alleles by CRISPR/Cas9
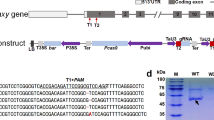
To identify potential cis-regulatory region that can regulate Waxy expression and affect grain ACs, Zhongzao35 (ZZ35) which is an early-flowering indica cultivar with a high grain AC (~ 25%) was selected for genome editing. Through Sanger sequencing, we obtained 2.7 kb cis-regulatory sequence immediately upstream the Waxy coding region in ZZ35, which covers the ~ 1.4 kb promoter region and a 1288 bp 5′untranslation region (5′UTR) with a 1132 bp 5′UTR-intron included. The sequence also confirmed ZZ35 Waxy belongs to Wxa allele.
To investigate the effect of 5′UTR-intron of Waxy and generate alleles with fine-tuned grain ACs, we designed 4 targets in promoter and 4 targets in 5′UTR-intron using CRISPR-P (http://crispr.hzau.edu.cn/CRISPR2/) for multiplex editing in the 2.7 kb cis-regulatory region (Fig. 1A). Using a full-fledged CRISPR/Cas9 system (Ma et al. 2015), we generated one construct vector with all 8 sgRNAs. This construct was introduced into ZZ35 to generate mutations resulted from random combination among 8 editing targets. Twelve first-generation transgenic plants (T0) were identified by PCR genotyping. In T1 generation, sixteen homozygous lines containing deletion variants in the cis-regulatory region of Waxy were obtained. For phenotype screening, the sixteen T1 alleles were planted in Hainan, China, for the measurement of grain ACs. Probably due to the destroyed gene expression, those alleles which contained deletions covering the transcription start site (TSS) exhibited a same phenotype: extreme reduced grain ACs. After eliminating alleles with phenotypic redundancy, here we presented genotype and phenotype (grain ACs) of eight new Waxy alleles, which were named Waxy precise adjustment 1 to 8 (WP1 to WP8, Fig. 1B). Compared with the grain AC of ZZ35 (25.03%), phenotype of the eight alleles could be divided into two groups. WP1, WP4, WP5, and WP6 were all glutinous rice with ACs less than 5%. All these four lines carried large-scale deletions spanning promoter and 5′UTR of Waxy. WP2, WP3, WP7, and WP8 showed slightly reduced grain ACs. WP3 contained a 394 bp deletion in 5′UTR-intron and a single-base deletion in the promoter region; WP2 contained a 407 bp deletion replaced by a 14 bp short sequence (that, non-homologous substitution, NHS) in 5′UTR-intron, and two short deletions in promoter; WP7 carried a 1049 bp deletion in promoter, and 2 single-base deletions in 5′UTR-intron; and WP8 carried a 537 bp deletion in promoter, 2 single-base deletions, and a 95 bp deletion which was replaced by a 28 bp sequence in 5′UTR-intron (Fig. 1A).

Generating novel Waxy alleles by genome editing of cis-regulatory region. A Schematic of the cis-regulatory region of Waxy and genotype of T1 alleles. 2.7 kb sequence immediately upstream the coding region was targeted by eight sgRNAs (purple arrows). Pink region: 5′UTR-intron; orange region: exon1; yellow region: promoter. Deletions (–), insertions ( +), and substitutions ( >) in eight Waxy alleles generated by editing were indicated by numbers or letters. B Grain amylose contents (ACs) of ZZ35 and T1 homozygous progeny of the eight alleles. Mature grains of nine individuals of each line were mixed together for the measurement in two technical replications. Bars represent standard deviation
Cis-regulatory region editing of Waxy achieved fine-tuning of grain ACs
Among the weak phenotype group of WPs, WP2 and WP3 have very similar genotypes. However, WP2 showed the most significant reduced grain AC, while there was no significant AC difference between WP3 and ZZ35. To verify the effect of cis-regulatory region editing on fine-tuning of grain ACs, WP2 and WP3, accompanied with a glutinous allele WP1 as control, were selected for further analysis. T2 generation lines of WP1, WP2, and WP3 were planted in Shanghai, China, and grain cooking quality-related phenotypes were measured. Compared with ZZ35, the grain AC of WP2 decreased by ~ 3 percentage points (25.6% in ZZ35 wild type and 22.7% in WP2), and the gel consistency (GC) increased by 2 mm (Fig. 2A, B). Parameters related to Rapid Visco Analysis (RVA) were also changed in WP2 (Fig. 2C–G). In WP3, all these traits exhibited no significant differences with ZZ35.

