
Abstract
Root system architecture (RSA) has positive effects on wheat growth and its yield performance. As root features are difficult to manipulate through conventional breeding strategies, marker-trait association (MTA) could be helpful for the improvement of RSA. In the present study, 112 durum wheat genotypes were investigated for several root system features as well as some agronomic traits. The population was genotyped using a 15K SNP and a total of 3321 markers were used in the association analysis. A total of 581 significant marker-trait associations were identified in all of the 14 chromosomes. The percentages of phenotypic variation (R2) of measured traits varied between 8.76 and 24.81%. Out of 581 associated markers, 125 loci were linked with multiple traits. The most significant associations on measured traits were detected for genome B compared to genome A (61% vs 39%). Also, major associated loci existed on chromosomes 1A, 1B, 2B, 3B, and 5B, which could be considered in the future breeding programs to manipulate relevant traits using marker-assisted selection procedures. Among associated SNP markers, most markers were related to the number of days to spike heading (181), anthesis (53), booting (69), physiological maturity (41), water use efficiency (109), and transpiration efficiency (24). Furthermore, the large number of QTLs (167 in total) for RSA and agronomic traits were detected. The highest numbers of QTLs were related to WUE (23), DAS (23), DB (21), and DA (20) than other traits. Among the detected QTLs, 16 QTLs for RSA overlapped with different agronomic traits, as well as 6 QTLs co-located with other RSA traits in at last one trait. These results are helpful for better understanding the genetic basis of root system features and agronomic traits. Furthermore, these results could be valuable for facilitating pyramiding of the ideal alleles using the MAS approach for favorable plant-type and high water use efficiency in the future durum wheat breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among cereal crops, wheat is a widespread species with a major contribution to the human diet, providing dietary proteins, carbohydrates, fiber, calcium, zinc, fats, and energy (Mwadzingeni et al. 2017; Boukid et al. 2019). Durum wheat (Triticum turgidum var. durum Desf.) is an allotetraploid species (2n = 4x = 28) with the genomic constitution of AABB, comprising the full diploid complement of chromosomes from T. urartu Ghandilyan (A genome) and Aegilops speltoides Tausch (putative B genome). It has been reported that the durum wheat production is approximately 40 million tons in 2016 (Giunta et al. 2019).
Root system architecture (RSA)––the hidden half of a plant––which plays a vital role in crop growth, anchors the aboveground plant tissues, uptake of water and nutrients necessary of the deep layers of the soil, and is the biosynthetic site of phytohormones required for plant development and even yield (Ma et al. 2017; Ju et al. 2018). During the first green revolution, the development of the new wheat varieties has mainly been achieved based on selection for above-ground organs, whereas the roots were mainly neglected (Waines and Ehdaie 2007). The main reason for the neglect of selecting root system in breeding programs is due to the difficulty of assessing field-grown plants for these features, especially when dealing with a large number of genotypes required for association analysis or quantitative trait loci (QTL) mapping (Cane et al. 2014). Nonetheless, quantifying RSA is important to achieve high productivity in wheat because yield performance is influenced by the accessibility and availability of nutrients and water, which are heterogeneously distributed in field soil (Ehdaie et al. 2016; Ren et al. 2017).
A study on phenotypic variability of RSA and its relationship with genetic diversity in the durum wheat germplasm provides the various perspectives for utilizing unexploited traits that can be used to increase the yield of durum cultivars across a range of environmental conditions. Lack of the high-throughput methods for phenotypic investigation of the root features is the main challenge in the breeding programs that can offer a proxy for field performance; therefore, developing marker-assisted selection (MAS) will support breeders to select the desirable root traits followed by the selection of the best genotypes with an efficient acquisition of water and nutrients from soils (Ren et al. 2017). Indeed, having molecular markers for the root system and architecture features would be a powerful tool for direct selecting for desired root traits in wheat breeding programs. Nowadays, there is increasing interest in genetic dissection of RSA to focus on the associations among genes and root traits. Several types of molecular markers, such as amplified fragment length polymorphisms (AFLP), simple sequence repeats (SSR), diversity arrays technology (DArT), sequence-tagged sites (STS), and single-nucleotide polymorphisms (SNP), have been developed for dissecting molecular variability and association analysis in durum wheat germplasm (e.g., Siosemardeh et al. 2012; Can et al. 2014; Hu et al. 2015; Baloch et al. 2017; Huang et al. 2018). Of these systems, SNPs are the most common polymorphisms among individuals of various crops (Deschamps and Campbell 2010). The accessibility of SNP genotyping platforms would simplify the genetic dissection of important traits and the application of marker-assisted and genomic selection (Trebbi et al. 2011). SNPs are the most frequent form of DNA variations and they can be converted into genetic markers amenable to high-throughput assays (Varshney et al. 2009). Hence, SNPs are the valuable markers to use in assessing population structure, genetic diversity, and association analyses in numerous organisms (Hu et al. 2015).
Association analysis is an approach based on linkage disequilibrium (LD) and is used to dissect the relationship between phenotypic and genotypic data (Flint-Garcia et al. 2003). Several advantages, such as archive to a higher density of molecular data in a short time and development of markers with minimum costs, have resulted in association analysis being used as an alternative method to the mapping of QTLs and genes in many crops (Mackay and Powell 2007; Zhu et al. 2008). Although many reports are available on the population structure patterns and association analysis in durum wheat germplasm (Hu et al. 2015; Baloch et al. 2017; Mwadzingeni et al. 2017; Huang et al. 2018; Sukumaran et al. 2018; Ruiz et al. 2018), there are few reports on root trait-marker associations in this cereal crop (Maccaferri et al. 2016; Alahmad et al. 2019). Hence, the main goal of the present study is to disclose associations between the root system features with the SNP markers in the core collection of durum wheat.
Materials and methods
Plant materials and growing conditions
A total of 112 durum wheat genotypes collected from different regions of Iran and various countries (Table 1) were evaluated for phenotypic and genotypic diversity in the root system and architecture features (Table 2). These materials consisted of landraces and cultivated genotypes that were preserved in the gene bank at the Ilam University. Seeds of each genotype were planted in polyvinyl chloride (PVC) tubes in a greenhouse at the Department of Agronomy and Plant Breeding, Ilam University, Ilam, Iran (Latitude 33° 39′ N, Longitude 46° 22′ E) during 2015–2016. In this work, we measured root system traits at two stages––the seminal roots (7-day-old seedlings) and advanced root growth (35-day-old seedlings)––along with some agronomic traits.
