
Abstract
Previous studies in maize have identified three quantitative trait loci (QTLs) coding for high oleic acid content (HOAC) (QTLs oleic6-1, oleic6-2, and oleic6-3) at bins 6.04–6.05, proximal to the DGAT1-2 gene. The aims of this work were (i) to discover new markers for linkage disequilibrium (LD) and haplotype analysis distal to DGAT1-2, (ii) to develop a new DGAT1-2 PCR probe to detect the allele determining HOAC (F469 insertion), (iii) to conduct cluster analysis for kernel traits, and (iv) to assess genetic diversity, LD, and association analysis for kernel traits with the DGAT1-2 PCR probe and 13 markers previously mapped near DGAT1-2 in the Argentine temperate maize collection of 111 inbred lines. The results showed high haplotype diversity distal to DGAT1-2 and relatedness between the inbred line LP199 (with HOAC) and the Non-Stiff Stalk line W22 (reference genome). The frequency of F469 was low (20%). F469 was clustered with flint-grain type characteristics, whereas HOAC was associated with F469 across linear models. Genetic diversity at bins 6.04–6.05 was high (0.62), whereas LD extent was low (r2 ≤ 0.45). This low extent of LD indicates a high level of recombination and no LD between DGAT1-2 and markers flanking QTLs oleic6-1 to 3. Nevertheless, the significant LD between markers flanking those QTLs and the cosegregation of F469 with nc009 (markers flanking QTLs oleic6-1 and 2) during inbred line conversion suggest that these QTLs might contribute to HOAC in the breeding collection. However, further studies are needed to precise mapping at bins 6.04–6.05 for breeding purposes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maize kernel oil is known for its high content of linoleic acid (18:2) and oleic acid (18:1) (Singh et al. 2014). The maize oil content in breeding populations is a variable trait that results from the selection for both low and high oil content (Laurie et al. 2004). The fatty acid composition in maize is also variable: as the oil percentage increases, the degree of unsaturation decreases (Lofland et al. 1954). In food and industrial applications, such as for the production of heart-healthy, stable cooking, frying oil, increasing oleic acid content by 50–65% is a target trait value (White et al. 2007).
The long continued increase in oil content in maize has been explained by the epistasis occurred among quantitative trait loci (QTLs) (Dudley 2008). Fine mapping in a BC5S1 population developed from the low- and high-oil-content inbred lines PH09B and ASKC28IB1 has allowed the identification of qHO6 at chromosome six bin 6.04 as responsible for the high oil and high oleic acid content (HOAC) (Zheng et al. 2008). Further studies demonstrated that qHO6 encodes an acyl-CoA:diacylglycerol acyltransferase (DGAT1-2, GenBank: CS727016.1, GRMZM2G169089), at which a phenylalanine insertion at position 469 (F469) results in the functional allele responsible for the increase of 61% in oleic acid contents in maize kernels (Zheng et al. 2008). By means of biparental mapping, three QTLs called ole6-1 (bin 6.04), ole6-2 (bin 6.04), and ole6-3 (bin 6.05) have been mapped proximal to DGAT1-2 (Yang et al. 2010). Interestingly, QTL ole6-3 colocalizes with another DGTA gene (GRMZM2G130749), but with a smaller additive effect than ole6-1. In contrast, association mapping has shown contradictory results regarding the association of DGAT1-2 with oleic acid content, depending on the maize population (Beló et al. 2008; Chai et al. 2012). The fact that bins 6.04 and 6.05 colocalize with QTLs and DGTA genes involved in HOAC indicates the potentiality of this region for marker-assisted selection (MAS) for high oil and oleic acid contents.
The high oil content in the maize kernel has been associated with higher embryo oil concentration and higher relative proportion of the oil-rich embryo. However, high oil genotypes have also been observed to lead to a significant reduction in the kernel size (Dudley and Lambert 2003). Fortunately, the finding that the DGAT gene is associated with increased oil concentration within the embryo with no changes in seed weight is promising because it allows direct selection of the high-oil DGAT allele (Zheng et al. 2008).
The Argentine temperate maize collection of 111 inbred lines (collection A group) mostly comprises orange flint germplasm locally developed from historic lines and temperate inbred lines with Iowa Stiff Stalk Synthetic (SS) background (with no known Non-Stiff Stalk (NSS) or Iodent components) (Lee and Tracy 2009), which has shown high diversity at the molecular level (Olmos et al. 2016). Although the phenotypic characterization of the entire collection is limited, previous studies have identified contrasting genotypes at different traits for the development of a biparental mapping population (Olmos et al. 2016; Estermann et al. 2017). Current donors of high oil and oleic acid contents belong to the Argentine x Caribbean Derived Stock (ACDS) genetic background, and are currently being used to improve the oil composition by means of conventional breeding and MAS. Preliminary work on the conversion of the elite inbred lines LP29 and LP214 (with low oleic acid content) with the inbred line donors L1445 and P.3525 (08.3525) (with HOAC) from collection A demonstrated that BC1F2-derived lines with a homozygous segment of F469 and alleles from the microsatellite markers nc009/umc1014 (which target the same locus), from either donor, show higher oleic acid content (54–57.8%) than the general average (49.87%) in a clear additive manner (Ruiz et al. 2010). However, the relationship between kernel oil content and composition- and grain yield-related traits in collection A is still unknown.
Based on the above, the aims of the present study were as follows: (i) to discover new markers for linkage disequilibrium (LD) and haplotype analysis distal to DGAT1-2 by using two inbreds with contrasting contents of oleic acid (LP199 and B98) for linkage disequilibrium (LD) and haplotype analysis in the context of seven reference genomes (B73, B104, EP1, F7, PH207, CML247, and W22), (ii) to develop a new DGAT1-2 PCR probe to detect the allele determining HOAC (F469 insertion), (iii) to conduct cluster analysis for oleic acid content and kernel trait descriptors, and (iv) to assess genetic diversity, LD, and association analysis for kernel traits of elite commercial lines with the DGAT1-2 PCR probe and 13 markers previously mapped near DGAT1-2 in the Argentine temperate maize collection of 111 inbred lines (collection A).