CRISPR/Cas9-created Waxy allele WP2 resulted in fine-tuned grain ACs and cooking quality. A Grain ACs of ZZ35 and T2 progeny of three genome editing lines. B Grain gel consistency (GC) of different T2 plants. C–J RVA-related parameters of different T2 plants. BDV, breakdown value; PKV, peak viscosity value; CSV, consistence value; CPV, cold paste value; HPV, hot paste value. H Grain moisture contents (MCs) of different T2 plants. Mature grains of nine plant individuals of each line were mixed together for the measurement in three technical replications. Bars represent SD. Two-tailed t-test (*, P < 0.05; **, P < 0.01, compared with ZZ35). I Appearance of milled rice of ZZ35 and T2 progeny of three genome editing lines. J Photos from scanning electron microscopy showed the changes in morphology of starch granules in WP2. Round and loosely packed starch granules were observed in ZZ35 and WP3, compared with the starch granules in WP2 with polygonal tendency. Polygonal granules with sharp edges were exhibited in WP1. Bars = 20 μm and 5 μm for the 800 × and 3000 × magnifications, respectively
Only the appearance of WP1 grains was obviously different from ZZ35 (Fig. 2I), but observations from scanning electron microscopy revealed differences in the microstructure of starch granules among these alleles. Starch granules of ZZ35 are round and loosely packed. In contrast, the starch granules of the glutinous allele WP1 are polygonal and sharp. Consistent with the grain AC changes, the starch granules in WP2 endosperm exhibited sharper edges than that in ZZ35, while starch granules of WP3 showed no obvious changes (Fig. 2J). These results indicated grain ACs in WP2 was fine-tuned.
The plant height, panicle length, and culm length of WP1, WP2, and WP3 were shorter than ZZ35, indicating the reduced plant growth of the three editing lines. However, the growth of WP1 and WP2 showed no significant difference with that of WP3, an allele with no significant changes in grain ACs. The result suggested that the impact on plant growth were probably caused by transgenic experiment, while the mutations in the cis-regulatory region of Waxy may not cause the difference in plant growth (Fig. 3A, B).

Gross morphology of editing lines and Waxy expression change caused by a mutation in 5′UTR-intron of WP2. A Appearance of mature plants of T2 progeny of three genome editing lines. B The plant growth related phenotypes of mature plants. C qRT-PCR analyses revealed the altered expressions of Waxy in T2 progeny of three genome editing lines. Shelled seeds in 10 DAP were used for analysis (three biological and three technical replicates). OsActin1 was used as internal control and data (relative to ZZ35) was presented as mean ± SD. D Effects of mutations in genome edited alleles WP1 to WP3 analyzed by dual-luciferase reporter system. 2 kb cis-regulatory sequence of each allele was cloned into the promoter of FLuc. Length of bars represent logarithm (base 2) of the relative luciferase activity of FLuc (relative to RLuc and empty vector). Bars represent standard deviation. Two-tailed t-test (**, P < 0.01, compared with ZZ35). pro-ZZ35: the WT 2 kb sequence immediately upstream the coding region of Waxy in ZZ35. del-WP1/WP2/WP3: 2 kb sequence constructed by importing the mutation (1435 bp deletion, 407 bp NHS and 394 bp deletion in WP1, WP2 and WP3, respectively) into pro-ZZ35 and supplementing with upstream sequences
A variation in 5′UTR-intron resulted in fine-tuned Waxy activity
Gene expression levels of Waxy in the T2 plants were detected by qRT-PCR with seeds at 10 days after pollination (DAP). The mRNA of Waxy in WP1 was almost undetectable, suggesting that the large deletion covering TSS destroyed Waxy expression. The expression level of Waxy in WP2 reduced to 0.66-fold of that in ZZ35, while the Waxy transcriptional activity of WP3 decreased only slightly (92.58% relative to ZZ35) (Fig. 3C). These results indicated fine-tuning of Waxy expression occurred in WP2 but not in WP3.