Key root system features and agronomic traits phenotype
For measuring the seminal root traits, a pot experiment was performed. Seeds of each genotype were kept at 4 °C for 48 h. Four seeds from each genotype were sown under 2 cm mixture of sand and soil in a ratio of 1:3 into plastic pots (10 cm diameter, 20 cm height). All genotypes were germinated and their seedlings were grown in controlled conditions with an optimal growing photoperiod cycle (16 h day and 8 h night) and temperature (25 °C day and 20 °C night). The pots were arranged in a randomized complete block design with two replications. Ten days after sowing, the number of seminal roots (SRN) was recorded when the first leaf emerged (Zadoks et al. 1974). Seminal root traits were measured 10 days after planting. For this purpose, in each pot, shoots were cut at the base then pots were emptied and the roots washed carefully from soil. In the second experiment, the seedling root traits were measured 5 weeks after planting. For this purpose, four seeds of each genotype were planted into polyvinyl chloride (PVC) tubes (65 cm long, 10 cm diameter) filled with a mixture of sand and soil in a ratio of 1:3. All tubes were arranged into a randomized complete block design with three replications. Plants were irrigated twice weekly with tap water. At the three-leaf stage of seedling growth, in each tube, two plants were retained and others removed. At the end of the 35-day dry-down period, plants were uprooted, thoroughly washed, and their root system were subjected to manual/visual phenotyping (Table 2).
In the third experiment, several agronomic traits and root traits were measured at the whole plant stage. Similar to previous experiments, four seeds of each genotype were sown into PVC tubes filled with a mixture of soil and sand. The used tubes in this experiment characterized using 1 m long and 14 cm in diameter. All tubes were arranged into a randomized complete block design with three replications. After establishing of plants, seedling plants were thinned to one plant per tube. Plants were irrigated twice weekly with tap water. During growth, the several phonological and agronomic traits were recorded. Finally, each tube was emptied and roots washed carefully from the soil, and several root traits were measured. All measured root system features and agronomic traits are listed in Table 2.
DNA isolation and SNP genotyping
Genomic DNA of the 112 genotypes was isolated from fresh leaves following the method described by Doyle and Doyle (1990) with minor modifications. DNA quality and concentration were determined using electrophoresis on 1% agarose gel. SNP genotyping was performed by Trait Genetics GmbH (http://www.traitgenetics.com/en/), using a 15k Illumina SNP chip developed from the 90k iSelect chip described by Wang et al. (2014). SNPs with minor allele frequency (MAF) < 0.05 and missing values > 10% were removed from the subsequent analysis which left a set of 3321 polymorphic SNP markers.
Statistical analysis
Descriptive statistics were computed based on mean values for measured root system features and agronomic traits across all genotypes using XLSTAT software (XLSTAT 2017). The coefficient of variation (CV) was estimated by dividing the standard deviation (SD) to the mean value multiplied by 100 (Bashiri et al. 2017). Pearson correlation coefficients were used to determine the general relationship between measured trait pairs. Variability in agronomic traits and root system features was determined by a principal component analysis (PCA). Population structure analysis was computed based on a Bayesian model using STRUCTURE v2.3.4 software (Pritchard et al. 2000). To allocate the genotypes into sub-populations (K), a continuous series of K were tested from 1 to 10 in 10 independent runs using 10,000 burn-in iterations followed by 100,000 Markov Chain Monte Carlo (MCMC) iterations. Online available STRUCTURE HARVESTER (Earl and vonHoldt 2012) was used to determine the most likely number of K. TASSEL ver5.2.32 (Bradbury et al. 2007) was employed to compute the association analysis. Linkage disequilibrium (LD) was investigated using 3321 SNP markers with known map position across 14 durum wheat chromosomes. Pairwise LD was estimated using the squared allele frequency correlations (r2) between markers. Associations between all traits described above and SNPs were analyzed with a mixed linear model (MLM) by incorporating genotypes, phenotypes, the Q-matrix (Q), and the kinship matrix (K) [MLM (Q + K)]. A LOD value > 3 was used as a threshold P value for both SNP-marker-trait associations (Hwang et al. 2014).
Results
Phenotypic assessment
The phenotypic performance of the recorded root system features and agronomic traits for the durum wheat population under controlled growth conditions is shown in Table 3. In addition, distribution histograms of the measured traits are presented in Figs. S1–3. The value of the coefficients of variance (CV) for measured traits varied between 3.25 (the number of days to physiological maturity [DPM]) and 50.34% (transpiration efficiency [TE]). Eleven traits had CV values ≥ 30% (Table 3). Pearson correlation coefficient analysis on the 26 measured traits indicated a high association among some of the individual traits. The number of traits significantly associated to individual traits ranged from 2 to 10 at P ≤ 0.5 and P ≤ 0.01 (Table 3). For instance, root fresh weight (RFW) significantly correlated with all root traits except the number of seminal roots (NSR), maximum seminal root length (MRL), mean of seminal roots (MSR), and total seminal root length (TSR). Besides, a significant positive correlation was observed between grain yield (GY) with the number of grain per spike (GPS), grain size (GS), 100-grains weight (GW), water use efficiency (WUE), and TE (Table S1).
To account for these associations, multivariate traits were computed using principal component analysis (PCA), resulting in eight PCs with eigenvalues > 1. The number of root traits and agronomic characters allocated to an individual PC varied between 1 and 11, with PC7 and PC8 containing only grain filling period (GFP) and GW, respectively, and PC1 containing several root traits including RFW, root dry weight (RDW), volume (RV), length (RL), surface area (RSA), fineness (RF), diameter (RD), surface density (RSD), length density (RLD), specific length (SRL), and tissue density (RTD). Overall, eight components accounted for 83.51% of the total phenotypic variation (Table S2). Among these, the first two PCs (PC1 = 23.86% and PC2 = 15.24%) represented the highest contribution in explaining the total variation. Hence, the scatter plot was rendered based on these PCs. As shown in Fig. 1, all measured traits grouped into three clusters (CI–CIII). CI consisted of all agronomic traits along with seminal root features except GFP, CII consisted of all seedling root traits except RF and SRL, and two last root traits along with GFP grouped into CIII.

