
Abstract
The common flesh color of commercially grown watermelon is red due to the accumulation of lycopene. However, natural variation in carotenoid composition that exists among heirloom and exotic accessions results in a wide spectrum of flesh colors. We previously identified a unique orange flesh watermelon accession (NY0016) that accumulates mainly β-carotene and no lycopene. We hypothesized this unique accession could serve as a viable source for increasing provitamin A content in watermelon. Here we characterize the mode of inheritance and genetic architecture of this trait. Analysis of testcrosses of NY0016 with yellow and red fruited lines indicated a codominant mode of action as F1 fruits exhibited a combination of carotenoid profiles from both parents. We combined visual color phenotyping with genotyping-by-sequencing of an F2:3 population from a cross of NY0016 by a yellow fruited line, to map a major locus on chromosome 1, associated with β-carotene accumulation in watermelon fruit. The QTL interval is approximately 20 cM on the genetic map and 2.4 Mb on the watermelon genome. Trait-linked marker was developed and used for validation of the QTL effect in segregating populations across different genetic backgrounds. This study is a step toward identification of a major gene involved in carotenoid biosynthesis and accumulation in watermelon. The codominant inheritance of β-carotene provides opportunities to develop, through marker-assisted breeding, β-carotene-enriched red watermelon hybrids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Watermelon is a major vegetable crop, consumed throughout the world. Watermelon flesh color is a principle fruit trait attributed to carotenoids produced and accumulated in chromoplasts of the flesh cells (Tadmor et al. 2005). Carotenoids are bioactive organic pigments naturally produced in plants, but not in mammalian cells. These compounds are considered important vitamin precursors and antioxidant molecules, beneficial in reducing the risk of cardiovascular diseases and cancer (Gerster 1997; Bramley 2000; Giovannucci et al. 2002).
Watermelon accessions have various flesh colors controlled by several genes. Major flesh colors are white, salmon yellow, orange, red, and canary yellow (Gusmini and Wehner 2006). Salmon yellow watermelon flesh color results from accumulation of pro-lycopene (tetra-cis-lycopene), orange from pro-lycopene and rarely from β-carotene, red from lycopene, and canary yellow watermelon results from accumulation of small amounts of xanthophylls and β-carotene (Tadmor et al. 2005; Bang et al. 2010).
In contrast with heirloom watermelons that show wide color variation, most modern watermelon cultivars have red flesh, reflected by the bright red pigment lycopene. Like in tomato, lycopene is the predominant carotenoid, constituting 70–90% of total carotenoids in red flesh watermelons, with an average of 48.2 mg lycopene per kilogram fresh weight (Perkins-Veazie et al. 2006). The carotenoids phytofluene, phytoene, β-carotene, cis β-carotene, lutein, neurosporene, and zeta-carotene may also be found in the flesh (Tadmor et al. 2005). The lycopene molecule is a symmetrical tetraterpene assembled from eight isoprene units, and although it is a valuable antioxidant, it lacks vitamin A activity. The orange carotenoid pigment β-carotene is the primary natural source of vitamin A. β-Carotene is biosynthesized through two cyclization events promoted by the enzyme lycopene beta-cyclase (LCYB), which creates beta-rings at both ends of the molecule. When consumed, β-carotene is cleaved by the intestinal enzyme β-carotene 15,15′-monooxygenase into two molecules of vitamin A.
Several studies have dealt with inheritance of flesh color in watermelon (Porter 1937; Poole 1944; Shimotsuma 1963; Henderson 1989; Henderson et al. 1998). Wehner (2007) summarized the flesh color inheritance: canary yellow is dominant to all but white flesh, which is epistatic to canary yellow. A dominance series of red flesh to orange (pro-lycopene) to salmon yellow, in that order, has been established. Canary yellow is epistatic to coral red flesh (Henderson et al. 1998). Still, the genetics of watermelon flesh coloration and metabolic pathways are complex and have not been completely resolved.
Genetic populations derived from a cross between different inbred parental lines commonly produce wide variation in flesh colors, and the flesh colors often differ in the pericarp, mesocarp, endocarp, and in carpel walls surrounding the seeds or the vascular tissue of the watermelon fruit. Thus, it is likely that several genes and biochemical pathways affect pigment expression in different watermelon fruit tissues (Ronen et al. 2000; Tadmor et al. 2004, 2005; Lewinsohn et al. 2005a, 2005b). Bang et al. (2007, 2010) studied inheritance of canary yellow and red watermelon flesh color and used biochemical information and gene sequence data to identify a SNP in lycopene beta-cyclase (LCYB), distinguishing between canary yellow and red flesh watermelons.
In addition to inheritance studies of flesh color in watermelon, a few QTL mapping projects have been published. Hashizume et al. (2003) found two flesh color QTL in a biparental population segregating for red, canary yellow, and white flesh. Two QTL mapping studies of a population including red, canary yellow, and pale yellow flesh identified a single QTL on chromosome 4 (Liu et al. 2015, 2016). The genetic basis of β-carotene accumulation in watermelon flesh has not yet been studied.
Previously, we discovered an orange watermelon accession (NY0016) that accumulates β-carotene as its major carotenoid (Tadmor et al. 2005). To study the inheritance of β-carotene accumulation, we crossed NY0016 to both red and yellow cultivars. The resulting hybrids accumulated both parental line carotenoids. A genetic population segregating for yellow and orange flesh was densely genotyped to construct a SNP-based genetic map and identify a QTL associated with watermelon flesh color and further develop a single nucleotide polymorphism (SNP) marker useful for the fortification of watermelon cultivars with β-carotene.