Material and methods
Plant material
The allelic variation near the DGAT1-2 region was assessed in the collection A provided by the maize breeding program of the National Institute of Agricultural Technology (INTA), Argentina. Collection A mostly includes orange flint lines from Argentina and reference US lines that include diverse genotypes which are parents of the biparental mapping population or source donors of desirable agronomic traits such as oil composition and oil content (Olmos et al. 2016). The population structure of collection A has been defined by four discrete subpopulations: P465, ACDS, LP299-2-related lines, and Iowa Stiff Stalk Synthetic and their derivates (BS13-BSSS), as well as by mixed germplasms (Olmos et al. 2016). The ID, pedigree information, grain type, known combined ability, and population structure of collection A are summarized in Supplementary Table 1. The allelic variation of the DGAT1-2 PCR-based probe was analyzed in the context of 13 markers previously mapped at bins 6.04 and 6.05 in collection A (Olmos et al. 2014a, 2016). In addition, the PCR-based probe for DGAT1-2 was genotyped in the set of 27 maize inbred line founders of the nested association mapping (NAM) population (Yu et al. 2008), imported in 2011 by INTA Pergamino (Pergamino, Buenos Aires, Argentina) from the US Department of Agriculture (USDA).
New markers for the DGAT1-2 region were searched for by selecting two contrasting maize inbred lines for HOAC: the Argentine LP199 (HOAC donor) and the US B98 (low oleic acid content control) (Supplementary Table 1). The allelic diversity of the new markers was analyzed in the context of available sequence data from the seven maize reference genomes (B73, B104, EP1, F7, PH207, CML247, and W22) available at the https://www.maizegdb.org and https://www.ncbi.nlm.nih.gov databases. These reference genomes belong to different heterotic patterns (Liu et al. 2003; van Heerwaarden et al. 2012) as follows: B73 (SS), B104 (SS), EP1 (mixed), F7 (mixed), PH207 (Iodent), CML247 (tropical or semitropical), and W22 (NSS line).
DGAT1-2 PCR-based probe
The conditions and procedures for DNA isolation, PCR, marker development, sequencing, alignment, and identification of novel polymorphisms were as previously described (Estermann et al. 2017).
The sequence of DGAT1-2, GRMZM2G169089_T01, was downloaded from the Gramene database (http://ensembl.gramene.org/). A PCR sequence-tagged site (STS) designated DGAT1-2 PCR was developed to target the F469 InDel allele. The resulting primers amplified a 152-bp fragment in the B73 reference sequence and a 155-bp fragment in the remaining inbreds (Supplementary Fig. 1). Primers were uploaded to the MaizeGDB (http://archive.maizegdb.org/cgi-bin/displayproberecord.cgi?id=3152625) and NCBI databases (http://www.ncbi.nlm.nih.gov/probe/?term=Pr032756446). DGAT1-2 was then genotyped in collection A and in the 26 NAM founder maize inbred lines.
In addition, the presence of DGAT1-2 alleles in the Argentine collection was confirmed by direct sequencing, using the STS primers and the protocol explained below. All 28 nucleotide sequences were uploaded to GenBank, which provided the PopSet: 914244630 accession number (http://www.ncbi.nlm.nih.gov/popset/914244630). Sequences were aligned and compared with the sequences of the seven maize reference genomes and the cDNA of high oil inbred lines (accession AW360460, Illinois High Oil and ASKC28IB (Zheng et al. 2008)) deposited at https://www.maizegdb.org.
Marker discovery
Three intergenic regions distal to DGTA1-2 comprising unique HapMap insertion-deletion markers (InDels) were targeted to be amplified in the two selected inbred lines: LP199 and B98. The InDel markers with single-nucleotide polymorphisms (SNPs) were retrieved from the legacy maize HapMap 1 project (hapmap_SNPs_log2ml.HP1-091207) available at https://www.panzea.org/. The alleles and B73 RefGen_v3 coordinates were as follows: (1) PZE06104611841 (-------/CCTGGCG, 104,626,046 bp) 2) PZE06104824646 (----/ACCC, 104,838,851 bp), and 3) PZE06104825234 (------/CATACA, 104,839,439 bp), respectively. The local accession of B73, # B73-05-6081 (Olmos et al. 2014b), was also included as a control size check.
All amplified nucleotide sequences, variations, and B73 RefGen_v3 coordinates were uploaded to GenBank (http://www.ncbi.nlm.nih.gov/genbank/), which provided the corresponding nucleotide accession number for each of the three sets of variations: (1) KU248822 (LP199)/KU248823 (B98), (2) KU248824 (LP199)/KU248825 (B98), and (3) KU248826 (LP199)/KU248827 (B98), respectively. The STS PCR probe p2526 amplified variation features described in the nucleotide sequences KU248824 and KU248825.
The PCR STS designated p2526 was originated from a fragment amplified to be sequenced at region 2 near the DGAT1-2 region. The PCR product to be sequenced showed a rich allelic variation when run on agarose gel electrophoresis, indicating its utility for agarose gel genotyping. In silico mapping revealed that the p2526 PCR probe (forward: GGCAACCGTTGAAGAGAGTC, reverse: AGGATCGTCTGGGGAACTTT) matches a transposon-like sequence and that the PCR amplicon carries additional polymorphisms, such as a microsatellite with a CAA motif, which explains its rich allelic variation.
Simple sequence repeat (SSR) genotyping
SSR markers previously mapped at bins 6.04 and 6.05 were selected (Olmos et al. 2016). All SSR primer sequences are available at MaizeGDB (http://www.maizegdb.org/). SSR physical coordinates were obtained by comparing the sequence similarity of the primer set with the Blastn program (Altschul et al. 1990) implemented at the Gramene interface (http://ensembl.gramene.org/tools.html) against the B73 RefGen_v3 reference genome.
Data analysis
Genetic diversity
The PowerMarker software V3.0 (Liu and Muse 2005) was used to calculate major allele frequencies, residual heterozygosity (observed heterozygosity), and average gene diversity (expected heterozygosity) of the 15 markers genotyped at bins 6.04 and 6.05 in collection A.
LD analysis
Pairwise LD was calculated for (i) the 14 genotyped markers in collection A, including the DGAT1-2 PCR probe, which targets the F469 InDel allele, and excluding umc1014, which targets the same locus as nc009, and (ii) the 39 SNP and InDel markers discovered near DGAT1-2, using the alignment of the Argentine LP199 (HOAC) and the US B98 (low oleic acid content) inbred lines in the context of the other seven available genome sequences of maize inbred lines. To visualize LD patterns among SSR loci, SSR markers were ordered according to their known genomic position. LD was calculated with Lewontin’s D’ and the Pearson correlation coefficient (r2), using the pairwise analysis of TASSEL v5.2.39 (Bradbury et al. 2007). To import SSR data into TASSEL v5.2.39, only the four most common homozygous alleles were coded using the nucleotide symbols (A, C, G, T) in a diploid hapmap format, whereas heterozygous states were converted to missing data. The nominal level of LD was defined at r2 = 0.20 (Gaut and Long 2003). The significance of LD in the different datasets was assessed with a P value test using TASSEL v5.2.39 default settings; the significance level was set up at P ≤ 0.01. LD was graphically represented using TASSEL v5.2.39 software by displaying r2 and P values in the upper and the lower triangles of the 2D plot, respectively. In addition, for the 39 SNPs and InDels, LD (r2), and haplotype block analysis were calculated and displayed using Haploview software version 4.2 (Barrett et al. 2005). Block structure was defined with the four-gamete rule, whose blocks are formed by consecutive markers where only three gametes are observed.