PCA-based biplot-based first two PCs. a Distribution of the 112 durum wheat genotypes based on the two first components. b Circle of correlations for a PCA conducted on the 112 durum wheat genotypes and 26 traits. NSR the number of seminal roots, MRL length of maximum seminal root, MSR average of seminal root, TSR sum of seminal root length, RFW root fresh weight, RDW, root dry weight, RL root length, RSA root surface area, RF root fineness, RD root diameter, RSD root surface density, SRL specific root length, RLD root length density, RTD root tissue density, RV root volume, DAS days to appearance of spike, DA days to anthesis, DB days to booting, DPM days to physiological maturity, GFP grain filling period, GPS number of grains per spike, GW 100-grains weight, GY grain yield, WUE water use efficiency, TE transpiration efficiency
Population structure and genetic relationships
SNP markers were used for estimating the genetic structure of the durum genotypes using the Bayesian clustering model implemented in the STRUCTURE software and an ad hoc statistics (ΔK) calculation was used to estimate the true number of sub-populations. The maximum value of ΔK was obtained at K = 3, and consequently, genotypes fell into three sub-populations with 14, 35, and 63 genotypes, respectively (Fig. 2). Each of these sub-populations (SPI–SPIII) consisted of diverse genotypes originating from different regions, so that grouping of genotypes was not in accordance with their origins. The genetic divergence among the three sub-populations was estimated through pair-wise FST, which varied between 0.03 for SPII and 0.80 for SPI.

Population structure based on 112 durum wheat genotypes and 3321 SNP markers. Each colored bar per genotype estimates the membership fraction to each of the three populations. See Table 1 for codes of genotypes
LD decay analysis
In order to identify differences in intra-chromosomal LD across all 14 chromosomes, the average r2 values between pairs of markers were classified into five groups (intervals) based on the genetic distance between them (Table 4). The LD analysis showed a difference in the r2 values of the chromosome groups. The highest LD was for markers with distance less than 5 cM. For genetic distances less than 5 cM, the highest LD was observed on 6A chromosome, while the lowest was for 1B and 6B chromosomes. For marker pairs with an inter-marker distances more than 50 cM, chromosomes 5A, 4B, and 3A presented higher LD than others. The plots of the LD decay for all 14 durum wheat chromosomes are illustrated in supplementary Fig. S4. LD analysis was also conducted at the whole genome level. To visualize the extent of LD in the whole genome, average LD decay was derived so that intra-chromosomal r2 values were plotted against the genetic distance (cM) between markers (Fig. S4).
Marker-trait associations
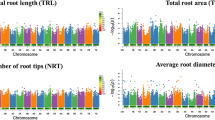
An overview and additional information of marker-trait associations (MTAs) under the MLM model are shown in Table S2. The result indicated 581 significant associations in total for 26 agronomic and root system traits. Large numbers of associated SNP markers, 181, 109, 53, 41, and 26, were found for the number of days to appearance of spike (DAS), WUE, the number of days to booting (DB), the number of days to anthesis (DA), DPM, and TE, respectively. In contrast, only one SNP marker was significantly associated with RV, RF, MRL, MSR, TSR, and RFW, and no significant SNPs were found for NSR, RSA, and RSD (Fig. 3a). The most significant association for measured traits were detected for chromosomes B relatives to chromosomes A (61% vs. 39%) (Fig. 3b). Among all of the 14 chromosomes, 5A and 2B embraced the most significant SNPs (Fig. 3c, d). Most of these significant SNPs (73%) can describe individually over 10–24% of the total phenotypic variation. Also, 12 trait-SNP association pairs indicated a R2 higher than 15%. Furthermore, there were 125 SNPs associated with multiple traits (two to five) (Table S4), while a total of 456 unduplicated SNPs were found for 26 root system and agronomic traits (Table S3).

Summary of marker-trait associations (MTAs). a Number of MTAs for each trait. b Pie chart showing the percentage of MTAs in different genomes. c Pie chart showing the percentage of MTAs in the different A chromosomes. d Pie chart showing the percentage of MTAs in the different B chromosomes
MTAs for seminal root traits
Association analysis was done for four seminal root traits across the first seedling stage (7-day-old seedlings). Only one duplicated and unduplicated SNP markers, Kukri_c62142_683 and Ku_c106533, were significantly associated with MRL, MSR, and TSR, respectively. These SNPs were located on 2A and 1B chromosomes, respectively. No significant SNP marker was found for the NSR trait (Table S3).
MTAs for seedling root traits
The results of the association analysis for this stage indicated that 52 SNP markers were significantly associated with the measured traits at the second stage of seedlings growth (35-day-old seedling) (Table S3). One significant SNP marker, wsnp_Ex_c10550_17231294, was duplicated for RV and RF. The marker explained 11.92 and 13.53% of the total phenotypic variation for these traits, respectively, and was located on chromosome 7B. Only one SNP marker, Excalibur_c74397_238, was significantly associated with RFW and this marker was located on chromosome 4A. Eight markers in total were found to be significantly associated with RL. The percentage of phenotypic variation explained by these markers (Marker R2) ranged from 10.36 (BS00089894_51) to 24.74% (wsnp_Ex_rep_c109742_92411838 and wsnp_Ku-c21316_31053745). Apart from SNP marker Excalibur_c35713_106, which was located on chromosome 6B, other markers were located on chromosome 1A. For RD, three significant markers were detected on chromosomes 2B (RFL_Conting4856_71) and 7B (Kukri_c50071_1084 and Excalibur_c21739_688). Six SNPs were found to be significantly associated with RTD. Marker R2 for these SNPs ranged from 9.73 (RAC875_c34512_685 and RAC875_c34512_493) to 15.50% (BS000068200_51). These markers were located on 2B and 5B chromosomes. A total of 13 SNPs were significantly associated with RLD. The highest R2 values were observed for SNP markers wsnp_Ex-rep_c109742_92411838 and wsnp_Ku-c21316_31053745 (24.81%) while the lowest were for BS00089894_51 (10.36%). The SNPs associated with RLD were located on 1A, 4A, and 6B chromosomes. Thirteen different SNPs were significantly associated with SRL and they had the same R2 value (varied between 10.27 and 11.84). These markers were located on 2A, 6A, 2B, 3B, 4B, and 7B chromosomes. Six SNPs were significantly associated with RDW, and all of them were located on 2B and 5B chromosomes. Of these, SNP marker BS000682_51 had the highest phenotypic effect of R2 = 15.50%.