Materials and methods
Plant materials and field experiment
Parental lines of the mapping population were the β-carotene accumulating orange fleshed inbred accession NY0016 and the canary yellow inbred accession Early Moon Beam (EMB). An F2:3 mapping population was developed through self-pollination of the NY0016 × EMB F1 and 87 F2 plants. Three additional lines were used in F1 testcrosses to evaluate the inheritance of β-carotene concentration: the lycopene accumulating, scarlet red-fleshed cultivar, Dixilee (DL); the coral red-fleshed Charleston Gray (CG); and the pro-lycopene accumulating salmon yellow accession Orange Flesh Tender Sweet (OFT) (Fig. 1). For QTL validation, three F2 populations advanced from the above testcrosses (NY0016 × EMB, NY0016 × CG, and NY0016 × DL) were used. Field experiments were conducted during summer 2016 and summer 2017 at the Newe Ya’ar experiment farm (32° 43′ 05.4″ N 35° 10′ 47.7″ E). For the mapping experiment, ten plants per F2:3 family, dependent upon seed germination, and two plots of ten plants from the parents and F1 were sown on plastic-covered beds. Row-to-plant density was 190 × 50 cm. QTL validation experiment included ~ 100 plants per F2 that were subjected to pre-planting genotyping. These plants were sown in trays and grown in nursery greenhouse for 30 days until transplanting at the field. Leaf samples were taken from each F2 plant at 14 days after sowing and used for DNA extraction and genotyping. Each plant was tagged at the tray and this ID was also used at the field to associate genotype with phenotype. Growing regime followed common practices for open-field watermelon growing in this region.

Flesh color and β-carotene concentration of four testcrosses of NY0016. Photographs of longitudinal cross-sections of the parents and F1 hybrids are shown on the left and bar plots of β-carotene and lycopene concentrations (μg/g fresh weight) on the right for testcrosses of NY0016 by: a the canary yellow cultivar, Early Moon Beam (EMB); b the scarlet red cultivar, Dixilee (DL); c the salmon yellow accession Orange Flesh Tender Sweet (OFT); and d the coral red cultivar, Charleston Gray (CG)
Phenotyping
For the mapping experiment (NY0016 × EMB F2:3), ten F3 individuals per F2:3 family were phenotyped for flesh color allowing the determination of segregation or fixation. All fruits were harvested at maturity (~ 72 days post sowing), cut open longitudinally and visually scored for flesh color. Individual F3 fruit flesh color was scored as either yellow (Y), orange (O), or a mixture (Mix) of yellow and orange and then numerically recoded as a 0 (Y), 0.5 (Mix), or 1 (O). The average of all F3 individuals from each F2 was used as the fruit color rating of the F2:3 families for QTL mapping. F2:3 families were also coded for flesh color using categorical scale, as follows: Y = all Yellow, Seg = segregating Y, Mix and O, O = all Orange and Mix = all Mix. Pictures of all 782 fruits across the experiment were taken, allowing further confirmation of flesh color phenotypes during data analysis. Flesh color was also measured (three reads per fruit) using a hand colorimeter (Minolta Sensing Inc., Minolta Chroma Meter Model CR-400, Osaka, Japan) on representative fruits from F2:3 families that were fixed for flesh color. Batches of flesh tissue (from 5 to 10 fruits) were collected from these fixed F2:3 families, immediately frozen in liquid nitrogen and stored at −80 °C. Frozen tissue samples were used for carotenoid composition analysis using high-pressure liquid chromatography (HPLC). Carotenoids were extracted in hexane:acetone:ethanol (2:1:1, v/v/v) mixture and separated on a 4-μm column using a Waters 2695 HPLC apparatus equipped with a Waters 2998 PDA detector (Milford, MA) as described previously (Tadmor et al. 2005). Carotenoids were identified by their characteristic absorption spectra and distinctive retention time. Quantification was performed by integrating the peak areas with standard curves of authentic standard and the Waters Empower Pro3 chromatography software. For qFC.1 QTL validation, 1–2 fruits per F2 plant were harvested at maturity for phenotypic analysis. Fruits were cut open longitudinally and visually scored for flesh color. Images were taken from all fruits.
DNA isolation for GBS
Total genomic DNA was extracted from young leaf tissue of the F2 plants and parents using the GenEluteTM Plant Genomic DNA miniprep kit (Sigma, St. Louis, MO). The quality of the DNA was analyzed by ND-1000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE) and electrophoresis on a 1.0% agarose gel. The concentration of DNA was estimated using a Qubit® 2.0 Fluorometer (Life Technologies, Singapore) with a Qubit® dsDNA BR Assay Kit (Life Technologies, Eugene, OR).
Genotyping of mapping population
Library construction, sequencing, and SNP calling were performed at the Genomic Diversity Facility at Cornell University (Ithaca, NY). Genomic DNA was digested with the restriction enzyme ApeKI to construct 96-plex libraries following GBS protocols modified from Elshire et al. (2011) and sequenced on an Illumina HiSeq 2000/2500. The GBS pipeline of TASSEL v3.0.174 (Glaubitz et al. 2014) was used to align the 100 bp single-end reads to the Charleston Gray genome (version 1; downloaded from ftp://www.icugi.org/pub/genome/watermelon/WCG/v1/) and to call SNPs. Marker summary statistics were calculated with TASSEL v5.2.30 (Bradbury et al. 2007) and SNPs with more than two alleles, 90% missing data or 100% heterozygosity were removed from the dataset. Using TASSEL, the remaining SNPs were imputed with the WindowLD algorithm of the FSFHap imputation plugin (Swarts et al. 2014) and then recoded and filtered with the parental genotypes in the ABH plugin, removing SNPs where the parental genotype is missing, ambiguous or heterozygous. All remaining analysis was completed in the QTL package (Broman et al. 2003) of R v3.3.1 (R Development core team 2016). Sets of SNPs with identical segregation patterns were binned to a single representative per set as redundant markers provide no additional information and slow the analysis. Deviations from expected Mendelian segregation ratios were identified with a χ 2 test and false discovery rate (FDR) multiple testing correction (Benjamini and Hochberg 1995). Severely distorted SNPs (FDR < 0.001) and single distorted SNPs in a region following expected segregation ratios were removed because they are most likely to be genotyping errors. In addition, SNPs with more than 20% missing data were removed to ensure high map quality.