Kernel type descriptors
Public descriptors of maize inbred lines were retrieved from the database of the Instituto Nacional de Semillas–INASE- (Argentine Seed Institute). Cross information was retrieved for only 39 out of the 111 previously genotyped maize inbred lines from collection A. These 39 genotypes were diverse and included in the four main subpopulations: the P465-, ACDS, LP299-2-, and BS13-BSSS-related lines, respectively (Olmos et al. 2016). The selected descriptors were as follows: prolificacy, hundred kernel weight, kernel length, kernel width, kernel thickness, kernel volume, and endosperm color.
Characterization of the oleic acid content
Field experiments were conducted at the Pergamino Experimental Station of INTA, Argentina (33° 56′ S, 60° 34′ W) on a Typic Argiudoll soil, during the 2012–2013 growing season on two sowing dates: October 10th (experiment 1) and November 5th (experiment 2). A total of 120 inbreds (87 inbreds from collection A and the remaining from promising elite inbred lines and control checks) were distributed in a completely randomized block design with three replicates. Water deficit was always prevented by means of sprinkler irrigation, used to keep the uppermost 1 m of soil near field capacity throughout the cycle. Experiments were kept free of pests, weeds, and diseases. Three to five plants per inbred line and sowing date were self-pollinated to collect ear samples at physiological maturity for oil characterization. Composite kernel samples from the middle part of the spikes of each experimental unit were collected for analysis. Ten kernels were finally selected and germs were removed. Fatty acid composition was analyzed using gas chromatography as described by Percibaldi et al. (1997). The content of oleic acid (C18:1) was expressed as the percentage of total fatty acids. A mixed linear model procedure, with SOLUTION and REML options, implemented in the SAS program (SAS Institute Inc. 2009), was used to estimate heritability and variance components of oleic acid content means.
Phenotypic variability and association analysis
Oleic acid content and kernel characteristics, along with the number of DGAT1-2 HOAC allele copies carried by inbreds, were analyzed by means of principal component analysis (PCA), using the program Infostat (Di Rienzo et al. 2011). In addition, association analysis was performed using TASSELv5.2.39, the genotypic matrix of 14 markers genotyped at bins 6.04 and 6.05 in collection A used for LD analysis, and the phenotypic matrix with kernel type descriptors and oleic acid content. To detect associations between markers and traits, the following four linear models were applied: (i) GLM without population structure (Q) as a cofactor, (GLM), (ii) GLM with population structure as a cofactor (GLM+Q), (iii) a mixed linear model (MLM) involving a family structure as a cofactor (MLM K), and (iv) a MLM with a combination of population and family structure as cofactors (MLM K+Q). Quantile–quantile plots were represented for marker-trait associations across models using TASSEL v5.2.39. Population and family structures were included in the MLM model as fixed or random effects, respectively (Yu et al. 2006). Population structure was represented by the Q matrix selected at the most likely k = 4 subpopulation, as previously described (Olmos et al. 2016). Family structure was represented by the kinship matrix (K) generated using the pairwise identity by state distance implemented in TASSEL v5.2.39. A significant threshold of 1% after Bonferroni multiple test correction was applied (Bradbury et al. 2007). The denominator in the Bonferroni correction was the total number of markers tested (14).
Results
Marker discovery
Comparison between LP199 and B98 allowed retrieving a total of 20 markers (11 SNPs and nine InDels) (Supplementary Table 2), as follows:
Region 1 amplified 341 bp in B73 (coordinates 104,625,741–104,626,081). The target HapMap1 marker PZE06104611841 (-------/CCTGGCG) was polymorphic between LP199 and B98, but had a motif pattern different from that described (-------/TGGCGCC) (KU248822, LP199; KU248823, B98). Three additional SNPs were additionally found.
Region 2 amplified 233 bp in B73 (coordinates 104,838,663–104,838,895). The target HapMap1 marker PZE06104824646 (----/ACCC) was not polymorphic (KU248824, LP199; KU248825, B98). However, a large 202-bp InDel allele was found a few bases downstream PZE06104824646, with B98 carrying the large 202-bp insertion. Besides, more markers, three SNPs and three InDels (15, 16, and 38 bp long), were found. The Gramene builder blast at region 2 found similarity with (1) a Type II Transposon Class DNA/PIF-Harbinger, (2) a tandem repeat consensus ACA (at the 15-bp InDel size), and (3) two close low-complexity elements, one of which was at the 38-bp InDel. The Blastn search of the B98 fragment against the Plant Miniature inverted-repeat transposable elements (MITEs) database (Chen et al. 2013) matched a MITE element (sequence SQ225171203) at the 202-bp insertion.
Region 3 amplified 287 bp in B73 (coordinates 104,839,355–104,839,641). The target marker PZE06104825234 (------/CATACA) was not polymorphic (KU248826, LP199; KU248827, B98). However, four InDels (2, 4, 5, and 13 bp in length) and five SNPs were found.
Considering only the LP199 and B98 genotypes, the rates of InDels and SNPs in the three regions were 1/341–1/114, 1/58–1/78, and 1/72–1/57 bp for regions 1, 2, and 3, respectively. Thus, the average rates of InDels and SNPs between LP199 and B98 in the 861 bp length were 1/96 and 1/78, respectively.
When the genomic sequences from LP199 and B98 were compared with B73, B104, EP1, F7, PH207, CML247, and W22, a total of 39 SNPs and InDels were found (Supplementary Table 2). Twenty-three out of these 39 markers were identified as HapMap 2 markers, whereas the remaining 16 were novel polymorphisms. Inbreds B73, B104 and PH207 shared the same haplotype along the three regions, whereas two pairs of lines (B98-F7 and LP199-W22) differed in only one SNP and another pair (EP1-CML247) differed in two SNPs.
Genetic diversity
The analysis of the DGAT1-2 PCR probe and the 14 previously genotyped markers (Supplementary Table 3) showed that 11 markers were positioned proximal whereas two markers were positioned distal to the STS DGAT1-2 probe. Average major allele frequency, gene diversity, and heterozygosity were 0.51, 0.62, and 0.05, respectively. The SSR markers umc2317 and nc009 reached the lowest and highest gene diversity, respectively (0.17 and 0.83, respectively) (Supplementary Table 4). STS p2526 had the greatest number of allelic variations (10) and a high level of gene diversity (0.79). In addition, SSR umc1979 showed the highest heterozygosity (0.14).