MTAs for agronomic traits
In total, 500 SNP markers were significantly associated with the measured phenological and agronomic traits at the whole plant stage (Table S3). One hundred eighty-one markers were identified to be significantly associated with DAS. The coefficient of determination (R2) was ranged from 8.76 to 19.4%. These DAS-associated SNPs were located on all of the 14 chromosomes except 3A and 4A. The SNP marker wsnp_Ex-c21092_30220342 explained the highest phenotypic variation (19.44%). DA was significantly associated with 53 SNP markers. These markers were distributed on 12 of the 14 chromosomes, 1A, 2A, 5A–7B. The lowest and highest R2 were observed for SNP markers Excalibur_c3730_2098 (9.3%) and Tdurum_conting11613_329 (21.6%), respectively. In total, 69 SNPs were found to be significantly associated with DB. For these markers, the R2 coefficients were varied between 8.9 (wenp_Ku_c22358_32187765) and 17 (Excalibur_c12996_775 and BS00109319_51). These markers were located on all of the 14 chromosomes except 3A and 4A. Forty-one SNP markers showed significant association with DPM and all of them were distributed on chromosomes 1A, 2A, 5A, 7A, 1B, 2B, 3B, 6B, and 7B. The R2 coefficients ranged from 9.14 (Tdurum_conting51640_847) to 18.14 (wsnp_Ex_c21092_30220342). Unlike other phonological traits, only two SNP markers, Ku_c32100_105 and BS00067983_51, were detected to be significantly associated with GFP. These GFP-associated SNPs were located on chromosome 6B. The association analysis for GPS revealed seven significant SNP markers on chromosomes 6A, 1B, 2B, and 4B. Of these, SNP marker BS00104364_51 showed the highest value of R2 (13.34%). For GS, six significant SNPs were detected on chromosomes 5A, 7A, 3B, and 5B. The coefficients of determination for these markers ranged from 9.57 (IACX6007) to 10.84% (RFL_Conting3811_3709). In total, 15 and 17 SNP markers were indicated to be significantly associated with GW and GY, respectively. The GW-associated SPNs were located on chromosomes 3A, 7A, 2B, 3B, 4B, 5B, 6B, and 7B. Of these, six SNPs, Excalibur_c19499_948, BobWhite_c9711_71, Kukri_c94033_432, TA002682-0717, BobWhite_c13098_526, and Excalibur_c28715_447, showed a R2 > 15%. The GY-associated SNP markers mainly were located on chromosome 2B (9 SNPs); however, the chromosomes 3A, 5A, 6A, 1B, 3B, and 4B were included 1, 1, 1, 1, 2, and 2 SNP markers, respectively. The R2 for GY-associated SNPs ranged from 9.69 (RAC875_c7540_366) to 14.81% (wsnp_Ex_c7203_12370983). For TE, 26 significant SNP markers were identified. These markers were located on chromosomes 2A, 3A, 6A, 2B, 3B, 4B, and 6B with a range of R2 between 9.50 and 17.21%. The highest R2 belonged to SNP marker BobWhite_c1196_297 and was located on chromosome 3B. Furthermore, association analysis for WUE showed 109 significant SNPs on chromosomes 1A, 2A, 3A, 5A, 6A, 2B, 3B, 4B, and 5B. The R2 values ranged from 9.41 to 22.49% and the SNP markers wsnp_Ex_c7203_12370983 and Excalibur_c3171_416 showed the highest and lowest values, respectively.
GWAS-QTL detection
Genome-wide association analysis (GWAS) revealed 167 significant QTLs across the different chromosomes (Table 5). From genome viewpoint, B genome had more QTLs compared to A genome (102:65). The range of detected QTLs varied between 2 and 20, and the minimum and maximum numbers were related to chromosomes 4A and 3B, respectively. In total, we identified 23 and 143 QTLs for RSA and agronomic traits, respectively. Among 23 QTLs, 16 QTLs overlapped with QTLs for agronomic traits. Of these, 4, 3, 3, 2, 2, 1, and 1 QTLs were identified on chromosomes 3B, 2B, 5B, 2A, 6A, 1A, and 7B, respectively. Furthermore, six QTLs co-located with other RSA traits in at least one trait. Among 143 QTLs for agronomic traits, 38 QTLs co-located with other traits. The highest numbers of QTLs were related to WUE (23), DAS (23), DB (21), and DA (20) than other traits; however, there are no QTLs for NSR, RSA, and RSD.
Discussion
Durum wheat is one of the most important cereal crops in the world. The knowledge on genetic bases of complex traits in this crop provides an opportunity for future breeding programs (Mwadzingeni et al. 2017). To complement the growing need for such knowledge, the present work disclosed the population structure and association between genomic regions with several root system features, phonological and agronomic traits in a diverse population of durum wheat genotypes. The phenotypic evaluation revealed a high coefficient of variance (CV ≥ 30) for MRL, MSR, TSR, RFW, RDW, RF, SRL, RTD, RV, GY, TE, and WUE (Table 3). These results suggest that there is a broad genetic potential that these genotypes can be used to mine alleles for improvement of root system architecture and grain yield into elite durum wheat lines. Our results are in line with Cane et al. (2014) and Sabiel et al. (2017), who also found a high variation for root system traits as well as grain yield and other related traits. Furthermore, El Hassouni et al. (2018) revealed that grain yield has a strong association with root ratio and root angle under drought stress conditions. Hence, this finding may provide new insights for future studies by focusing on the root features. Besides, the highly significant and positive correlations (Table S1) were observed among root traits and agronomic characters, confirming the value of the data in the present MTA analysis.
Root system features and agronomic traits commonly reveal a high degree of phenotypic diversity, and this may confuse the identification of genotypic variation in these traits (Fitter 2002). Multivariate approaches provide efficient classifiers to capture phenotypic diversity and can recognize distinct groups related to the regional origin and functional plant adaptation to different environments (Bodner et al. 2013). In the present study, principal components analysis (PCA) showed that measured root and agronomic traits captured almost all of the variability (83.51%), indicating that the measured traits here were efficient in classifying distinctive groups among the tested genotypes (S2 Table). The biplot-based PCA indicated characteristics groups of traits and distribution of genotypes based on the two first PCs (Fig. 1). As shown by the angles among traits’ vectors, all seminal root features were positively correlated with grain yield and other phonological and agronomic traits (except GFP). Also, these traits positively correlated with PC2 axis. All of the seedling root traits showed a positive correlation with each other and also displayed a strong association with the PC2 axis. Formerly, these interrelationships were confirmed by coefficients correlation matrix for studied traits. Hence, suitable genotypes can be chosen based on the high values of PC1 and PC2. Accordingly, the top-right quarter was mostly occupied by desirable genotypes. Likewise, several studies showed that PCA is an efficient multivariate approach to intercept relationships among measured trait and selection of superior genotypes (Ruiz et al. 2014; Khalili et al. 2013; Phung et al. 2016; Pour-Aboughadareh et al. 2017; Ahmadi et al. 2018a, b, c).