Genetic map construction
The filtered binned SNPs were used to construct a genetic linkage map for the F2 population in R/qtl (Broman et al. 2003). SNPs were formed into linkage groups with a minimum logarithm of odds (LOD) score of 7 and a maximum recombination fraction of 0.35. The greedy algorithm of the orderMarkers function was used to determine the marker order requiring the minimal number of crossovers to explain the observed data. The Lander-Green algorithm (Lander and Green 1987) was used to estimate the linkage map and genetic distances were derived from recombination fractions using the Kosambi mapping function (Kosambi 1943).
QTL mapping
Multiple QTL mapping (MQM) of flesh color using Haley-Knott regression (Haley and Knott 1992) was performed in R/qtl (Broman et al. 2003). An optimal model of highest penalized LOD score (Manichaikul et al. 2009) was chosen from forward and backward selection of models from null to ten QTL with interactions using the function stepwiseqtl (Zeng et al. 1999; Broman and Sen 2009). Penalties for model selection and genome-wide LOD significance thresholds were determined with 1000 permutations of the scantwo function. Flesh color was heteroskedastic with non-normal residuals so non-parametric QTL mapping (scanone with model “np,” Kruskal and Wallis 1952; Kruglyak and Lander 1995) was used to confirm QTL. Genome-wide single marker analysis of un-imputed SNP data was performed for initial QTL mapping using the GLM function in TASSEL v5.2.30 (Bradbury et al. 2007). Candidate genes were identified within the 1.5-LOD interval of QTL by comparison to the genome annotation of the Charleston Gray cultivar (http://cucurbitgenomics.org/organism/4).
Development of CAP assay for HB-SNP
Reference-genome (Charleston Gray; http://cucurbitgenomics.org/organism/4) sequence surrounding the QTL peak marker was used for primers design (forward: ACACTGTGGATGCTTCAAAG, reverse: CCTCTTCCAGCTGCTCATC). DNA from NY0016, EMB and two selected F2 plants, corresponding to F3 families fixed for yellow and orange flesh, were used for PCR followed by sequencing of the amplicons. These four sequences were aligned and the A/G polymorphism at position Chr1: 8,973,472 bp was confirmed to distinguish between the parental alleles and between corresponding segregants. A CAP assay based on selective digestion of the amplicon with Eam1104I was used to analyze NY0016×EMB F2 population and confirmed against the GBS data.
Genotyping of the qFC.1 validation material
Three CAP markers were used for genotyping of qFC.1 QTL validation populations: the HB-SNP CAP marker was used as described above for genotyping the NY0016 × EMB F2 population. A slightly modified CAP assay targeting this locus was used to genotype NY0016 × CG and NY0016 × DL populations. This assay was developed around the G/A polymorphism at position Chr1: 9,047,499 that distinguish between NY0016 and the red lines (CG and DL). Primers for this marker are as follows: forward: CTTCGCTCCTCTCTCCATC and reverse: CGCTGATTTGTGAGAACGC. A 390-bp PCR product selectively digested with Alw26I to distinguish between parental alleles on agarose gel. The third marker that was used for the validation experiment was the CAP assay at the LCYB gene (ClCG04G004090, chr4), according to protocol adopted from Bang et al. (2007). PCR primers are the following: forward: ATCTTGCTCGACCTTATGCG and reverse: CTCTCCAGTCCATAAACACC. PCR product of 319 bp was selectively digested using BsaHI restriction enzyme to distinguish between red and non-red alleles on agarose gel.
Results
Flesh color and carotenoid composition
To evaluate the mode of inheritance of high β-carotene accumulation in watermelon fruit, we crossed NY0016 with yellow and red-fleshed tester accessions. Parents and F1s were grown in parallel, evaluated for flesh color and measured for carotenoids composition. Codominant inheritance of flesh color in the cross between “Early Moon Beam” (EMB), a canary yellow heirloom cultivar, and NY0016, was supported by multiple lines of evidence. First, visual observation and hue measurements of the F1 hybrid were intermediate between the parental hues. Second, although little orange pigment was visible and β-carotene concentrations were very low in the F1 (1.13 μg/g FW), both orange and yellow pigments were clearly produced (Fig. 1a, Table S1). The F1 hybrid of a cross with the scarlet red-fleshed cultivar Dixilee displayed a codominant phenotype with red and orange pigments (Fig. 1b). Codominant inheritance of lycopene and β-carotene was also supported by the intermediate β-carotene and lycopene concentrations and hue of the F1 compared with parental phenotypes (Fig. 1b, Table S1). Another testcross was performed with the orange fleshed heirloom cultivar, “Orange Fleshed Tender Sweet” (OFT), which accumulates pro-lycopene as the main fruit pigment. While visual observation and color measurement did not show a prominent difference between the parents and F1 (as all are orange fleshed), clear codominance was found for β-carotene content as the F1 was intermediate to the parents (Fig. 1c, Table S1). The fourth testcross was with the coral red-fleshed cultivar Charleston Gray. Codominant inheritance of flesh color was visually observed and supported by the intermediate concentrations of lycopene and β-carotene in the F1 compared to the parental lines (Fig. 1d, Table S1).
Segregation of the high β-carotene trait in F2 and F2:3 populations
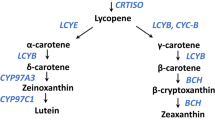
The optimal population for genetic mapping of flesh color required selection of the most appropriate cross to obtain simple segregation, while avoiding interactions with additional genes affecting carotenoid composition. Considering the general framework of the carotenoid biosynthetic pathway (Fig. 2), we determined that the best parent to cross with NY0016 would be the canary yellow accession, EMB, where the coloring pigments are mainly xanthophylls, β-carotene down-stream carotenoids (Fig. 2). Both NY0016 and EMB are homozygotes to the non-red allele at the LCYB gene. In such a cross, the segregation is expected to reflect only the variation related to partial or complete inhibition of the flux at the β-carotene hydroxylation stage, leading to the accumulation of β-carotene. Conversely, segregants of a cross with a red-fleshed accession would also segregate to LCYB allelic variation, which inhibits the carotenoid biosynthesis flux at the lycopene cyclization stage (Bang et al. 2007) and could potentially interact with β-carotene accumulation, making the analysis more complex. The cross between NY0016 and the canary yellow line EMB was first evaluated on a small scale for phenotypic segregation at the F2 generation (Fig. S1A). Segregants included yellow, orange, and fruits expressing a mixture of these colors in ratios not significantly different from expectations under a single codominant gene model (χ 2 test, p value = 0.85).