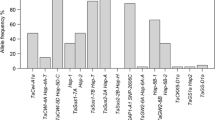
The low-oleic-acid content DGAT1-2 allele (152 bp), which corresponded to the phenylalanine deletion, had one of the greatest major allele frequencies (0.81). The HOAC allele, which was mostly homozygous, occurred in only 25 out the 111 inbred lines (23%): P465, LP662 (heterozygous), LP122, LP199, LP1044, LP122-2, LP124 (heterozygous), LP1512, LP126, LP5708, LP1411, LP562, LP869, A485, L1445, L58, LP598 (heterozygous), LP661, (LP562x3584)-1-39 and (LP562x3584)-1-53 (both heterozygous), 08.3525, 08.3538, 08.3590, 28 (heterozygous), and 34. The HOAC allele was present in LP199 but absent in B98, in agreement with the known performance in oil composition of these inbred lines, as well as present in four inbred lines developed as HOAC donors by conventional breeding: L1445, 08.3525, 08.3538, and 08.3590. The screening of the STS DGAT1-2 probe in the NAM founder inbred line set revealed that the HOAC allele was present only in the Oh7B, Ky21, M162W (temperate), IL14H (sweet), and Tzi8 (tropical) inbred lines, whereas the Tx303 (mixed structure) inbred line carried a heterozygous allele (Supplementary Fig. 1). The STS DGAT1-2 probe product was then confirmed by direct sequencing (GenBank PopSet: 914244630), and thus, the occurrence of homoplasy among the PCR products can be discarded. In addition, the GenBank PopSet: 914244630 partial sequences were then compared with the reference maize genome and high oil inbred line cDNA sequences (Supplementary Fig. 3). The HOAC allele was absent not only in B73 but also in all maize reference genomes (B104, EP1, F7, PH207, CML247, and W22) and present in the Illinois High Oil and ASKC28IB1 (Pioneer, USA) cDNA accessions. In addition, in a partial cds spanning 411 bp length, six markers were found nearby the phenylalanine insertion (---/TTC): C/T, T/C, C/T, ---/TTC, T/C, G/A, and G/A. The nearest two SNP (T/C and G/A), 98 and 134 bp, respectively, were linked to the DGAT1-2 HOAC allele and located outside the ASKC28IB1 mRNA stop codon, with the allele C of T/C carried by inbred lines LP122, LP199, LP122-2, and AW360460, and allele A of G/A carried by inbred lines LP122, LP199, LP122-2, and LP1044.
LD analysis
Multi-allelic D′ and r2 values between pairwise markers within collection A ranged from 0 to 1 and from 1.31E−33 to 0.45, respectively (Fig. 1, Supplementary Table S5). Lower values of r2 coefficients were found, and only 13% were statistically significant (P ≤ 0.01). Only six pairwise comparisons (umc1979-nc009, p2526-umc1857, nc009-umc1857, umc1857-umc1614, umc2319-umc2318, and phi452693-nc013) were above the nominal level of 0.2 and resulted statistically significant. In contrast, D′ values showed greater variation: 24% of the pairwise marker comparisons were greater than 50% of the theoretical maximum. The LD plot (Fig. 1) showed that DGAT1-2 had statistically significant LD only with umc1918 (r2 = 0.10), which locates distal to DGAT1-2. Among significant values, the highest LD extent was found between the pairs umc1979-nc009/umc1014 (r2 = 0.45) and umc1857-umc1614 (r2 = 0.23), which flank distal and proximal, respectively, to QTLs ole6-1 and ole6-2; however, there was no LD between flanking markers. In contrast, another region with LD was found between markers flanking the QTL ole6-3 interval: umc1614-nc013 (r2 = 0.14) and umc2055-nc013 (r2 = 0.19), and also between phi452693 and nc013 (r2 = 0.21), which flanks proximal to QTL ole6-3.

Pairwise LD at the bin segment 6.04–6.05 within collection A. r2 = upper triangle, p values = lower triangle. Strengths and significance of LD between markers are indicated by a color scheme
Multi-allelic r2 values for the 39 SNPs and InDels near DGTA1-2 ranged from 0.00 to 1.00 (Fig. 2). Region 3 (from marker 27 to marker 38) was a segment that exhibited almost complete LD (Fig. 2). Three blocks of LD, with three (block 1), eight (block 2), and seven haplotypes (block 3), respectively, were found. Despite the relative low number of genotypes and markers analyzed, the allelic diversity along the three regions had a particular LD pattern along the 861 bp that shifted from a hot spot of recombination to complete LD that did not depend on physical distance. The ones responsible for this pattern were the pairs LP199-W22 and EP1-CML247, which share distinct haplotypes at different physical positions (Supplementary Table 2).

Haplotype and LD analysis of three sequenced regions distal to the DGAT1-2 gene. The numbers in each box correspond to the LD (r2) values between markers. Strengths of the LD between markers are indicated by the following color scheme: r2 = 0 (white), 0 < r2 < 1 (shades of gray), r2 = 1 (black)
Phenotypic variability and association analysis
The combined analysis of variance across environments for oleic acid content detected highly significant differences (P < 0.0001) between the genotypes, with a general mean of 34.2% (Table 1). Genotype (G) and the G × E interaction were also highly significant (P < 0.0001). However, the component of variance for G was larger than the G × E interaction component (92 vs. 3%) and the resulting heritability for oleic acid content was high (0.97). The promising inbred lines 8.3525, 8.3538, and 8.3590 (whose pedigree derived from the donor LP199) had greater oleic acid content (above 60%) than LP199 (47.23%), indicating that these inbred lines would carry some other source of variation contributing to increasing their oleic acid content.
The kernel characteristics were variable, and the range of minimum and maximum values was as follows: prolificacy (1–3), hundred kernel weight (200–350), kernel length (8–11), kernel width (7–10), kernel thickness (3–6), kernel volume (210–550), and endosperm color (1–5) (Supplementary Table 6).