Familial relatedness and population structure can result in false positives in GWAS (Maulana et al. 2018). Therefore, when GWAS is performed, these parameters need to be considered in the model. In this study, the genetic structure was investigated by STRUCTURE analysis. Results from the clustering of genotypes indicated that our panel is structured into three major groups. These results will be suitable for tracking potential parents that could be useful for future breeding programs (Mwadzingeni et al. 2017). It has been reported that the identification of SNPs associated with traits varies depending on the used model and traits (Gurung et al. 2014). To MAT analysis, although the general linear model (GLM) and the mixed linear model (MLM) are two statistical approaches that are used to identify SNPs associated with measured traits, the MLM model, due to its ability to control false positives, was selected to perform population structure and K matrix. Association analysis of important root system features and agronomic traits in durum wheat genotypes revealed that the MLM model was effective in detecting significant MTAs. This finding is consistent with the findings of Hu et al. (2015), Huang et al. (2018), and Maulana et al. (2018). Therefore, SNP markers identified based on this model may be explored by candidates for future studies through marker-assisted selection (MAS). In the present study, a total of 581 significant SNP markers associated with studied root and agronomic traits were detected in all of the 14 chromosomes (Table S3). This will add to previously identified genomic regions influencing similar or complementary traits (Le Gouis et al. 2012; Edae et al. 2013; Hu et al. 2015; Mwadzingeni et al. 2017; Huang et al. 2018; Sukumaran et al. 2018). The explained coefficient of determination for the detected associations was high (range 8.76–24.80%), revealing numerous genes contributing a very huge amount to a quantitative trait. A large number of associations indicated a moderate variation (R2 ≤ 15%) for each trait (S3 Table); hence, this may be attributed to markers exhibiting rare alleles, small quantitative effect, and or complex allelic interactions (Debibakas et al. 2014). Moreover, the most significant associations were observed between SNP markers and some of the agronomic traits such as DAS, DB, DA, DPM, WUE, and TE (Fig. 3a), suggesting that these SNPs are specifically distributed across the whole genome of durum wheat (Sabiel et al. 2017). As a result, more associated markers were observed in genome B than A (356 vs. 225) (Fig. 3b). This contradicts what was reported previously by Peng et al. (2011); genome A has more genes controlling important agronomic traits. Likewise, Mwadzingeni et al. (2017) and Maulana et al. (2018) reported that chromosome B harbors a large number of associated markers with various agronomic traits.
Many SNP markers (125 SNPs) revealed significant association with more than one trait (Table S4), which would be reflected co-localized marker–trait associations (Sun et al. 2015) and might be the result of pleiotropy or co-expression caused by expression quantitative trait loci (eQTLs) (House et al. 2014). Moreover, Sun et al. (2015) reported that this situation provides effective strategies to improve several traits simultaneously. In the present study, for instance, seven SNP markers––Excalibur_c5064_765, Excalibur_c6807_1155, Excalibur_rep_c67411_210, Kukri_c6552_4243, RAC875_c7540_366, wsnp_Ex_c7203_12370983, and wsnp_Ex_rep_c68194_66973531––showed simultaneously high significant associations with TE, WUE, and GY. These SNPs demonstrate great importance in genetic control of grain yield along with transpiration and water use efficiency and can be used for the MAS for the grain yield in durum wheat. Furthermore, we found that three loci––BS00094057_51, RAC875_c24767_132, and wsnp_CAP12_rep_c4278_1949864––had a strong association with WUE and GY. One SNP marker, BobWhite_c1196_297, showed significant association with traits DAS, TE, WUE, and GY. The SNP marker BS00048754_51 was closely associated with four agronomic and one root trait, DAS, DB, GY, and SRL. In general, this information is quite helpful for MAS and marker-assisted improvement (MAI) in durum wheat.
Among RSA features, RLD, SRL, and RTD are very important. Root extension and distribution can be expressed as RLD (Gao et al. 2010). This trait is one of the crucial parameters required for the uptake of nutrients and water by a plant (Pierret et al. 2000). RTD is one of the most important root traits which is related to several features of plant growth and survival. Genotypes with high RTD devote the considerable assimilates to build up one unit of root volume. Thus, this enables plants to extend their root system, resulting in high acquisition capacities for belowground resources (Wahl and Ryser 2000; Ahmadi et al. 2018b). We identified 13 SNP loci associated with RLD. Out of these, nine SNPs were distributed on chromosome 1A. Also, our results detected six significant markers associated with RTD, which were mainly distributed on chromosome 5B (Table S3). Previously, it has been reported that chromosomes 1A and 5B harbor a region controlling several root features and agronomic traits such as root length, the number of main roots, plant height, seeds per spike, and spike length (Edae et al. 2013; Cane et al. 2014; Mwadzingeni et al. 2017; Sabiel et al. 2017; Maulana et al. 2018). In addition to RLD and RTD, SRL is another important root trait, and a higher amount of this parameter shows a higher proportion of fine root length. Accordingly, genotypes with a fine root system achieve a higher SRL and exploit a higher volume of soil per unit of root biomass (Ryser 2006). Despite the important roles of these features, estimation of RLD, RTD, and SRL is hard in the field. Hence, the identification of marker associated with these parameters is an indirect way to select suitable genotypes for these traits. Our results indicated 13 significant markers associated with SRL traits, and mainly they were distributed on chromosomes 2A and 4B (Table S3). These SNPs demonstrate significant roles in genetic control of RSA and can be combined with other root trait-associated SNP markers for MAI of the wheat root system. Similarly, Cane et al. (2014) and Bai et al. (2013) revealed that most of the genome regions conferring root features were in chromosomes 1B, 2A, 5A, and 6A.
Moreover, among agronomic traits, the most significant associations were detected for DAS (181 SNPs) followed by WUE (109 SNPs), DB (69 SNPs), DA (53 SNPs), and DPM (41 SNPs), respectively (Table 3). Of these, WUE is a crucial trait in breeding for drought tolerance (Wehner et al. 2016). Indeed, this parameter dissects the amount of biomass production per unit water supplied. Therefore, WUE is not highly appropriate for yield improvement but gives valuable information on water deficiency tolerance (Blum 2009; Robredo et al. 2007). In this study, we found that SNP markers associated with WUE were distributed on chromosomes 1A, 2A, 3A, 5A, 6A, 2B, 3B, 4B, and 5B, which might reveal that different genes regulate WUE. Similarly, Lewien et al. (2016) using 750 recombinant inbred lines of spring wheat detected several genomic regions for WUE on chromosomes 1A, 6A, and 4B. Furthermore, we detected 15 and 17 associated SNP markers for GW and GY on different chromosomes (Table S3). Out of these SNPs, most loci were distributed on chromosome 2B. Because genotypes with high grain weight and grain yield are valuable for wheat breeding, the effectiveness of these regions could be very helpful for durum breeding programs. In accordance with this result, an earlier study on durum wheat identified the main controlling regions for GW and GY on chromosome 2B (Maccaferri et al. 2008; Sukumaran et al. 2018).