Biochemical pathway of carotenoid synthesis with relevant cultivars listed next to the predominant carotenoid found in their flesh
Next, we phenotyped 87 F2:3 families that served as the mapping population for flesh color. Ten fruits per F2:3 family were visually evaluated for flesh color at mature stage and families were then classified as fixed-orange, fixed-yellow, fixed-mix, or segregating (Table S2). The segregation across the F2:3 families was quantitatively different from the frequencies calculated at the F2 (Fig. S1B). The family design allowed us to better define F2 heterozygosity through segregation at the F2:3, compared to the definition of mixed flesh color as in heterozygous F2 fruits. The observed F2:3 frequencies significantly deviated from the codominant single gene model suggesting possible epistasis involved. QTL mapping was performed to study the genetic architecture of flesh color in more depth.
Genotyping-by-sequencing, map construction, and QTL mapping
GBS of the F2 population (N = 87) generated almost 288 million reads which resulted in 32,364 SNPs with an average depth of 14.5 reads per site. Imputation with FSFHap reduced the percentage of missing data from 18.9 to 8.9%. A final high-quality set of 3160 filtered SNPs was subjected to clustering based on segregation pattern and 1078 binned SNPs were used for genetic map construction (Table S3) and QTL mapping. Most of the genome was well-covered with average SNP spacing of 0.3 Mb but large gaps of 2.1–6.9 Mb remained. There was no significant segregation distortion (FDR < 0.05) after multiple testing correction (Table S3).
The 1078 binned SNPs formed 11 linkage groups, in accordance with the watermelon 11 chromosomes (Guo et al. 2013), of 42 to 234 cM for a total map length of 1827.8 cM (Figs. S2 and S3). Linkage groups consisted of 36 to 152 SNPs separated by an average genetic distance of 1.7 cM. Large gaps remained in the final map on linkage groups (named by chromosome) 2 and 6. Our map size of 1827.8 cM is comparable to previous intra-variety genetic maps of C. lanatus, which have ranged from 1096 to 3955 cM (Sandlin et al. 2012; Reddy et al. 2014; Meru and McGregor 2016; Shang et al. 2016). Bin marker genetic positions were aligned with physical positions on the Charleston Gray genome (www.icugi.org). Good co-linearity was obtained and genome-wide variation in recombination frequencies across the genome is visible (Fig. S4).
Genome-wide mapping resulted in identification of a single major QTL (qFC.1; P value = 2.8 × 10−9) on chromosome 1, explaining 37 and 57% of the variation in flesh color by MQM and by single marker analysis, respectively (Fig. 3). This QTL was confirmed using non-parametric QTL mapping. The 1.5-LOD confidence interval was from 53.5 to 74 cM on the linkage map with the peak LOD score at 60 cM (Fig. 3b). The most significant SNP in the single marker analysis, at 8,973,472 bp (hereafter, HB-SNP), was within the 1.5-LOD interval of the QTL identified through MQM. Segregation of HB-SNP, the QTL peak marker, at the F2 fitted 1:2:1 Mendelian ratio (22:41:22; chi-square p value = 0.69), however, the HB-SNP marker association with phenotypic color classes distribution across F2:3 families was not symmetric (Fig. 3c, d); all 22 F2:3 families generated from F2 individuals that were homozygotes to the EMB allele of HB-SNP (genotype = A) were fixed for yellow flesh. However, F2 individuals homozygous for the NY0016 allele at the HB-SNP (genotype = G) produced varying F2:3 phenotypes; 59% of the F2:3 families within this genotypic group were fixed for orange flesh, 36% were segregating, and one F2:3 family (5%) was fixed for the mixed (O + Y) phenotype (Fig. 3d). The same pattern is shown for F2 individuals heterozygous at this marker; only 66% of the F2:3 families in this genotypic group expressed the expected phenotype and segregated for flesh color, 22% were fixed for yellow flesh, 10% were fixed for orange flesh, and one F2:3 family (2%) was fixed for the mixed (O + Y) phenotype. These results together with the observed segregation across all F2:3 families (Fig. S1b), which deviated from a single gene model, suggest the possible involvement of another genetic factor in regulating β-carotene accumulation in this population.

SNP association with flesh color in an F2:3 population of watermelon segregating for orange and yellow flesh. a Genome-wide scan of flesh color trait using single marker analysis. Dashed gray line represent genome-wide significance threshold. b LOD profile of qFC.1 QTL using MQM analysis. c Analysis of variance for the genotypic effect of qFC.1 peak marker (SNP_8973472) based on the averaged numeric color scores of F2:3 families. Green diamonds illustrates the mean and confidence intervals. d Phenotypic proportions of flesh color categorical classes across F2:3 families for each genotype at the qFC.1 QTL peak marker (SNP_8973472)
Carotenoids composition was analyzed for 45 F2:3 families with fixed uniform flesh color (yellow or orange). As expected, β-carotene concentration differentiated between the yellow and orange groups and explained the observed color variation (Fig. 4a). We used this quantitative data to perform whole-genome mapping of β-carotene concentration. A single QTL was detected at the same position as qFC.1 on chromosome 1 (Fig. 4b). HB-SNP, at the qFC.1 QTL peak, explained 66% of β-carotene variation across the fixed F2:3 families (Fig. 4c), providing direct link between this variant and the biochemical shift toward β-carotene accumulation.