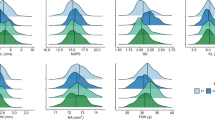
The PCA in a set of 37 inbreds from collection A revealed a pattern of genetic differentiation associated with the kernel characteristics (Fig. 3). The first two principal components (PCs) explained 41.6% (PC1) and 17.4% (PC2) of the variation of the kernel characteristics and the DGAT1-2 HOAC allele. In the corresponding biplot (Fig. 3), oleic acid content is located in the same PC1 quadrant (I) as the DGAT1-2 HOAC allele and endosperm color. Meanwhile, PC2 split oleic acid content, DGAT1-2 HOAC allele, and endosperm color from kernel length, width, and volume as well as from kernel weight, thickness, and prolificacy. The inbred lines LP222, LP199, LP 869, LP1044, LP122-2, and LP1411 (all from the ACDS genetic background) clustered together in quadrant I and shared a HOAC allele, whereas their kernel weight and kernel volume were almost all (except LP869) below the general means of 285 g and 360 mm3, respectively. Quadrant I also comprised inbreds LP116, LP125-R, LP613, and LP561, which carried a low-oleic-acid-content allele but were clustered closer to the PC axes. The remaining inbreds that carried one copy of the HOAC allele, i.e., LP662 and LP124, were clustered in quadrant IV separated by PC2. These two inbred lines were then split from the remaining genotypes and variables by PC1. As a result, increased oleic acid content was positively associated with the DGAT1-2 HOAC allele and endosperm color intensity and lower kernel weight and volume, whereas increased plant prolificacy reduced the performance of oleic acid content and kernel weight.

Principal component analysis of oleic acid content, kernel characteristics and the number of FFS (F469) allele copies in a set of 37 inbreds from collection A
Association analysis indicated a reduction of significant marker-trait association after accounting for population structure in the GLM model. Thus, the single GLM model associated two markers, p2526 and DGAT1-2 with four kernel traits: p2526 with endosperm color, hundred kernel weight, kernel volume and oleic acid content, and DGAT1-2 with hundred kernel weight and oleic acid content. After population structure correction on the GLM + Q model, the two markers umc1614 and DGAT1-2 were only associated with endosperm color and oleic acid content, respectively. Finally, both MLM models had similar results with the DGAT1-2 marker, which was consistently associated with oleic acid content across the MLM K and MLM K + Q models. The R2 values at GLM models were inflated for all associated traits, and a minimal R2 and allele estimate values were obtained for oleic acid content at the MLM Q + K model. Histograms of the distribution of oleic acid content means and residuals and Q-Q plots across models and traits are presented in Fig. 4 and Supplementary Fig. 2, respectively. A deviation from the identity line was observed for almost all traits, in particular for those statistically significant. Correction for population structure reduced the observed –Log10(P value) at GLM + Q, which was not affected by MLM adjusted models.

Histograms of the distribution of a oleic acid content means, b residuals for the MLM K, and c MLM K + Q models, respectively, measured in a set of 87 inbreds from collection A
Despite the limited mapping panel size and marker density and the likely occurrence of false positives at GLM models, the significant association of alleles was compared with SSR haplotypes and multivariate results. Thus, the DGAT1-2 HOAC allele was associated with a reduction in kernel weight, accordingly to the separate PC1 split between kernel weight with the phenylalanine insertion at position 469 allele (i.e., F469, which is equivalent to the FFS notation) and oleic acid content (Fig. 3). Also, the transposable element at STS p2526 (412-bp allele) was taken as a reference allele for the calculation of allele estimates in the association analysis and the 209-bp allele from STS p2526 had a positive effect on HOAC. Finally, at the GLM + Q model (Table 2), endosperm color was associated with umc1614, with the 114- and 117-bp alleles having positive allele estimates over the reference (121-bp allele). Although umc1614 was not in LD with DGAT1-2 (Fig. 1), a correlation between the 114- or 117-bp allele from umc1614 (Supplementary Table 3) and traits was revealed at cluster analysis. All inbred lines clustered at PC1 quadrant (I) shared similar features like HOAC, the FFS allele, the flint P465 or ACDS background, and the orange or red endosperm color indicating stratification. These results might indicate either that those false positives were not corrected at the GLM models or that false negatives were over controlled at MLM models.
Discussion
Diversity of DGAT1-2 and kernel characteristics
The phenylalanine insertion F469 of the diacylglycerol acyltransferase DGAT1-2 (GRMZM2G169089) is an ancestral allele present in teosinte and the maize progenitor, Zea mays ssp. parviglumis (Zheng et al. 2008). However, the screening of the high-oil allele F469 in the NAM parent founder revealed that this is absent in most modern temperate, tropical, and specialty corn inbred lines. The main forces that could have originated the loss of the high-oil DGAT1-2 allele in modern inbred lines were the narrow genetic base of US hybrid corn and the consequent genetic drift (Chai et al. 2012). Besides, the low frequency of the F469 insertion in US temperate inbreds indicates that this a more recent mutant selected by domestication or breeding (Zheng et al. 2008). Additionally, the low frequency of the F469 insertion prevented the association mapping from identifying this locus since, after the filtration of the low alleles required to run trait-association models, low-frequency alleles become undetectable (Yu et al. 2006; Beló et al. 2008; De La Fuente et al. 2013).
In our work, the prevalence of the F469 allele in certain groups of inbred lines from the historic P465 and ACDS genetic and composite background, with mainly orange and red flint grain type, suggests at least two origins of the F469 allele. Also, a coupling between genetic background, oil content, grain type, and yield during the past breeding history of the Argentine maize collection is clear. The origin and genetic background of Argentine flint modern germplasm still constitute a controversial subject. Results based on previous SSR genotyping suggest that modern flint germplasms of Argentina have been admixtured from indigenous maize populations of South America as well as from Italian maize varieties introduced by immigration (Olmos et al. 2014a). In relation to oil content and yield, commercial maize dent germplasms have shown, in general, a negative correlation between oil content and yield (Laurie et al. 2004; Singh et al. 2014). Moreover, a strong additive correlation has been found between kernel oil content and other agronomic traits such as the percentage of lodged and broken plants (Rosulj et al. 2002). Due to the positive correlation between oil and oleic acid content (Lofland et al. 1954; Zheng et al. 2008), it seems possible that, besides desirable characteristics of flint grains, both traits have shaped the maize inbred lines released by the public sector of Argentina. However, uncoupling of oil and oleic acid content and yield is possible by biotechnology. For instance, the production of transgenic maize with the maize DGAT1-2 HOAC allele (Zheng et al. 2008) and of transgenic maize with a fungal DGAT2 gene has allowed increasing kernel oil content with no yield penalty (Oakes et al. 2011). The breeding of elite maize with higher grain oil content is based on two strategies: increasing the relative proportion of the oil-rich embryo within the grain or enhancing oil synthesis and accumulation in embryonic structures (Barthole et al. 2012). Thus, the DGAT that catalyzes the last reaction in the biosynthesis of triacylglycerol appears to be a promising target to increase the oil content in maize embryos. The comparative analysis of the high oil inbred lines with the controls B73 and Mo17 showed that the high oil inbred lines had a correlated response of reduced kernel weight and endosperm volume but increased germ and germ oil and volume (Lambert 2000). Our results indicate that the ACDS genetic background is a source of HOAC positively associated with the F469 allele but with an overall low kernel size performance. Promising inbred line donors of HOAC, such as L1445 and P.3525 (08.3525), which carry a homozygous copy of the F469 allele, have not yet been fully characterized for the grain yield- and yield-related components. A decrease in kernel size is expected with an increase in total oil concentration, unless a selection index that would put more weight on other traits such as germ and endosperm size is used (Lambert 2000). Thus, further phenotyping of the Argentine maize collection with agronomic traits related to grain quality and yield and the application of biotechnology will allow a better understanding and the possibility to take full advantage of the genetic reservoir and variability to develop specialty corn.