The large number of QTLs (167 in total) for RSA and agronomic traits evidenced in our study underlines the complexity of the genetic control of these traits already at each growth stage.
The numerous studies have revealed striking differences as to the role of specific QTLs on specific traits when the genetic dissection was based upon QTL and association mapping (Maccaferri et al. 2008; Maccaferri et al. 2011; Graziani et al. 2014). Therefore, a more comprehensive search for novel haplotypes governing the key root system and agronomic features in durum wheat should deploy larger and more genetically diverse panels such as landraces or genotypes which derived from wild relatives more likely to carry novel alleles for these features conferring adaptation to diverse environments especially drought conditions (Cane et al. 2014). The use of high-density SNP maps coupled with sequencing information will facilitate the identification of novel QTLs and in some case may also provide valuable clues on the possible candidates (Trebbi et al. 2011).
Our results revealed that approximately 10% (16:167) of RSA-QTLs simultaneously overlapped with agronomic traits including phonological (days to booting, anthesis, and physiological maturity), agronomic (water use efficiency and transpiration efficiency), and grain yield or its components (100-grains weight, number of grains per spike, and grain size) (Table 5), thus showing a notable evidence to the implications of the root system variability at seedling growth stage on the adult performance of durum wheat. This result is in accordance with Christopher et al. (2013) and Cane et al. (2014), which reported that several root system architectures have overlapped with agronomic traits in both bread and durum wheats.
Conclusions
This study detected a total of 581 highly significant SNP marker-trait associations. Some SNPs were associated (125) with multiple traits, suggesting a certain pleiotropy in the RSA and agronomic trait of durum wheat. These associations specifically were distributed across the genome, with a considerable number in B (356) than in A (225) genome. The results further approve the strong genetic control and the possibility of genetic improvement of the studied traits. Also, major associated loci existed on chromosomes 1A, 1B, 2B, 3B, and 5B, which could be used in durum wheat breeding programs to manipulate related traits using MAS procedures. In addition, the large number of QTLs (167 in total) for RSA and agronomic traits evidenced in our study, so that among them 16 QTLs for RSA overlapped with different agronomic traits. Therefore, our results contribute to understanding the genetics and breeding for key RSA features and agronomic traits.
References
Ahmadi J, Pour-Aboughadareh A, Fabriki-Ourang S, Mehrabi AA, Siddique KHM (2018a) Screening wild progenitors of wheat for salinity stress at early stages of plant growth: insight into potential sources of variability for salinity adaptation in wheat. Crop Pasture Sci 69:649–658
Ahmadi J, Pour-Aboughadareh A, Fabriki-Ourang S, Mehrabi AA, Siddique KHM (2018b) Screening wheat germplasm for seedling root architectural traits under contrasting water regimes: potential sources of variability for drought adaptation. Arch Agron Soil Sci 64:1351–1365
Ahmadi J, Pour-Aboughadareh A, Ourang SF, Mehrabi AA, Siddique KHM (2018c) Wild relatives of wheat: Aegilops–Triticum accessions disclose differential antioxidative and physiological responses to water stress. Acta Physiol Plant 40:90
Alahmad S, El Hassouni K, Bassi FM, Dinglasan E, Youssef C, Quarry G, Aksoy A, Mazzucotelli E, Juhasz A, Able JA, Christopher J, Voss-Fels KP, Hickey LT (2019) A major root architecture QTL responding to water limitation in durum wheat. Front Plant Sci 10:436
Bai C, Liang Y, Hawkesford MJ (2013) Identification of QTLs associated with seedling root traits and their correlation with plant height in wheat. J Exp Bot 64:1745–1753
Baloch FS, Alsaleh A, Shahid MQ, Çiftçi V, E. Sáenz de Miera L, Aasim M, Nadeem MA, Aktaş H, Özkan H, Hatipoğlu R (2017) A whole genome DArTseq and SNP analysis for genetic diversity assessment in durum wheat from central Fertile Crescent. PLoS One 12:e0167821
Bashiri H, Cheghamirza K, Arji I, Mahmodi N (2017) Assessing genetic diversity of Pyrus spp. in the central Zagros mountains based on morphological characters. Genet Resour crop Evol 64:391
Blum A (2009) Effective use of water (EUW) and not water-use efficiency (WUE) is the target of crop yield improvement under drought stress. Field Crop Res 112:119–123
Bodner G, Leitner D, Nakhforoosh A, Sobotik M, Moder K, Kaul HP (2013) A statistical approach to root system classification. Front Plant Sci 4:2–15
Boukid F, Dall’Asta M, Bresciani L, Mena P, Del Rio D (2019) Phenolic profile and antioxidant capacity of landraces, old and modern Tunisian durum wheat. Eur Food Res Technol 245:73–82
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Bramley H, Turner NC, Siddique KHM (2013) Water use efficiency. In: Kole C (ed) Genomics and breeding for climate resilient crops. Springer Science & Business Media, pp 225–269
Cane MA, Maccaferri M, Nazemi G, Salvi S, Frncisa R, Colalongo C, Tuberosa R (2014) Association mapping for root architectural traits in durum wheat seedlings as related to agronomic performance. Mol Breeding 34:1629–1645
Christopher J, Christopher M, Jennings R, Jones S, Fletcher S, Borrell A, Manschadi AM, Jordan D, Mace E, Hammer G (2013) QTL for root angle and number in a population developed from bread wheats (Triticum aestivum) with contrasting adaptation to water-limited environments. Theor Appl Genet 126:1563–1574
Debibakas S, Rocher S, Garsmeur O, Toubi L, Roques D, D’Hont A, Hoarau JY, Daugrois JH (2014) Prospecting sugarcane resistance to sugarcane yellow leaf virus by genome-wide association. Theor Appl Genet 127:1719–1732
Deschamps S, Campbell M (2010) Utilization of next-generation sequencing platforms in plant genomics and genetic variant discovery. Mol Breed 25:553–570
