
Abstract
The Nicotiana tabacum × N. africana cross is a semilethal cross, where greater than 99 % of progeny die in the cotyledonary stage due to an interspecific lethality reaction. The low frequencies of surviving plants are mixtures of maternal tobacco haploids and aneuploid F1 hybrids. For effective use of surviving haploids in a breeding program, an efficient method is needed to distinguish them from aneuploid hybrids during the seedling stage. The first objective of this research was to investigate the use of N. africana engineered with a green fluorescent protein (gfp) transgene for distinguishing haploids from other surviving plants derived from this cross. Results demonstrated gfp expression to be a useful phenotypic marker for separation of aneuploid hybrids at the seedling stage. Microsatellite marker genotyping, flow cytometry determinations, and chromosome counts resulted in identification of progeny that exhibited intermediate DNA amounts and variable chromosome numbers donated by the N. africana parent. Three individuals with near-haploid DNA content were identified that carried N. africana markers and n = 24 + 2 or n = 24 + 3 chromosomes. The results present evidence of genetic instability in progeny from the N. tabacum × N. africana cross and suggest that, in contrast to previous belief, chromosome elimination may play at least a partial role in the generation of haploids from this cross. Additionally, microsatellite marker genotyping demonstrated a role for a gene or genes located near the end of N. tabacum chromosome H (linkage group 11) in the interspecific lethality reaction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Haploid plants possess the gametic chromosome number (n) and are useful in plant breeding programs because they can be converted to doubled haploids which can be self-pollinated to produce inbred lines in a dramatically reduced amount of time as compared to conventional methods of inbreeding. Tobacco, Nicotiana tabacum L. (2n = 48), has been used as a model system to develop and study doubled haploid breeding methods. For example, anther or microspore culture can be used to efficiently produce a large number of haploid plants for this species (Bourgin and Nitsch 1967; Nitsch and Nitsch 1969). Introduction of deleterious variation using this process (reviewed by Wernsman 1992) has limited the utility of this method in practical breeding programs, however. Haploid plants of both maternal and paternal origin occur spontaneously at low frequencies in progeny of N. tabacum × N. tabacum crosses (de Nettancourt and Stokes 1960; Burk 1962; Lewis and Rose 2011). Burk et al. (1979) found that N. tabacum could also yield low frequencies of maternal haploids when pollinated with the African species N. africana [Merx. and Buttler] (2n = 46). This is a semilethal cross, and >99 % of the progeny die at the seedling stage due to an interspecific lethality reaction. Surviving plants are mixtures of haploids and aneuploid F1 hybrids (Burk et al. 1979; Gerstel et al. 1979; Kramer and Reed 1988). Because of the high number of seeds (~3000) produced per pollination, this method is somewhat attractive for tobacco haploid isolation. For efficient use in a breeding program, however, a system for distinguishing plants of the two groups at the seedling stage is needed. While leaf shape, trichome size and density, and stomatal measurements can help distinguish haploids from aneuploid hybrids early in plant development (Flowers et al. 1967; Burk et al. 1979; Reed 1993), this process can still be considered an art and many tobacco breeding programs have not adopted the N. africana-based system of haploid production for this reason.
A visual seed or seedling marker would be useful for distinguishing maternal haploids from aneuploid N. tabacum × N. africana F1 hybrids early in seedling development. Methods previously used for tobacco involve a recessive yellow seedling marker (yg) (Burk 1962); the TMV-resistance gene, N (de Nettancourt and Stokes 1960); a rootless Rac − mutant genetic stock (Pelletier et al. 1987); and a transgenic purple seedling trait (Lewis and Rose 2011). Some of these visible markers are not useful in applied breeding programs, however, because of the required genetics of elite cultivars. Expression of green fluorescent protein (GFP) (Tsien 1998) might also be used as a dominant seedling marker. If N. africana were engineered to express visible levels of GFP, aneuploid F1 hybrids from the N. tabacum × N. africana cross would likewise be expected to express the trait, while maternal N. tabacum haploids would not. The first objective of this research was to genetically engineer N. africana to express GFP, and to evaluate a genetic stock expressing this trait for its utility in separating maternal haploids from aneuploid F1 hybrids from N. tabacum × N. africana interspecific crosses.