Diversity of genomic regions near DGAT1-2
The high diversity within the Argentine maize collection was confirmed by the high rate of SNPs and InDels discovered between the two contrasting inbred lines LP199 and B98. Thus, the intergenic rate of SNP and InDel variation hereby found was slightly higher than the reported SNP rate (1/104 bp) between randomly sampled sequences of maize loci (Tenaillon et al. 2001) but lower than that reported (1/44 bp) in the first-generation haplotype map of maize NAM founders (Gore et al. 2009). InDels larger in size were clearly frequent in the small genomic segment analyzed, providing, with linked SNPs, highly divergent haplotypes useful for mapping and MAS application (Tian et al. 2008; Guo et al. 2016). In addition, the comparison with reference genomes belonging to distinct heterotic patterns revealed interesting relationships. That was the case of the sharing of almost all the haplotype length between the HOAC inbred line LP199 from the ACDS background and the low-oleic-acid-content inbred line W22 with NSS background (Plewa and Weber 1975). This type of close genetic relationship of Argentine flint inbreds with dent germplasms has been previously observed (Olmos et al. 2014b). This finding reveals new evidence of the complex origin of the Argentine Composite II as a result of the introgression of US dent germplasms that might have occurred from the 1950s through the late 1980s during the early development of the public Argentine maize collection (Olmos et al. 2014b). Furthermore, it is also in agreement with the suggested ancestry of US flint elite commercial inbred lines (developed after 1985) from open-pollinated landraces (developed before the 1930s), where NSS was proposed as the main component of flint ancestry besides SS and Iodent genetic background (van Heerwaarden et al. 2012).
Previously, in collection A, we detected low extent of LD and a high ratio of linked to unlinked SSR locus pairs in significant LD (Olmos et al. 2016). Here, LD and diversity analysis were carried out with markers near DGAT1-2 in the context of 13 SSR markers previously mapped at bins 6.04 and 6.05 and genomic regions positioned distal to DGAT1-2. As a result, a low but significant extent of LD and greater gene diversity were found, indicating a high recombination rate around this position. None of the markers flanking the DGAT1-2 gene (STSs p2526 and umc1979) was in LD with DGAT1-2, showing the lack of a single segment comprising the F469 allele that can be targeted for MAS. However, by means of association analysis at the GLM model, the 209-bp allele from STS p2526 was associated with HOAC besides the HOAC DGAT1-2 allele, but the association was not consistently maintained after correction for population and relatedness. The consistency of association analysis is usually verified by conducting different comparative approaches, where MLM models usually yield a lower number of significant markers than GLM due to the P value correction of the spurious association caused by family and population structure (Yu et al. 2006). In addition, GLM analysis by TASSEL routinely produces not only a lower number of significant markers than MLM but also lower P (Weng et al. 2011; Pandit et al. 2017). However, GLM is carried out because MLM can produce false negatives by overcompensating relatedness and population structure (Zhao et al. 2011). Thus, in the present study, the transposon-like insertion (412-bp allele of STS p2526) was present in low and HOAC inbred lines as well as in inbred lines carrying the FFS allele. However, the 209-bp allele from STS p2526 with positive additive effect on HOAC was present in almost all inbred lines carrying the FFS allele, whereas the 412-bp allele was only present in HOAC inbred lines 8.3538 and 8.3590. However, further studies are needed to elucidate any effect of STS p2526 alleles upon the DGAT1-2 expression level, considering the potential cis-regulatory activity of transposons on transcription factor binding sites (Wittkopp and Kalay 2012). In addition, the presence of a transposable element at STS p2526 may contribute to the greater allelic variation of STS p2526 and perhaps DGAT1-2 activity. In that matter, transposons in plants have shown to be abundant, highly polymorphic, stable, and generally associated with gene regions (Casa et al. 2000; Thornsberry et al. 2001; Guillet-Claude et al. 2004; Camus-Kulandaivelu et al. 2008).
The integration of traits in the process of converting an elite variety by using backcross breeding and MAS needs minimizing the residual non-recurrent parent germplasm (i.e., linkage drag) at the chromosomal proximity of the QTL or candidate gene (Peng et al. 2014). Linkage drag can be reduced with flanking markers as closely linked to the introgressed gene as possible (Hospital 2001). Thus, as a result of association analysis, the 209-bp alleles from the closest marker STS p2526 probe (almost 2000 bp apart) significantly associated with HOAC can be useful to select for and against linkage drag distal to DGAT1-2, depending on donors, during conversion of low oleic acid content inbred lines. In addition, the SSR umc1105 distal to DGAT1-2 (Yang et al. 2010), not included in this study, seems to be the preferable flanking marker to select against the donor fragment at that position. The fact that the FFS allele of DGAT1-2 was consistently associated with increased HOAC across the GLM and MLM models and that markers flanking QTLs oleic6-1, oleic6-2, and oleic6-3 (Yang et al. 2010) were not associated with HOAC in collection A indicate that DGAT1-2 would be the only responsible for the phenotypic variation observed. However, preliminary work on the conversion of inbred lines for HOAC demonstrated that the cosegregation of the F649 insertion from DGAT1-2 with favorable alleles from microsatellites nc09/umc1014 from HOAC inbred lines L1445 (128/125 bp) and 8.3525 (120/119 bp) (both from ACDS background) successfully increased the oleic content of BC1F2-derived lines up to 54–57.8% (Ruiz et al. 2010). Because the nc009/umc1014 locus flanks distal to a large chromosomal region with QTLs ole6-1 (Q8-umc1979-nc010) and ole6-2 (phi031/nc010-umc1105) in a 2,368,345-bp region delimited by the umc1105- phi031/nc010 interval (Yang et al. 2010), it is possible that DGAT1-2 with favorable alleles from QTLs ole6-1 and ole6-2 would be partially responsible for HOAC in collection A. In addition, further results of the weak but significant LD between and among markers flanking these QTLs observed in collection A indicate the shaping of linkage during the history of inbred line development.