In WP2, besides a 407-bp NHS variation in the 5′UTR-intron, there were another two short deletions in the promoter region. To test whether the 407-bp NHS caused fine-tuned Waxy expression, the activities of cis-regulatory region variant were examined with a dual-luciferase reporter assay in rice protoplast. Based on the WT cis-regulatory region of Waxy in ZZ35, we created three 2 kb chimeric sequences harboring the 407 bp NHS in WP2 (del-WP2), 394 bp deletion in WP3 (del-WP2), and 1435 bp deletion in WP1 (del-WP2), respectively. The full 2 kb sequence immediately upstream of Waxy coding region in ZZ35 (pro-ZZ35) was used as control. These four 2 kb fragments were cloned into luciferase reporter vectors. Compared with the pro-ZZ35, the luciferase activity driven by del-WP2 was reduced by 7.3 times, and that of del-WP3 was only reduced by 3.5 times (Fig. 3D). The luciferase activity of del-WP1 was almost the same as that of the blank vector. These results were consistent with the expression level of Waxy in different alleles, indicating that the 407 bp NHS in 5′UTR-intron caused the fine-tuning of Waxy expression.
Furthermore, to test whether the 407 bp NHS in 5′UTR-intron of WP2 has effect in other genetic backgound, we embedded the 407 bp NHS in to a 2 kb fragment based on the cis-regulatory region of Nipponbare (Nip, Wxb) for the dual-luciferase reporter assay. Consistent with the lower Wxb activity, the luciferase activity induced by wild-type 2 kb cis-regulatory fragment of Waxy in Nipponbare (Wxb) decreased to 0.05 of that in ZZ35(Wxa). The luciferase activity was further reduced by 2 times when the 407 bp NHS was imported to Wxb (Fig. 4B), suggesting the 407 bp NHS had effects on Wxb.

The 407 bp NHS variation in 5′UTR-intron can also affect Wxb expression. A Schematic of the genetic variations between the cis-regulatory region of ZZ35 (Wxa) and Nip (Wxb). 2 kb cis-regulatory region with a total of 16 natural variations is divided into four segments (V1–V4). Deletion is represented in yellow; SNV is showed in blue. B Effects of different cis-regulatory sequences on gene activity analyzed by dual-luciferase reporter system. 2 kb cis-regulatory sequence of each allele was cloned into the promoter of FLuc. The length of bars represents logarithm (base 2) of the relative luciferase activity of FLuc (relative to RLuc and empty vector). Bars represent standard deviation. Statistical analysis was performed by two-tailed t-test (**, P < 0.01, compared with ZZ35). WT-ZZ35/Nip: the wild-type 2 kb sequence immediately upstream the coding region of Waxy in ZZ35 or Nip; del-WP2(ZZ35)/(Nip): 2 kb sequence constructed by importing the 407 bp NHS in WP2 allele into WT-ZZ35/Nip and supplementing with upstream sequence; ZZ35V*NipV.*: 2 kb chimeric sequence combined by the fragments of V1 to V4 amplified from the corresponding materials (ZZ35 or Nip)
In addition, we scanned the variations between the 2 kb cis-regulatory sequence of Waxy in ZZ35 and Nipponbare. Besides the major variation between Wxa and Wxb (G/T polymorphism in the first splicing site), there are another 15 variations between ZZ35 and Nipponbare, including a (CT)n polymorphism, (TAAT) n polymorphism, and 13 single nucleotide variants (SNVs) (Fig. 4A). To evaluate the genetic effects of these variations, we divided them into four variation segments (V1–V4): V1 containing a (CT)n polymorphism site and two SNVs, V2 containing the SNV in the first splicing site, V3 containing a (TAAT) n polymorphism site and one SNV, and V4 containing 11 SNVs. Effects of chimeric fragments of V1–V4 segments which were cloned from ZZ35 or Nipponbare were tested by dual-luciferase reporter assay. All chimeric fragments containing V2 from ZZ35 induced the luciferase activity close to the activity induced by the full 2 kb cis-regulatory fragment of ZZ35. On the contrary, embedding any other segments from ZZ35 into Nipponbare segments cannot rescue the low luciferase activity value (Fig. 4B). This result indicated that these natural variations, except for the G/T polymorphism in the first splicing site, had few influences on Waxy activity.