Burk et al. (1979) suggested that the rate of maternal haploidy from the N. tabacum × N. africana cross reflects the rate of spontaneous parthenogenesis in this species (approximately 1 in 1000 seeds). Gynogenesis is a form of parthenogenesis that is stimulated by the presence of a sperm cell, whereby a haploid embryo develops in the absence of double fertilization. Some interspecific crosses appear to stimulate gynogenesis, and in most examples, fertilization of the polar nuclei occurs so that an endosperm is produced. Chromosome elimination, on the other hand, is also known to play a role in haploid development in a number of interspecific crosses involving crop plants (Subrahmanyam and Kasha 1973; Laurie and Bennett 1989; Riera Lizarazu et al. 1996; Gernand et al. 2005). In this genetic phenomenon, the chromosomes of one parental species become eliminated in the initial mitotic divisions following fertilization. In some cases, haploid or near-haploid plants have been identified that harbor addition chromosomes or introgressions donated by the wild parent (Laurie and Bennett 1989; Wilkinson et al. 1995; Riera Lizarazu et al. 1996; Chen et al. 1998; Kynast et al. 2001; Gernand et al. 2005; Li et al. 2009). A secondary objective of the research reported here was to determine whether evidence could be obtained to support a possible role of chromosome elimination in the generation of maternal haploids from N. tabacum × N. africana crosses.
Finally, as mentioned previously, the vast majority of progeny from the N. tabacum × N. africana cross do not survive because of an interspecific lethality mechanism that results in death of hybrids in the cotyledonary stage. Hybrid lethality is of one of several genetic systems that can contribute to reproductive isolation between species, and it can be manifested by zygote abortion soon after fertilization or cell death in hybrid seedlings after germination (Adachi 2001; Mino et al. 2002). Hybrid lethality is a commonly observed post-zygotic reproductive barrier in F1 hybrids from interspecific crosses between N. tabacum and species from section Suaveolentes, of which N. africana is a member (reviewed by Tezuka et al. 2012). The control underlying such genetic interactions is of interest, as it affects the evolution of species. Little is currently known about the mechanisms behind interspecific hybrid lethality in plants, however. Burk et al. (1979) and Gerstel et al. (1979) provided evidence to suggest a role for a gene or genes on chromosome H of the N. tabacum genome in the N. tabacum × N. africana lethality reaction. An additional objective of this research was to use microsatellite markers to gain insight on the genetic control of this lethality response, with specific interest on the role of genomic regions located on chromosome H of the N. tabacum genome.
Materials and methods
Transformation
Nicotiana africana was transformed using Agrobacterium tumefaciens according to a modification of the procedure of An et al. (1986). Agrobacterium strain GV3101 carried expression vector pBIN mgfp5-ER (Siemering et al. 1996; Haseloff et al. 1997) bearing the mgfp5-ER gene under the control of the CaMV 35S promoter and linked to the selectable marker gene nptII. This gfp version was optimized using mutations to result in improved expression and greater fluorescence in plants (Heim et al. 1994; Cubitt et al. 1995; Chiu et al. 1996; Tsien 1998; Siemering et al. 1996; Haseloff et al. 1997), and contains an endoplasmic reticulum (ER) targeting sequence which results in protein concentration within the lumen of the endoplasmic reticulum, instead of elsewhere in the cell (Haseloff et al. 1997).
After 2 days of co-cultivation with Agrobacterium, inoculated leaf disks were transferred to shoot regeneration medium comprised of MS inorganic salts supplemented with 1 mg L−1 benzylaminopurine, 0.1 mg L−1 α-naphthaleneacetic acid, 30 g L−1 sucrose, and 7 g L−1 agar. Also added were 100 mg L−1 kanamycin and 250 mg L−1 cefotaxime to select for transformed cells and to eliminate contaminating bacteria. Regenerated shoots were transferred to rooting medium consisting of MS inorganic salts plus 30 g L−1 sucrose and 7 g L−1 agar. Rooted plants were transferred to soil-filled, 8 cm × 8 cm plastic pots containing Fafard 2P Mix (Conrad Fafard, Inc, Agawam, MA) and were designated as R 0 transformants. N. africana plants homozygous for a stably expressing single transgene copy were identified through a process of self-pollination, Southern blotting, and evaluation of testcross progeny for non-segregation of the 35S:mgfp5-ER transgene and its phenotypic expression. Phenotypic expression of 35S:mgfp5-ER was determined by exposure of plants to a near ultraviolet (365 nm) handheld UVL-56 light (UVP, Inc, Upland, CA) in a darkened room.