Analysis of β-carotene concentration for F2:3 families fixed for yellow or orange flesh. a Comparison between the orange (O) and yellow (Y) families. Green diamonds illustrates the mean and confidence intervals. b Comparative whole-genome scans of flesh color (87 F2:3 families, upper pane) and β-carotene concentration (45 fixed F2:3 families, lower pane). Dashed gray line represent genome-wide significance threshold. c Effect of qFC.1 peak marker (SNP_8973472) on β-carotene concentration across 45 fixed F2:3 families
Validation of qFC.1 and epistasis with LCYB locus
To validate the effect of qFC.1 QTL on flesh color and β-carotene accumulation, a second season experiment was performed. Three F2 populations derived from crosses of NY0016 with the tester lines described above (EMB, CG, and DL, Fig. 1, Table S1) were analyzed for the genotype-phenotype association between the HB-SNP and flesh color. One hundred twenty F2 plants, of the orange × yellow (NY0016 × EMB) F2 population, were genotyped using the HB-SNP marker and 1–2 fruits per plant were visually phenotyped for flesh color. Strong association was found (p = 1.5 × 10−15) between HB-SNP segregation and flesh color. In consensus with the results obtained with the smaller F3 mapping population (Fig. 3c, d), non-symmetric segregation pattern was observed also on this F2 population; while homozygotes to the EMB allele had yellow flesh in 95% of the plants (22/23), only 52% (14/27) of the homozygotes to the NY0016 allele were orange and the rest were mostly mix orange-yellow (Fig. 5a). This result validates the QTL effect and the possible involvement of a second locus, essential for β-carotene accumulation in this cross. We further tested the QTL effect in two orange × red crosses. Since lycopene is upstream to β-carotene in the carotenoids pathway, it is expected that red flesh (reflecting blockage of the carotenoid biosynthesis metabolic flux at the lycopene stage) is epistatic over orange (accumulation of high β-carotene). Analysis of orange × red populations, therefore, allowed us to test these assumed epistatic relations and to evaluate the effect of qFC.1 under red-fleshed background. Plants in two orange × red F2 populations (CG and DL, Fig. 1, Table S1) were genotyped using the HB-SNP marker and LCYB marker at the lycopene β-cyclase gene, which was previously reported to be associated with segregation of red flesh phenotype (Bang et al. 2007). In the NY0016 × CG F2 population, 103 plants were genotyped with the two markers and phenotyped for flesh color. LCYB marker showed complete co-segregation with red flesh as all 26 plants that were homozygotes for the CG allele (red parent) had uniform red flesh and all other genotypes at this marker were either yellow, orange, or mixed-color. This association also confirmed the epistasis of red locus over qFC.1 as the segregation of the HB-SNP across these 26 uniform red plants fitted the expected Mendelian 1:2:1 ratio. We then tested the association of the HB-SNP with flesh color on the non-uniform red plants. This group (n = 77) is composed of orange, yellow, and fruits that showed non-uniform mixture of red, orange, and yellow segments. We therefore visually scored the presence of orange and yellow colors in the flesh independently as two different traits, irrespective of the combined flesh color. Association of HB-SNP was highly significant for both phenotypes. Yellow flesh color was observed in all plants homozygotes for the CG allele and was at low frequency in plants homozygotes for the NY0016 allele (p = 6.3 × 10−7, Fig. 5b). Presence of orange flesh color was conversely in high frequency in plants homozygote for NY0016 and almost absent in plants homozygotes for CG allele (p = 3.9 × 10−7, Fig. 5b). In the second orange × red cross, NY0016 × DL, 89 F2 plants were genotyped and phenotyped. Complete association was found between the LCYB marker and uniform red flesh across 25 plants that were homozygote for the DL (red parent) allele. As in the NY0016 × CG population, the effect of qFC.1 was masked by the effect of red genotype at the LCYB locus. Across the remaining non-uniform red plants (n = 64), we found significant association of the QTL with yellow (p = 6.8 × 10−6) and orange (p = 1.2 × 10−11) compositions (Fig. 5c).

Association of HB-SNP with flesh color in three F2 populations. Mosaic plots showing proportions of flesh color categorical classes at the different LCYB and HB-SNP genotypes: a 120 F2 plants of the NY0016 × EMB cross. Yellow-orange stripes represent mixed flesh color. EMB—homozygote to EMB allele, H—heterozygote, NY0016—homozygote to NY0016 allele. b 103 F2 plants of the NY0016 × CG cross. CG—homozygote to CG allele. c 89 F2 plants of the NY0016 × DL cross. DL—homozygote to DL allele
Annotation of genomic sequence at the QTL interval
The 20.5 cM 1.5-LOD confidence interval corresponded to a 2.4-Mb region of chromosome 1, which included 184 annotated genes on the Charleston Gray genome (Table S4). The most obvious candidate gene for association with β-carotene accumulation is the β-carotene hydroxylase gene (β-OHase, Fig. 2), where reduced activity is expected to be correlated with accumulation of its substrate. However, the two annotated homologs of the gene encoding this enzyme (ClCG01G002410, ClCG05G008380) are not located within the qFC.1 QTL confidence interval. Only one carotenoid biosynthesis structural gene was found within the QTL confidence interval, the gene encoding phytoene synthase (PSY), the first rate-limiting enzyme in the pathway. This gene is located ~ 1 Mbp away from the QTL peak. Annotations within the QTL interval identified also 12 putative transcription factors that are potential candidates, some of which are located at the QTL peak region (Table S4).