Thus, despite the high availability of marker density and the size of the mapping population, no single mapping method ensures the detection at the genome wide level of all sources of favorable alleles for oil content and composition at chromosome 6 and other chromosomal regions (Beló et al. 2008). The fact that all Pioneer-derived lines from collection A (family LP299-2), except LP317, had values of oleic acid content below the general mean (34.2%) indicates that they would not be derived from the Pioneer HOAC PH1B5 and PH7DD (Beló et al. 2008). Thus, the incorporation of additional sources of HOAC will further increase diversity and provide more tools for precise QTL mapping and exploitation of different genetic sources for oleic acid improvement.
Conclusion
Our work provides new markers distal to DGAT1-2, useful for fine mapping and selecting against donor segments to avoid linkage drag during inbred line conversion. The high recombination rate observed near the QTL oleic6-3 (which colocalized with the other DGAT GRMZM2G130749) and the lack of linked markers with HOAC at this locus indicate the need to increase the marker density at this region to explore this source of variation for breeding purposes. The successful conversion for HOAC of BC1F2-derived lines up to 54–57.8% (Ruiz et al. 2010) with the DGAT1-2 PCR probe and nc009/umc1014 markers flanking the large QTL oleic6-1 and QTL oleic6-2 interval suggests that all contribute to the conversion for HOAC without linkage drag penalty. The clear association of HOAC with orange-red endosperm color and low-kernel weight and volume indicates the need to make further efforts to uncouple the linkage of HAOC with these traits to shape new elite maize lines with high oil and oleic acid contents and different kernel trait characteristics.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/s0022-2836(05)80360-2
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265. https://doi.org/10.1093/bioinformatics/bth457
Barthole G, Lepiniec L, Rogowsky PM, Baud S (2012) Controlling lipid accumulation in cereal grains. Plant Sci 185-186:33–39. https://doi.org/10.1016/j.plantsci.2011.09.002
Beló A, Zheng P, Luck S, Shen B, Meyer DJ, Li B, Tingey S, Rafalski A (2008) Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol Gen Genomics 279:1–10. https://doi.org/10.1007/s00438-007-0289-y
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Camus-Kulandaivelu L, Chevin L-M, Tollon-Cordet C, Charcosset A, Manicacci D, Tenaillon MI (2008) Patterns of molecular evolution associated with two selective sweeps in the Tb1-Dwarf8 region in maize. Genetics 180:1107–1121. https://doi.org/10.1534/genetics.108.088849
Casa AM, Brouwer C, Nagel A, Wang L, Zhang Q, Kresovich S, Wessler SR (2000) The MITE family heartbreaker (Hbr): molecular markers in maize. Proc Natl Acad Sci U S A 97:10083–10089
Chai Y, Hao X, Yang X, Allen WB, Li J, Yan J, Shen B, Li J (2012) Validation of DGAT1-2 polymorphisms associated with oil content and development of functional markers for molecular breeding of high-oil maize. Mol Breed 29:939–949. https://doi.org/10.1007/s11032-011-9644-0
Chen J, Hu Q, Zhang Y, Lu C, Kuang H (2013) P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res 42:D1176–D1181. https://doi.org/10.1093/nar/gkt1000
De La Fuente GN, Murray SC, Isakeit T, Park YS, Yan Y, Warburton ML, Kolomiets MV, Vendramin GG (2013) Characterization of genetic diversity and linkage disequilibrium of ZmLOX4 and ZmLOX5 loci in maize. PLoS ONE 8(1):e53973. https://doi.org/10.1371/journal.pone.0053973
Di Rienzo JA, Casanoves F, Balzarini MG, et al (2011) InfoStat
Dudley JW (2008) Epistatic interactions in crosses of Illinois high oil × Illinois low oil and of Illinois high protein × Illinois low protein corn strains all rights reserved. No part of this periodical may be reproduced or transmitted in any form or by any means, electr. Crop Sci 48:59–68. https://doi.org/10.2135/cropsci2007.04.0242
Dudley JW, Lambert RJ (2003) 100 generations of selection for oil and protein in corn. In: Plant breeding reviews. Wiley, pp 79–110
Estermann MA, Ugarte A, Pretini N, Vega CRC, Olmos SE (2017) Allele mining in the argentine public maize inbred line collection of two paralogous genes encoding NAC domains. Mol Breed 37. https://doi.org/10.1007/s11032-016-0602-8
Gaut BS, Long AD (2003) The lowdown on linkage disequilibrium. Plant Cell 15:1502–1506. https://doi.org/10.1105/tpc.150730
Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J, Ware DH, Buckler ES (2009) A first-generation haplotype map of maize. Science (80- ) 326:1115–1117. https://doi.org/10.1126/science.1177837
Guillet-Claude C, Birolleau-Touchard C, Manicacci D, Rogowsky PM, Rigau J, Murigneux A, Martinant JP, Barrière Y (2004) Nucleotide diversity of the ZmPox3 maize peroxidase gene: relationships between a MITE insertion in exon 2 and variation in forage maize digestibility. BMC Genet 5(19):19. https://doi.org/10.1186/1471-2156-5-19
Guo C, Du J, Wang L et al (2016) Insertions/deletions-associated nucleotide polymorphism in Arabidopsis thaliana. Front Plant Sci 7(1792)
Hospital F (2001) Size of donor chromosome segments around introgressed loci and reduction of linkage drag in marker-assisted backcross programs. Genetics 158:1363–1379
Lambert R (2000) High-oil corn hybrids. In: Specialty corns, second edition. CRC Press
Laurie CC, Chasalow SD, LeDeaux JR et al (2004) The genetic architecture of response to long-term artificial selection for oil concentration in the maize kernel. Genetics 168:2141–2155. https://doi.org/10.1534/genetics.104.029686
Lee EA, Tracy WF (2009) Modern maize breeding. Department of Genetics, University of Georgia, Athens
Liu K, Muse SV (2005) PowerMaker: an integrated analysis environment for genetic maker analysis. Bioinformatics 21:2128–2129. https://doi.org/10.1093/bioinformatics/bti282
Liu K, Goodman M, Muse S, Smith JS, Buckler E, Doebley J (2003) Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 165:2117–2128
Lofland HB, Quackenbush FW, Brunson AM (1954) Distribution of fatty acids in corn oil. J Am Oil Chem Soc 31:412–414. https://doi.org/10.1007/BF02639020