Discussion
Amylose contents is a typical quantitative trait that needs to be fine-tuned, as the excellent grain eating and cooking quality usually correlate with moderate ACs (Tian et al. 2009). Mining or creating diverse allelic variations is urgently needed for rice improvement, which now can be genetically engineered to regulate the expression of the ACs-associated gene Waxy. To date, there were only a few reports of CRISPR/Cas9-created alleles with fine-tuned Waxy expression (Huang et al. 2020; Zeng, et al. 2020). In this study, through genome editing in cis-regulatory region of Waxy, we found a 407 bp NHS inside the 5′UTR-intron caused the change of Wxa expression and fine-tuning of ACs. Embedding this variant into the cis-regulatory region of Wxb could also affect the report gene activity in rice protoplast, implying the general effect of the 407 bp NHS in rice varieties with different Waxy genotype. Our study provided a potentially useful allele, the 407-bp NHS in the cis-regulatory region of Waxy, which could be introduced into other rice cultivars to finely adjust rice grain ACs. Generation of new alleles containing both the 407 bp NHS variant in the 5′UTR-intron and the T variant in the splicing site, and follow-up phenotype analysis, is needed to further verify this hypothesis.
In plant, the 5′UTR-introns have been found to harbor specific features, which are different from 3′UTR introns and coding region-residing introns (Chung et al. 2006). Studies have verified that 5′UTR-introns could contain binding sites for transcription factors and regulate gene expressions. For example, in rice, transcriptional repressor Ghd7 directly binds to the 5′UTR-intron of ARE1 to regulate the nitrogen use efficiency (Wang et al. 2021). Deletions in the 5′UTR-introns could result in significant change of gene expression level (Chung et al. 2006; Vetrici et al. 2021). In olive cultivars, sequence polymorphisms in 5′UTR-intron of FDA2 gene were significantly associated with phenotypic variation (Salimonti et al. 2020). Here we found a 407 bp NHS (constituted with a 407 bp deletion and 14 bp non-homologous sequence, which may be caused by repairing process in genome editing) in the 5′UTR-intron of Waxy gene could fine-tune the expression level. Interestingly, another 394 bp deletion in WP3 allele, which was fully covered by the 407 bp NHS, had little effect on gene expression. This suggested that a functional cis-regulatory element may locate in the 13 bp non-overlapped sequence, or the 14 bp insertion alter gene activity by introducing new TF binding sites or disrupting spacing between existing TF binding sites. The exact mechanisms need further researches.
Conclusions
In summary, through genome editing in cis-regulatory region of Waxy under a multi-sgRNA-in-one strategy, we generated novel Waxy alleles in a limited number of T0 mutant lines, and got a series of lines with changed ACs. A new allele WP2 with fine-tuned ACs (decreased by 2.9% in the ZZ35 background) was obtained. The 407 bp NHS in 5′UTR-intron of Waxy in WP2 was demonstrated to affect the Waxy expression and ACs. Our result suggested that 5′UTR-intron of Waxy was a potential target for rice grain quality improvement.