Discussion
The inheritance pattern revealed here, codominance of orange flesh to scarlet red, coral red, and canary yellow flesh, are not consistent with the previous studies summarized by Wehner (2007) (Porter 1937; Poole 1944; Shimotsuma 1963; Henderson 1989; Henderson et al. 1998). These previous studies found canary yellow to be dominant to all but white flesh, and scarlet red dominant to orange flesh. Variation in inheritance of flesh color between our study and previous reports arise since the orange parental sources of flesh color variants differed between studies; the previous studies (Gusmini and Wehner 2006) orange (like OFT, Fig. 1) and salmon yellow fruited lines result from the accumulation of different amounts of pro-lycopene, while here we used the orange-fruited NY0016 that uniquely accumulates β-carotene (Tadmor et al. 2005).
Watermelon biparental families have been genotyped by GBS in recent years but have been hampered by the low coverage of GBS and the narrow genetic base of C. lanatus with identified markers limited to 266 and 389 SNPs (Lambel et al. 2014; Meru and McGregor 2016). The NY0016 parent used in the current study is atypical from most cultivars and therefore may represent genetic extreme of C. lanatus variation, leading to the higher number of informative SNPs (3160) identified between parental lines, which allowed the construction of relatively high-density map for this intra-specific cross.
A two-gene epistatic model is proposed here to explain the segregation of β-carotene accumulation in the mapping population. This model is supported by the observed frequencies across the F2:3 families, partial dominance at the main locus (qFC.1) interacting with partial dominance at a second locus resulting in the four observed phenotypic classes at the F2:3 families. This model would also explain the non-symmetric association between genotype and phenotype at the QTL peak marker where homozygote EMB allele show complete association with yellow phenotype while both heterozygotes and homozygotes for the NY0016 allele can produce different phenotypic classes, depending on the genotype at a second locus (Fig. 3c, d). Although our data suggests the action of epistasis, we were unable to detect a second locus using stepwise QTL mapping approach where qFC.1 was used as a cofactor, likely due to lack of sufficient statistical power caused by the small size of our mapping population (N = 87) (Carlborg and Haley 2004).
Only one structural carotenogenesis gene, PSY, was found within the 2.4 Mbp QTL confidence interval. PSY is the first enzyme in the carotenogenesis pathway and is not directly involved in the enzymatic steps where β-carotene is substrate or product. However, SNP in PSY gene was previously shown to enhance provitamin A carotenoids accumulation in cassava (Welsch et al. 2010) and a splicing mutation in PSY gene of habanero pepper was shown to cause orange coloration in fruits (Kim et al. 2010). These results indicate on the possible effect of variation in this gene on the activity of other structural enzymes in the carotenoids pathway. Therefore, while PSY is located at the distal part of our QTL confidence interval (1 Mb away from the QTL peak), it cannot be excluded as a candidate gene. Twelve putative transcription factors, that are also potential candidates, were identified within the QTL interval, some of which are located at the QTL peak region. Recent studies of gene expression during fruit maturation in watermelon support a central role of transcriptional regulation in determining carotenoid profiles. However, thus far, no transcriptional regulator of carotenoid accumulation was found in watermelon. Two recent studies examined carotenoid profiles and expression levels of carotenoid biosynthetic genes in different colored watermelon fruits at a series of developmental stages and found coordinated up and down-regulation at different stages by fruit color (Lv et al. 2015; Wang et al. 2016). The authors suggest that carotenoids profiles in mature watermelon fruits are determined through differential transcriptional regulation of carotenoid metabolic genes. Guo et al. (2015) completed a genome-wide comparative transcriptome analysis between red and white fleshed watermelon accessions and found significant expression level differences in several carotenoid biosynthetic genes indicating a role of complex regulatory networks during fruit development. Fruit carotenoids accumulation in various crops has been shown to be regulated mainly at the transcriptional level (Ronen et al. 1999, 2000), still, a carotenogenesis regulating transcription factor has not been identified yet as the major causal gene of fruit color variation.
A codominant cleaved amplified polymorphic (CAP) marker based on HB-SNP polymorphism was developed (Fig. S5) in order to further study the interactions of this QTL with other color genes and to start introducing the high β-carotene allele into elite red-fleshed lines. Using this marker, we validated the QTL in a second season experiment where three F2 populations were analyzed for association of HB-SNP with flesh color (Fig. 5). In addition to another experiment with F2 of the orange × yellow cross (NY0016 × EMB), the QTL was also validated on orange × red crosses. As expected, the red × orange crosses introduced more complexity to color segregation and expanded the color classes observed. Through genotyping of the locus associated with lycopene accumulation (LCYB, Bang et al. 2007) in parallel to the HB-SNP, we were able to demonstrate the epistasis between LCYB and qFC.1 QTL, where, as expected, homozygotes to the red allele at LCYB displayed uniform red flesh color and mask the effect of qFC.1 QTL. The significant effect of qFC.1 QTL on flesh color in these red × orange populations (Fig. 5) provide another independent validation for the involvement of this locus in watermelon β-carotene accumulation. The HB-SNP marker will further be used to construct a secondary mapping population aimed at mapping the epistatic locus using a selective phenotyping approach. Through genotypic selection of large number of F2 segregants, derived from the NY0016 × EMB cross, which are homozygous for the NY0016 allele at the HB-SNP, we will be able to analyze a population that is expected to segregate for yellow-orange flesh color phenotypes, mostly independent of qFC.1 QTL causal gene. This population is expected to provide sufficient statistical power to search for the putative second locus, involved in watermelon β-carotene accumulation.
Codominant inheritance of β-carotene with red and yellow carotenoid pigments was supported by chemical and genetic analyses. A major QTL for color and pigment accumulation was identified and mapped using a high-density genetic map in a population segregating for orange and yellow flesh. The QTL confidence interval does not include any obvious candidate gene; however, it includes several transcription factors, some of which are located at the QTL peak, suggesting the possible involvement of transcription factors in watermelon fruit β-carotene accumulation. Our results suggest involvement of second locus acting epistatically with the qFC.1 QTL.