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Earl DA, vonHoldt BM (2012) Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Edae EA, Byrne PF, Manmathan H, Haley SD, Moragues M, Lopes MS, Reynolds MP (2013) Association mapping and nucleotide sequence variation in five drought tolerance candidate genes in spring wheat. Plant Genome 6:1–13
Ehdaie B, Mohammadi SA, Nouraein M (2016) QTLs for root traits at mid-tillering and for root and shoot traits at maturity in a RIL population of spring bread wheat grown under well-watered conditions. Euphytica 211:17–38
El Hassouni K, Alahmad B, Belkadi B, Filali-Maltouf L, Hickey LT, Bassi FM (2018) Root system architecture and its association with yield under different water regimes in durum wheat. Crop Sci 58:1–16
Fitter A (2002) Characteristics and functions of root systems. In: Waisel Y, Eshel A, Beeckman T, Kafkafi U (eds) Plant roots: the hidden half, CRC Press, pp 15–32
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Gao Y, Duan A, Qiu X, Liu Z, Sun J, Zhang J, Wang H (2010) Distribution of roots and root length density in a maize/soybean strip intercropping system. Agric Water Manag 98:199–212
Giunta F, Pruneddu G, Motzo R (2019) Grain yield and grain protein of old and modern durum wheat cultivars grown under different cropping systems. Field Crop Res 230:107–120
Gould SH (1955) The methods of Archimedes. Am Math Mon 62:473–476
Graziani M, Maccaferri M, Royo C, Salvatorelli F, Tuberosa R (2014) QTL dissection of yield components and morphophysiological traits in a durum wheat elite population tested in contrasting thermo-pluviometric conditions. Crop & Pasture Sci 65:80–95
Gurung S, Mamidi S, Bonman JM, Xiong M, Brown-Guedira G, Adhikari TB (2014) Genome-wide association study reveals novel quantitative trait loci associated with resistance to multiple leaf spot diseases of spring wheat. PLoS One 9:e108179
Hajabbasi MA (2001) Tillage effects on soil compactness and wheat root morphology. J Agric Sci Technol 3:67–77
House MA, Griswold CK, Lukens LN (2014) Evidence for selection on gene expression in cultivated rice (Oryza sativa). Mol Biol Evol 31:1514–1525
Hu X, Ren J, Ren X, Huang S, Sabiel SAI, Luo M, Nevo E, Fu C, Peng J, Sun D (2015) Association of agronomic traits with SNP markers in durum wheat (Triticum turgidum L. durum (Desf.)). PLoS One 10:e0130854
Huang S, Sun L, Hu X, Wang Y, Zhang Y, Nevo E, Peng J, Sun D (2018) Associations of canopy leaf traits with SNP markers in durum wheat (Triticum turgidum L. durum (Desf.)). PLoS One 13:e0206226
Hwang EY, Song Q, Jia G, Specht JE, Hyten DL, Costa J, Cregan PB (2014) A genome-wide association study of seed protein and oil content in soybean. BMC Genome 15:1
IBPGR (1985) Revised descriptor list for wheat (Triticum spp.). International Board for Plant Genetic Resources. Rome, Italy
Ju C, Zhang W, Liu Y, Gao Y, Wang X, Yan J, Yang X, Li J (2018) Genetic analysis of seedling root traits reveals the association of root trait with other agronomic traits in maize. BMC Plant Biol 18:171
Khalili M, Pour Aboughadareh A, Naghavi MR (2013) Screening of drought tolerant cultivars in barley using morpho-physiological traits and integrated selection index under water deficit stress condition. Advanced Crop Science 3:462–471
Le Gouis J, Bordes J, Ravel C, Heumez E, Faure S, Praud S, Galic N, Remoue C, Balfourier F, Allard V, Rousset M (2012) Genome-wide association analysis to identify chromosomal regions determining components of earliness in wheat. Theor Appl Genet 124:597–611
Lewien M, Carter A, Pumphrey M, Talbert L, Akhunov E (2016) Nested association mapping of water use efficiency in spring wheat (Triticum aestivum L.) using carbon isotope discrimination analysis and remote sensing traits. International Annual Meetings, 6-9 November, Phoenix, Arizona
Ma J, Luo W, Zhang H, Zhou XH, Qin NN, Wei YM, Liu YX, Jiang QT, Chen GY, Zheng YL, Lan XJ (2017) Identification of quantitative trait loci for seedling root traits from Tibetan semi-wild wheat (Triticum aestivum subsp. tibetanum). Genome 60:1068–1075
Maccaferri M, El-Feki W, Nazemi G, Salvi S, Angela Cane M, Colalongo MC, Stefanelli S, Tuberosa R (2016) Prioritizing quantitative trait loci for root system architecture in tetraploid wheat. J Exp Bot 67:1161–1178
Maccaferri M, Sanguineti MC, Corneti S, Ortega JLA, Salem MB, Bort J, DeAmbrogio E, del Moral LFG, Demontis A, el-Ahmed A, Maalouf F, Machlab H, Martos V, Moragues M, Motawaj J, Nachit M, Nserallah N, Ouabbou H, Royo C, Slama A, Tuberosa R (2008) Quantitative trait loci for grain yield and adaptation of durum wheat (Triticum durum Desf.) across a wide range of water availability. Genetics 178:489–511
Maccaferri M, Sanguineti MC, Demontis A, El-Ahmed A, del Moral LG, Maalouf F, Nachit M, Nserallah N, Ouabbou H, Rhouma S, Royo C, Villegas D, Tuberosa R (2011) Association mapping in durum wheat grown across a broad range of water regimes. J Exp Bot 62:409–438
Mackay I, Powell W (2007) Methods for linkage disequilibrium mapping in crops. Trends Plant Sci 12:57–63
Mahanta D, Rai RK, Mishra SD, Raja A, Purakayastha TJ, Varghese E (2014) Influence of phosphorus and biofertilizers on soybean and wheat root growth and properties. Field Crop Res 166:1–9
Maulana F, Ayalew H, Anderson JD, Kumssa TT, Huang W, Ma XF (2018) Genome-wide association mapping of seedling heat tolerance in winter wheat. Front Plant Sci 9:1272
Mwadzingeni L, Shimelis H, Rees DJG, Tsilo TJ (2017) Genome-wide association analysis of agronomic traits in wheat under drought stressed and non-stressed conditions. PLoS One 12:e0171692
Newman EI (1966) A method for estimating the total length of roots in a sample. J Appl Ecol 3:139–145
Paula P, Pausas JG (2011) Root traits explain different foraging strategies between resprouting life histories. Oecologia 165:321–331
Peng J, Sun D, Nevo E (2011) Wild emmer wheat, Triticum dicoccoides, occupies a pivotal position in wheat domestication process. Aust J Crop Sci 5:1127–1143
Phung NTP, Mai CD, Hoang GT, Truong HTM, Lavarenne J, Gonin M (2016) Genome-wide association mapping for root traits in a panel of rice accessions from Vietnam. BMC Plant Biol 16:64