Oakes J, Brackenridge D, Colletti R, Daley M, Hawkins DJ, Xiong H, Mai J, Screen SE, Val D, Lardizabal K, Gruys K, Deikman J (2011) Expression of fungal diacylglycerol acyltransferase2 genes to increase kernel oil in maize. Plant Physiol 155:1146–1157. https://doi.org/10.1104/pp.110.167676
Olmos SE, Delucchi C, Ravera M et al (2014a) Genetic relatedness and population structure within the public Argentinean collection of maize inbred lines. Maydica 59:16–31
Olmos SE, Lorea R, Eyhérabide GH (2014b) Genetic variability within accessions of the B73 maize inbred line. Maydica 59:298–305
Olmos SE, Lia VV, Eyhérabide GH (2016) Genetic diversity and linkage disequilibrium in the Argentine public maize inbred line collection. Plant Genet Resour Characterisation Util 15:515–526. https://doi.org/10.1017/S1479262116000228
Pandit E, Tasleem S, Barik SR, Mohanty DP, Nayak DK, Mohanty SP, Das S, Pradhan SK (2017) Genome-wide association mapping reveals multiple QTLs governing tolerance response for seedling stage chilling stress in Indica Rice. Front Plant Sci 8(552)
Peng T, Sun X, Mumm RH (2014) Optimized breeding strategies for multiple trait integration: I. Minimizing linkage drag in single event introgression. Mol Breed 33:89–104. https://doi.org/10.1007/s11032-013-9936-7
Percibaldi NM, Borrás FS, Robutti JL (1997) Método rápido de extracción y mutilación para determinar ácidos grasos en mejoramiento genético de aceite de maíz. In: Actas VI Congreso Nacional de Maíz. AINBA, Pergamino, Buenos Aires. Argentina, pp 5–8
Plewa MJ, Weber DF (1975) Monosomic analysis of fatty acid composition in embryo lipids of ZEA MAYS L. Genetics 81:277–286
Rosulj M, Trifunovic S, Husic I (2002) Nine cycles of mass selection for increasing oil content in two maize (Zea mays L.) synthetics. Genet Mol Biol 25:449–461
Ruiz M, Olmos S, Percibaldi N, et al (2010) Conversión de líneas elite de maíz a versiones alto oleico asistida por marcadores moleculares
SAS Institute Inc (2009) SAS/STAT 9.2 User’s Guide. User’s Guid SAS Inst Inc, Cary, NC 8640
Singh N, Vasudev S, Yadava DK, et al (2014) Oil improvement in maize: potential and prospects. In: Maize: nutrition dynamics and novel uses. Division of genetics, Indian Agricultural Research Institute, New Delhi, India, pp 77–82
Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS (2001) Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proc Natl Acad Sci U S A 98:9161–9166. https://doi.org/10.1073/pnas.151244298
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289. https://doi.org/10.1038/90135
Tian D, Wang Q, Zhang P, Araki H, Yang S, Kreitman M, Nagylaki T, Hudson R, Bergelson J, Chen JQ (2008) Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature 455:105–108. https://doi.org/10.1038/nature07175
van Heerwaarden J, Hufford MB, Ross-Ibarra J (2012) Historical genomics of North American maize. Proc Natl Acad Sci U S A 109:12420–12425. https://doi.org/10.1073/pnas.1209275109
Weng J, Xie C, Hao Z, Wang J, Liu C, Li M, Zhang D, Bai L, Zhang S, Li X (2011) Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (Zea mays l.) inbred lines. PLoS One 6:e29229. https://doi.org/10.1371/journal.pone.0029229
White PJ, Pollak LM, Duvick S (2007) Improving the fatty acid composition of corn oil by using germplasm introgression. Lipid Technol 19:35–38. https://doi.org/10.1002/lite.200600009
Wittkopp PJ, Kalay G (2012) Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet 13:59–69
Yang X, Guo Y, Yan J, Zhang J, Song T, Rocheford T, Li JS (2010) Major and minor QTL and epistasis contribute to fatty acid compositions and oil concentration in high-oil maize. Theor Appl Genet 120:665–678. https://doi.org/10.1007/s00122-009-1184-1
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551. https://doi.org/10.1534/genetics.107.074245
Zhao K, Tung C-W, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, McClung AM, Bustamante CD, McCouch SR (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2:467. https://doi.org/10.1038/ncomms1467
Zheng P, Allen WB, Roesler K, Williams ME, Zhang S, Li J, Glassman K, Ranch J, Nubel D, Solawetz W, Bhattramakki D, Llaca V, Deschamps S, Zhong GY, Tarczynski MC, Shen B (2008) A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat Genet 40:367–372. https://doi.org/10.1038/ng.85
Acknowledgments
This research was conducted at the National Institute of Agricultural Technology (INTA) Pergamino station, Argentina, and financed by PICT 2009 N° 0036 (ANPCyT) and INTA projects. We thank all staff members that participated during laboratory and field analyses.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Supplementary Fig. S1
Allelic variation of the DGAT1–2 PCR probe within the NAM inbred lines resolved in 6% denaturing polyacrylamide gel. Insertion = HOAC allele FFS (F469) (156 bp), deletion = low-oleic allele F:S (152 bp). (PNG 530 kb)
Supplementary Fig. S2
QQ plots for marker-trait associations across models: (A) GLM, (B) GLM + Q, (C) MLM K, (D) MLM K + Q. (PPTX 187 kb)
Supplementary Fig. S3
Alignment of Zea mays subsp. mays voucher DGAT1–2 (DGAT1–2) gene, partial cds. From collection A, reference genomes and cDNA sequences from the HOAC AW360460 and EU039830 alleles. Sequences were aligned with ClustalW and compared with the P465 flint inbred line durum carrying an FFS allele. Six markers nearby the phenylalanine insertion (---/TTC) were found: C/T, T/C, C/T, ---/TTC, T/C, G/A, and G/A. (DOCX 36.7 kb)
Supplementary Table 1
(DOCX 85.5 kb)
Supplementary Table 2
(XLSX 16.8 kb)
Supplementary Table 3
(XLSX 32.4 kb)
Supplementary Table 4
(XLSX 10.7 kb)
Supplementary Table 5
(XLSX 20.8 kb)
Supplementary Table 6
(XLSX 603 kb)
Rights and permissions
About this article

Cite this article
Olmos, S.E., Trejo, J.M., Percibaldi, N.M. et al. Genetic diversity near the DGAT1-2 gene for high oleic acid content and kernel trait variation in a maize breeding collection. Mol Breeding 38, 98 (2018). https://doi.org/10.1007/s11032-018-0855-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-018-0855-5