The results of this study are a first step toward uncovering the genetic basis of β-carotene accumulation in watermelon with the ultimate goal of combining high levels of β-carotene and lycopene for improved nutrition. Based on annotations of the Charleston Gray genome at the QTL interval, we suggest that transcriptional regulation may play a key role in β-carotene accumulation in watermelon flesh. Thus, future analyses of this trait could create new insights into the transcriptional regulation of carotenogenesis genes, which would be of wide interest and could be beneficial to additional carotenoid accumulating agricultural crops.
References
Bang H, Davis AR, Kim S et al (2010) Flesh color inheritance and gene interactions among canary yellow, pale yellow, and red watermelon. J Am Soc Hortic Sci 135(4):362–368
Bang H, Kim S, Leskovar D, King S (2007) Development of a codominant CAPS marker for allelic selection between canary yellow and red watermelon based on SNP in lycopene β-cyclase (LCYB) gene. Mol Breed 20:63–72
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57:289–300
Bradbury PJ, Zhang Z, Kroon DE et al (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Bramley PM (2000) Is lycopene beneficial to human health? Phytochemistry 54:233–236
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Broman KW, Sen S (2009) A guide to QTL mapping with R/qtl. Vol. 46. Springer, New York
Carlborg O, Haley CS (2004) Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5:618–625
Elshire RJ, Glaubitz JC, Sun Q et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Gerster H (1997) The potential role of lycopene for human health. J Am Coll Nutr 16:109–126
Giovannucci E, Rimm EB, Liu Y, Stampfer MJ, Willett WC (2002) A prospective study of tomato products, lycopene, and prostate cancer risk. J Natl Cancer Inst 94:391–398
Glaubitz JC, Casstevens TM, Lu F et al (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9:e90346
Guo S, Sun H, Zhang H et al (2015) Comparative transcriptome analysis of cultivated and wild watermelon during fruit development. PLoS One 10:1–21
Guo S, Zhang J, Sun H et al (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45:51–58
Gusmini G, Wehner TC (2006) Qualitative inheritance of rind pattern and flesh color in watermelon. J Hered 97:177–185
Haley CS, Knott SA (1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69:315–324
Hashizume T, Shimamoto I, Hirai M (2003) Construction of a linkage map and QTL analysis of horticultural traits for watermelon [Citrullus lanatus (THUNB.) MATSUM & NAKAI] using RAPD, RFLP and ISSR markers. Theor Appl Genet 106:779–785
Henderson WR (1989) Inheritance of orange flesh color in watermelon. Cucurbit Genet Coop Rpt 12:59–63
Henderson WR, Scott GH, Wehner TC (1998) Interaction of flesh color genes in watermelon. J Hered 89:50–53
Kim OR, Cho MC, Kim BD, Huh JH (2010) A splicing mutation in the gene encoding phytoene synthase causes orange coloration in habanero pepper fruits. Molecules and Cells 30:569–574
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Eugenics 12:172–175
Kruglyak L, Lander ES (1995) A nonparametric approach for mapping quantitative trait loci. Genetics 139:1421–1428
Kruskal WH, Wallis WA (1952) Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47(260):583–621
Lambel S, Lanini B, Vivoda E et al (2014) A major QTL associated with Fusarium oxysporum race 1 resistance identified in genetic populations derived from closely related watermelon lines using selective genotyping and genotyping-by-sequencing for SNP discovery. Theor Appl Genet 127(10):2105–2115
Lander ES, Green P (1987) Construction of multilocus genetic linkage maps in humans. Proc Natl Acad Sci U S A 84:2363–2367
Lewinsohn E, Sitrit Y, Bar E et al (2005a) Not just colors—carotenoid degradation as a link between pigmentation and aroma in tomato and watermelon fruit. Trends Food Sci Technol 16:407–415
Lewinsohn E, Sitrit Y, Bar E et al (2005b) Carotenoid pigmentation affects the volatile composition of tomato and watermelon fruits, as revealed by comparative genetic analyses. J Agric Food Chem 53:3142–3148
Liu S, Gao P, Wang X et al (2015) Mapping of quantitative trait loci for lycopene content and fruit traits in Citrullus lanatus. Euphytica 202:411–426
Liu S, Gao P, Zhu Q et al (2016) Development of cleaved amplified polymorphic sequence markers and a CAPS-based genetic linkage map in watermelon (Citrullus lanatus [Thunb.] Matsum. and Nakai) constructed using whole-genome re-sequencing data. Breed Sci 66:244–259
Lv P, Li N, Liu H et al (2015) Changes in carotenoid profiles and in the expression pattern of the genes in carotenoid metabolisms during fruit development and ripening in four watermelon cultivars. Food Chem 174:52–59
Manichaikul A, Moon JY, Sen Ś et al (2009) A model selection approach for the identification of quantitative trait loci in experimental crosses, allowing epistasis. Genetics 181:1077–1086
Meru G, McGregor C (2016) Genotyping by sequencing for SNP discovery and genetic mapping of resistance to race 1 of Fusarium oxysporum in watermelon. Sci Hortic 209:31–40
Perkins-Veazie P, Collins JK, Davis AR, Roberts W (2006) Carotenoid content of 50 watermelon cultivars. J Agric Food Chem 54:2593–2597
Poole CF (1944) Genetics of cultivated cucurbits. J Hered 35(4):122–128
Porter DR (1937) Inheritance of certain fruit and seed characters in watermelons. Hilgardia 10(12):489–509
Development Core Team R (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Reddy UK, Nimmakayala P, Levi A, et al (2014) High-resolution genetic map for understanding the effect of genome-wide recombination rate on nucleotide diversity in watermelon. G3 (Bethesda, Md) 4:2219–30
Ronen G, Cohen M, Zamir D, Hirschberg J (1999) Regulation of carotenoid biosynthesis during tomato fruit development: expression of the gene for lycopene epsilon-cyclase is down-regulated during ripening and is elevated in the mutant Delta. Plant J 17:341–351