Materials and methods
Plant materials and cultivation
Zhongzao35 (short for ZZ35) is an indica rice variety bred by the China National Rice Research Institute by crossing Zhongzao22 and Jiayu253. It is an elite double-season early rice which is mainly planted in the lower reaches of the Yangtze River (https://www.ricedata.cn/variety/varis/605894.htm). A multi-sgRNA-in-one vector was constructed with editing targets in Waxy regulatory region by using pYLCRISPR/Cas9 system (Ma et al. 2015), which was then transferred into ZZ35 callus by Agrobacterium tumefaciens-mediated transformation using the strain EHA105 (Hiei et al. 1997). Rice transformants were grown in greenhouse in Shanghai, China (E:121.51, N:30.84), in normal rice-growing condition. Positive T0 transformants were identified by PCR. T1 plants were planted in Hainan, China (E: 109.18, N: 18.36), and T2 plants were planted in Shanghai.
Rice grain phenotype assays
The grain ACs and GCs were measured following the procedure described in NY/T 55–1987 and NY/T 83–2017, respectively. The moisture content (MC) and RVA related parameters (including breakdown value, BDV; cold paste value, CPV; hot paste value, HPV; consistence value, CSV) were measured following the procedure described in NY/T 1765–2009. Measurement of peak viscosity value (PKV) followed the procedure described in NY/T 1765–20,097.
For starch granules observation by scanning electron microscopy, rice grains were dried completely in 37 °C for 2 weeks and cooled. The grains were cut across the short axis with a surgical blade slightly, and the resulted surfaces were sputter coated with gold and observed by scanning electron microscopy (JEOL JSM-6360LV).
Reverse transcription and quantitative PCR analysis
Total RNA from shelled seeds of 10 DAP was extracted using EASYspin Plus Complex Plant RNA Kit (Aidlab, Beijing, China), treated with RNase-free DNase I, and used for cDNA synthesis with PrimeScript™ RT Master Mix (TaKaRa, Japan) according to the manufacturer’s instructions. qPCR was performed using TB Green® Premix Ex Taq™ II (TaKaRa, Japan) by LightCycler® 480 (Roche Diagnostics, Rotkreuz, Switzerland). Waxy gene was amplified with primers 5′-TCATCGAAGGCAAGACTGGT-3′ and 5′-TCCTGGTTCATGCAGTTCCT-3′. The OsACT1 gene was amplified (with primers 5′-CCTTCAACACCCCTGCTATG-3′ and 5′-TGAGTAACCACGCTCCGTCA-3′) as an internal control for normalization.
Protoplast transfection
Rice protoplasts used in this study were isolated following the procedure below. Briefly, stem and sheath tissues derived from seedlings 12 day after germination were cut into 1 mm strips, and incubated in enzyme solution for 4 h at 28 °C with gentle shaking (40–60 rpm) in darkness. Protoplasts were collected and washed by W5 solution and resuspended in MMG solution. For transient expression assays, 1 μg plasmid DNA was mixed with freshly prepared protoplasts (~ 1 × 107 cells) and equal volume PEG4000 solution (40%, W/V) (Sigma–Aldrich Biotech, St. Louis, MO), and cultured at 28 °C in the dark for 16 h.