Pierret A, Moran CJ, Mclachlan CB, Kirby JM (2000) Measurement of root length density in intact samples using x-radiography and image analysis. Image Anal Stereol 19:145–149
Pour-Aboughadareh A, Ahmadi J, Mehrabi AA, Etminan A, Moghaddam M, Siddique KHM (2017) Physiological responses to drought stress in wild relatives of wheat: implications for wheat improvement. Acta Physiol Plant 39:106
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ren Y, Qian Y, Xu Y, Zou C, Liu D, Zhao X, Zhang A, Tong Y (2017) Characterization of QTLs for root traits of wheat grown under different nitrogen and phosphorus supply levels. Front Plant Sci 8:2096
Robredo A, Perez-Lopez U, de la Maza HS, Gonzalez-Moro B, Lacuesta M, Mena-Petite A, Munzo-Rueda A (2007) Elevated CO2 alleviates the impact of drought on barley improving water status by lowering stomatal conductance and delaying its effects on photosynthesis. Environ Exp Bot 59:252–263
Ruiz M, Giraldo P, Gonzalez JM (2018) Phenotypic variation in root architecture traits and their relationship with eco-geographical and agronomic features in a core collection of tetraploid wheat landraces (Triticum turgidum L.). Euphytica 214:54
Ryser P (2006) The mysterious root length. Plant Soil 286:1–6
Sabiel SAI, Huang S, Hu X, Ren X, Fu C, Peng J, Sun D (2017) SNP-based association analysis for seedling traits in durum wheat (Triticum turgidum L. durum (Desf.)). Breed Sci 67:83–94
Schenk MK, Barber SA (1979) Root characteristics of corn genotypes as related to P uptake. Agron J 71:921–927
Siosemardeh A, Osmani Z, Bahraminejad B, Vahabi K, Roohi E (2012) Identification of AFLP markers associated with stress tolerance index in Sardari wheat ecotypes. J Agr Sci Tech 14:629–643
Sukumaran S, Reynolds MP, Sansaloni C (2018) Genome-wide association analyses identify QTL hotspots for yield and component traits in durum wheat grown under yield potential, drought, and heat stress environments. Front Plant Sci 9:81
Sun X, Du Z, Ren J, Amombo E, Hu T, Fu J (2015) Association of SSR markers with functional traits from heat stress in diverse tall fescue accessions. BMC Plant Biol 15:116–149
Trebbi D, Maccaferri M, de Heer P, Sorensen A, Giuliani S, Salvi S, Sanguineti MC, Massi A, va der Vossen EA, Tuberosa R (2011) High-throughput SNP discovery and genotyping in durum wheat (Triticum durum Desf.). Theor Appl Genet 123:555–569
Varshney RK, Spurthi N, Nayak S, May GD, Jackson SA (2009) Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 27:522–530
Wahl S, Ryser P (2000) Root tissue structure is linked to ecological strategies of grasses. New Phytol 148:459–471
Waines JG, Ehdaie B (2007) Domestication and crop physiology: roots of green-revolution wheat. Ann Bot 100:991–998
Wang S, Wong D, Forrest K, Allen A, Chao S, Huang BE, Maccaferri M, Salvi S, Milner SG, Cattivelli L, Mastrangelo AM, Whan A, Stephen S, Barker G, Wieseke R, Plieske J, International Wheat Genome Sequencing Consortium, Lillemo M, Mather D, Appels R, Dolferus R, Brown-Guedira G, Korol A, Akhunova AR, Feuillet C, Salse J, Morgante M, Pozniak C, Luo MC, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E (2014) Characterization of polyploidy wheat genomic diversity using a high-density 90000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796
Wehner G, Balko C, Ordon F (2016) QTL for water use related traits in juvenile barley. Agronomy 6:62
XLSTAT (2017) Data analysis and statistical solution for Microsoft excel. Addinsoft; Paris
Zadoks JC, Chang TT, Konzak CG (1974) A decimal code for the growth stages of cereals. Weed Res 14:415–421
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgments
The authors sincerely thank the Department of Gene Bank, Leibniz Institute of Plant Genetics and Crop Plant Research Gatersleben, Germany for making the genotypic data. Also, the authors are grateful to Dr. Marion Roder, from the Leibniz Institute of Plant Genetics and Crop Plant, for her fruitful comments on the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: AAM and AP; methodology: AAM, AP, and AH; investigation: AAM and SM. Software: AM and AP; writing—original draft: AAM and AP; writing—review and editing: AAM, AP, SM and AH.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Table S1
Pearson’s correlation matrix for 26 root and agronomic traits in 112 durum wheat genotypes (DOCX 22 kb)
Table S2
Variable loading scores of 26 root and agronomic traits and the proportion of variation of each principal component (DOCX 15 kb)
Table S3
Significant trait-SNP marker association pairs for measured root and agronomic traits in 112 durum wheat genotypes (XLSX 47 kb)
Table S4
The SNP marker loci associated with multiple traits in durum wheat genotypes (XLSX 18 kb)
Fig. S1
Frequency distribution of the measured traits. (A) number of seminal roots, (B) length of maximum seminal root, (C) average of seminal root length, (D) sum of seminal root length, (E) root fresh weight, (F) root dry weight, (G) root length, (H) root surface area, (I) root fineness, (J) root diameter, (K) root surface density, (L) specific root length (PNG 1191 kb)
Fig. S2
Frequency distribution of the measured traits. (A) root length density, (B) root tissue density, (C) root volume, (D) days to appearance of spike, (E) days to anthesis, (F) days to booting, (G) days to physiological maturity, (H) grain filling period, (I) number of grains per spike, (J) root diameter, (K) 100-grains weight, (L) grain yield (PNG 1279 kb)
Fig. S3
Frequency distribution of the measured traits. (A) water use efficiency and (B) transpiration efficiency (PNG 208 kb)
Fig. S4
Genome LD decay of whole genome (PNG 358 kb)
Rights and permissions
About this article

Cite this article
Mehrabi, A.A., Pour-Aboughadareh, A., Mansouri, S. et al. Genome-wide association analysis of root system architecture features and agronomic traits in durum wheat. Mol Breeding 40, 55 (2020). https://doi.org/10.1007/s11032-020-01136-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-020-01136-6