Ronen G, Carmel-Goren L, Zamir D, Hirschberg J (2000) An alternative pathway to β-carotene formation in plant chromoplasts discovered by map-based cloning of Beta and old-gold color mutations in tomato. PNAS 97:11102–11107
Sandlin K, Prothro J, Heesacker A et al (2012) Comparative mapping in watermelon [Citrullus lanatus (Thunb.) Matsum. et Nakai]. Theor Appl Genet 125:1603–1618
Shang J, Li N, Li N et al (2016) Construction of a high-density genetic map for watermelon (Citrullus lanatus L.) based on large-scale SNP discovery by specific length amplified fragment sequencing (SLAF-seq). Sci Hortic 203:38–46
Shimotsuma M (1963) Cytogenetical studies in the genus Citrullus: VII. Inheritance of several characters in watermelons. Jpn J Breed 13(4):235–240
Swarts K, Li H, Romero Navarro JA et al (2014) Novel methods to optimize genotypic imputation for low-coverage, next-generation sequence data in crop plants. Plant Genome 7. https://doi.org/10.3835/plantgenome2014.05.0023
Tadmor Y, Katzir N, King S, Levi A, Davis A, Hirschberg J (2004) Fruit coloration in watermelon: lessons from the tomato. In: Lebeda A, Paris HS (eds) Progress in cucurbit genetics and breeding research. Palacky University in Olomouc, Czech Republic, pp 181–185
Tadmor Y, King S, Levi A et al (2005) Comparative fruit colouration in watermelon and tomato. Food Res Int 38:837–841
Wang N, Liu S, Gao P et al (2016) Developmental changes in gene expression drive accumulation of lycopene and β-carotene in watermelon. J Am Soc Hortic Sci 141:434–443
Wehner TC (2007) Gene list for watermelon. Cucurbit Genet Coop Rep 30:96–120
Welsch R, Arango J, Bär C, Salazar B, Al-Babili S, Beltrán J, Chavarriaga P, Ceballos H, Tohme J, Beyer P (2010) Provitamin A accumulation in cassava (Manihot esculenta) roots driven by a single nucleotide polymorphism in a phytoene synthase gene. Plant Cell 22:3348–3356
Zeng ZB, Kao CH, Basten CJ (1999) Estimating the genetic architecture of quantitative traits. Genet Res 74:279–289
Acknowledgements
We gratefully acknowledge support from the “Center for the Improvement of Cucurbit Fruit Quality,” ARO, Israel. We also acknowledge partial support by US-Israel Binational Agricultural Research & Development (BARD) Senior Research Fellowship Program (USDA-BARD Agreement Number 58-6080-5-011F), and by USDA-National Institute of Food and Agriculture (NIFA), Specialty Crop Research Initiative (SCRI) grant number 2015-51181-24285. We also thank the USA National Watermelon Research and Promotion Board for their continual support; publication no. 211/2017 of the Agricultural Research Organization, Bet Dagan, Israel.
Funding
Funding for this research was provided by the Israeli Ministry of Agriculture Chief Scientist grant no. 20-01-0135.
Author information
Authors and Affiliations
Contributions
AG, AL, AM, and YT conceived and designed the study. LV, AM, and GT performed field experiments, phenotyping, and genotyping. AG, ZF, and SB analyzed the data. AG, SB, AL, WW, and YT wrote the paper. All authors discussed the results and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Key message
Major QTL regulating the accumulation of β-carotene in the fruit flesh was mapped to 2 Mbp intervals on chromosome 1 in watermelon.
Electronic supplementary material
ESM 1
Figure S1. Photographs of longitudinal cross-sections of representative individuals for each flesh color category and a pie chart of segregation ratios for the (a) F2 population and (b) F2:3 families. Figures S2-S3. Genetic linkage map of Citrullus lanatus with 1078 binned SNPs produced from an F2:3 population derived from experimental line NY0016 crossed with cultivar EMB. Linkage groups 1 through 6 are displayed in Figure S1 and linkage groups 7–11 in Figure S2. SNP positions (cM) are labelled to the left of each linkage group (labelled at the top of the figure) and the chromosome and physical position (bp) are to the right. The 1.5-LOD interval of qFlesh-1 is indicated with a pink vertical bar to the right of linkage group 1. Fig. S4. Plots of genetic (cM) versus physical distance (bp) of each SNP across the 11 watermelon chromosomes. Fig. S5. Gel electrophoresis of PCR with CAPS marker for the parents (EMB and NY0016), F1, and representative F2 individuals. Letters above the gel bands represent the translated genotype: E = Homozygote EMB, N = Homozygote NY0016, H = Heterozygote. (PDF 1735 kb)
Table S1
Variation in color and carotenoid concentrations in four testcrosses with NY0016 (XLSX 10 kb)
Table S2
Flesh color data for the F2:3 families in categorical and continuous scales (separated tabs), including pedigree, family ID (family); the family flesh color categories used for QTL mapping (flesh0–1), family categorical flesh ratings (seg) and the individual fruit colors (fruit1-fruit10). (XLSX 23 kb)
Table S3
1078 Binned SNPs information, including: SNP name (SNP), genetic position (cM) on each linkage group (LG), physical position (bp) on each chromosome (CS), results of a chi test for segregation distortion (P, neglog10P, and FDR), and the proportions of individuals with each genotype at that locus (AA, AB, BB, or missing). AA indicates individuals homozygous for NY0016 alleles, BB homozygous for EMB alleles or heterozygous individuals AB. (XLSX 111 kb)
Table S4
Annotated genes list within the 1.5-LOD interval (8.00 to 10.50 Mb on chromosome 1) for flesh color QTL (qFC.1). Gene information includes: identifier for watermelon gene (geneID), chromosome (CG_Chr), start position (bp), stop position (bp), and the functional annotation. Annotated candidates are highlighted. (XLSX 18 kb)
Rights and permissions
About this article

Cite this article
Branham, S., Vexler, L., Meir, A. et al. Genetic mapping of a major codominant QTL associated with β-carotene accumulation in watermelon. Mol Breeding 37, 146 (2017). https://doi.org/10.1007/s11032-017-0747-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-017-0747-0