Dual-luciferase assay
Cloning of the 2 kb cis-regulatory sequences: WT-ZZ35 (pro-ZZ35) and WT-Nip were 2 kb DNA sequences immediately upstream the Waxy coding region, amplified by Tks Gflex™ DNA Polymerase (TaKaRa, Dalian, China) with primers (Full-S: 5′-tcgacggtatcgataGCAGGCACTCAGCTCGCT-3′ and Full-R: 5′-gcggccgctctagaAGGTGGTTGTCTAGCTGTTGCT-3′) from the genomic DNA of ZZ35 and Nipponbare, respectively. del-WP1, del-WP2, and del-WP3 were composed of two fragments before and after the deletions in the respective genome editing alleles, and the length defects caused by deletions were supplemented by the upstream sequences to ensure the length of 2000 bp (± 10 bp). DNA fragments were amplified with primers: WP1-seg1 (5′-tcgacggtatcgataTGCGAATTCGGTCGTCG-3′ and 5′-ctgaataaCCCACGGCTGTAATAAGCTAG-3′); WP1-seg2 (5′-ccgtgggTTATTCAGATCGATCACATGCAT-3′ and Full-R); WP2-seg1 (5′-tcgacggtatcgataGAAAAGCGCAGGTAATTGAC-3′ and 5′-ccaaaataGGGAAACAAAGAATTATAAAC-3′); WP2-seg2 (5′-gtttcccTATTTTGGGTATAGGTCAAAGC-3′ and Full-R); WP3-seg1 (5′-tcgacggtatcgataGAAAAGCGCAGGTAATTGAC-3′ and 5′-ccctcctAGAGGGGAAACAAAGAATTATAAAC-3′); and WP3-seg2 (5′-tcccctctAGGAGGGTTTATTTTGGGTAT-3′ and Full-R). Chimeric fragments of V1–V4 segments were amplified with the different matching pairing primers (Full-S; V2-zz-R: 5′-ATACCTTGCAGATGTTCTTCCT-3′; V2-nip-R: 5′-ATAACTTGCAGATGTTCTTCCT-3′; V3-zz-F: 5′-GGAAGAACATCTGCAAGGTAT-3′; V3-nip-F: 5′-GGAAGAACATCTGCAAGTTAT-3′; V3-R: 5′-GCCTAACCAAACATAACGAAC-3′; V4-F: 5′-GTTCGTTATGTTTGGTTAGGCT-3′; Full-R) from genomic DNA of ZZ35 or Nipponbare.
Dual-luciferase assays were performed as described previously (Xu et al. 2013) using modified pGreenII-0800-LUC plasmids (Hellens et al. 2015). The 2 kb cis-regulatory sequences were cloned into the promoter region of firefly luciferase (FLuc) with pEASY®-Basic Seamless Cloning and Assembly Kit (TransGen, Beijing, China) according to the manufacturer’s instructions. The luciferase activity was detected with Dual Luciferase Reporter Assay Kit as the technical manual (Vazyme, Nanjing, China).
Data availability
All data supporting the results of this study are available within the article.
Abbreviations
- ACs :
-
Amylose contents
- BDV :
-
Breakdown value
- CPV :
-
Cold paste value
- CSV :
-
Consistence value
- DAP :
-
Days after pollination
- GC :
-
Gel consistency
- HPV :
-
Hot paste value
- MC :
-
Moisture content
- Nip :
-
Nipponbare
- PKV :
-
Peak viscosity value
- RVA :
-
Rapid Visco Analysis
- ZZ35 :
-
Zhongzao35
References
Chen K, Wang Y, Zhang R, Zhang H, Gao C (2019) CRISPR/Cas genome editing and precision plant breeding in agriculture. Annu Rev Plant Biol 70:667–697
Chung BY, Simons C, Firth AE, Brown CM, Hellens RP (2006) Effect of 5′UTR introns on gene expression in Arabidopsis thaliana. BMC Genomics 7:120
Gao H, Gadlage MJ, Lafitte HR, Lenderts B, Yang M, Schroder M, Farrell J, Snopek K, Peterson D, Feigenbutz L, Jones S, St Clair G, Rahe M, Sanyour-Doyel N, Peng C, Wang L, Young JK, Beatty M, Dahlke B, Hazebroek J, Greene TW, Cigan AM, Chilcoat ND, Meeley RB (2020) Superior field performance of waxy corn engineered using CRISPR-Cas9. Nat Biotechnol 38:579–581
Hellens RP, Allan AC, Friel EN, Bolitho K, Grafton K, Templeton MD, Karunairetnam S, Gleave AP, Laing WA (2015) Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 18:13
Hiei Y, Komari T, Kubo T (1997) Transformation of rice mediated by Agrobacterium tumefaciens. Plant Mol Biol 35:205–18
Hua K, Zhang J, Botella JR, Ma C, Kong F, Liu B, Zhu JK (2019) Perspectives on the application of genome-editing technologies in crop breeding. Mol Plant 12:1047–1059
Huang L, Li Q, Zhang C, Chu R, Gu Z, Tan H, Zhao D, Fan X, Liu Q (2020) Creating novel Wx alleles with fine-tuned amylose levels and improved grain quality in rice by promoter editing using CRISPR/Cas9 system. Plant Biotechnol J 18:2164–2166
Isshiki M, Morino K, Nakajima M, Okagaki RJ, Wessler SR, Izawa T, Shimamoto K (1998) A naturally occurring functional allele of the rice waxy locus has a GT to TT mutation at the 5-splice site of the first intron. Plant J 15:133–138
Kim MJ, Kim H, Shin JS, Chung CH, Ohlrogge JB, Suh MC (2006) Seed-specific expression of sesame microsomal oleic acid desaturase is controlled by combinatorial properties between negative cis-regulatory elements in the SeFAD2 promoter and enhancers in the 5′-UTR intron. Mol Genet Genom 276:351–368
Larkin PD, Park WD (2003) Association of waxy gene single nucleotide polymorphisms with starch characteristics in rice. Mol Breed 12:335–339
Liu L, Gallagher J, Arevalo ED, Chen R, Skopelitis T, Wu Q, Bartlett M, Jackson D (2021) Enhancing grain-yield-related traits by CRISPR-Cas9 promoter editing of maize CLE genes. Nat Plants. https://doi.org/10.1038/s41477-021-00858-5
Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, Wang B, Yang Z, Li H, Lin Y, Xie Y, Shen R, Chen S, Wang Z, Chen Y, Guo J, Chen L, Zhao X, Dong Z, Liu YG (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284
Miao C, Xiao L, Hua K, Zou C, Zhao Y, Bressan RA, Zhu JK (2018) Mutations in a subfamily of abscisic acid receptor genes promote rice growth and productivity. Proc Natl Acad Sci U S A 115:6058–6063
Mohammadi P, Castel SE, Brown AA, Lappalainen T (2017) Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res 27:1872–1884
Oliva R, Ji C, Atienza-Grande G, Huguet-Tapia JC, Perez-Quintero A, Li T, Eom JS, Li C, Nguyen H, Liu B, Auguy F, Sciallano C, Luu VT, Dossa GS, Cunnac S, Schmidt SM, Slamet-Loedin IH, Vera Cruz C, Szurek B, Frommer WB, White FF, Yang B (2019) Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat Biotechnol 37:1344–1350
Rodríguez-Leal D, Lemmon ZH, Man J, Bartlett ME, Lippman ZB (2017) Engineering quantitative trait variation for crop improvement by genome editing. Cell 171:470–480
Vetrici MA, Yevtushenko DP, Misra S (2021) Douglas-fir LEAFY COTYLEDON1 (PmLEC1) is an active transcription factor during zygotic and somatic embryogenesis. Plant Direct 5:e00333. https://doi.org/10.1002/pld3.333
Salimonti A, Carbone F, Romano E, Pellegrino M, Benincasa C, Micali S, Tondelli A, Conforti FL, Perri E, Ienco A, Zelasco S (2020) Association study of the 5′UTR intron of the FAD2-2 gene with oleic and linoleic acid content in Olea europaea L. Front Plant Sci 11:66
Shen L, Wang C, Fu Y, Wang J, Liu Q, Zhang X, Yan C, Qian Q, Wang K (2018) QTL editing confers opposing yield performance in different rice varieties. J Integr Plant Biol 60:89–93
Song X, Meng X, Guo H, Cheng Q, Jing Y, Chen M, Liu G, Wang B, Wang Y, Li J, Yu H (2022) Targeting a gene regulatory element enhances rice grain yield by decoupling panicle number and size. Nat Biotechnol. https://doi.org/10.1038/s41587-022-01281-7
Tian Z, Qian Q, Liu Q, Yan M, Liu X, Yan C, Liu G, Gao Z, Tang S, Zeng D, Wang Y, Yu J, Gu M, Li J (2009) Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. Proc Natl Acad Sci U S A 106:21760–21765
Wang ZY, Zheng FQ, Shen GZ, Gao JP, Snustad DP, Li MG, Zhang JL, Hong MM (1995) The amylose content in rice endosperm is related to the post-transcriptional regulation of the waxy gene. Plant J 7:613–622
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu JL (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947–951
Wang Q, Su Q, Nian J, Zhang J, Guo M, Dong G, Hu J, Wang R, Wei C, Li G, Wang W, Guo HS, Lin S, Qian W, Xie X, Qian Q, Chen F, Zuo J (2021) The Ghd7 transcription factor represses ARE1 expression to enhance nitrogen utilization and grain yield in rice. Mol Plant 14:1012–1023
Xu YZ, Kanagaratham C, Jancik S, Radzioch D (2013) Promoter deletion analysis using a dual-luciferase reporter system. Methods Mol Biol 977:79–93
Yano K, Morinaka Y, Wang F, Huang P, Takehara S, Hirai T, Ito A, Koketsu E, Kawamura M, Kotake K, Yoshida S, Endo M, Tamiya G, Kitano H, Ueguchi-Tanaka M, Hirano K, Matsuoka M (2019) GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture. Proc Natl Acad Sci U S A 116:21262–21267
Zeng D, Liu T, Ma X, Wang B, Zheng Z, Zhang Y, Xie X, Yang B, Zhao Z, Zhu Q, Liu YG (2020) Quantitative regulation of Waxy expression by CRISPR/Cas9-based promoter and 5'UTR-intron editing improves grain quality in rice. Plant Biotechnol J 18:2385–2387
Zeng F, Roslinsky V, Cheng B (2017) Mutations in the promoter, intron and CDS of two FAD2 generate multiple alleles modulating linoleic acid level in yellow mustard. Sci Rep 7:8284
Zhang C, Zhu J, Chen S, Fan X, Li Q, Lu Y, Wang M, Yu H, Yi C, Tang S, Gu M, Liu Q (2019) Wxlv, the ancestral allele of rice Waxy gene. Mol Plant 12:1157–1166
Zhang J, Zhang H, Botella JR, Zhu JK (2018) Generation of new glutinous rice by CRISPR/Cas9-targeted mutagenesis of the Waxy gene in elite rice varieties. J Integr Plant Biol 60:369–375
Zhang Y, Pribil M, Palmgren M, Gao C (2020) A CRISPR way for accelerating improvement of food crops. Nat Food 1:200–205
Acknowledgements
We gratefully acknowledge Prof. Yaoguang Liu for kindly providing the pYLCRISPR/Cas9 vector and Prof. Zhongnan Yang for providing the pGreenII-0800-LUC vector.
Funding
This work was supported by National Key R&D Program of China (2018YFA0900600).
Author information
Authors and Affiliations
Contributions
Q.Z. and S.Z. performed most of the experiment and data analysis. X.Y. and X.W. contributed to the phenotype analysis and rice planting. X.Z. and X.H. designed the experiments and wrote the paper. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Q., Zhang, S., Yu, X. et al. Fine-tuning grain amylose contents by genome editing of Waxy cis-regulatory region in rice. Mol Breeding 42, 72 (2022). https://doi.org/10.1007/s11032-022-01342-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-022-01342-4