Southern blotting and PCR testing
The presence of 35S:mgfp5-ER was determined by both Southern blotting and PCR. For Southern blotting, 12.5 µg of genomic DNA was digested with HindIII and was separated on a 0.8 % agarose gel at 16 V for 20 h, followed by blotting to a Genescreen Plus nylon membrane (Perkin-Elmer, Inc, Boston, MA) according to Sambrook et al. (1989). DNA was UV-cross-linked to bind the DNA to the membrane. A 800-bp gfp probe was isolated using forward primer 5′-CCTTAAGGATCCAAGGAGATATAACAATGA-3′ and reverse primer 5′-CCGGTTGAGCTCTTAAAGCTCATCATGTT-3′. The PCR product was gel-purified and labeled with α-32P-dCTP using a random primed DNA labeling kit (Roche Diagnostics Corporation, Indianapolis, IN). Prehybridization and hybridization with the radiolabeled probe were performed according to Sambrook et al. (1989) in PerfectHyb™ Plus Hybridization Buffer (Sigma-Aldrich, Inc, St. Louis, MO). The DNA blot was washed and exposed to X-ray film according to Sambrook et al. (1989).
PCRs to test for the presence of 35S:mgfp5-ER were performed using a 96-well Bio-Rad PTC-100® thermal cycler (Bio-Rad Laboratories, Inc, Hercules, CA) in 20 µl volumes containing 150 ng genomic DNA, 2 μL of 10X PCR buffer (New England Biolabs, Ipswich, MA), 2 μL of 20 mM MgSO4, 1.6 μL of 2.5 mM dNTPs, 0.8 μL of 5 µM forward mGFP5 primer (5′-CCTTAAGGATCCAAGGAGATATAACAATGA-3′), 0.8 μL of 5 µM reverse mGFP5 primer (5′-CCGGTTGAGCTCTTAAAGCTCATCATGTTT-3′), 0.2 μL of Taq DNA polymerase (5 U μL−1) (New England Biolabs, Ipswich, MA), and 9.6 μL of dH2O. Reaction parameters for gfp transgene detection were a single cycle for 2:30 min at 94 °C, followed by 30 cycles of 94 °C for 30 s, 65 °C for 60 s, 72 °C for 60 s, and a final extension period of 5 min at 72 °C. Reaction products were separated on a 1.5 % (w/v) agarose gels containing 0.15 µg mL−1 ethidium bromide in 1 × TAE buffer. Gels were run for 2 h at 100 V and were visualized using a FOTODYNE Transilluminator (FOTODYNE Inc, Hartland, WI).
N. tabacum × N. africana hybridizations
Flowers of burley tobacco cultivar TN 90LC were hand-pollinated with pollen from N. africana line GH12-195-3 identified to be homozygous at a single 35S:mgfp5-ER locus. TN 90LC was chosen for use as the female N. tabacum parent because of its homozygosity for recessive alleles at the yellow burley 1 (yb 1) and yellow burley 2 (yb 2) loci. The yb 1 yb 1 yb 2 yb 2 genotype results in an easily distinguished chlorophyll deficiency in the stems and leaf midveins that becomes increasingly obvious about 40 days post-germination (Stines and Mann 1960). The yellow burley phenotype permits easy recognition of maternal haploids (yb 1 yb 2 genotype; yellow burley phenotype) from aneuploid F1 interspecific hybrids (green stem and leaf midvein phenotype) (Wernsman et al. 1989; Lewis and Rose 2011).
Approximately 65,990 seeds from the N. tabacum × N. africana 35S:mgfp5-ER interspecific cross were germinated in 16 cm × 22 cm plastic pans containing Fafard 2P Mix and maintained in a laboratory growth room at 26 °C with 18-h/6-h dark/light conditions. Pans were seeded to an average density of approximately 480 seeds per 10 dm2. After 20–25 days, all surviving plants were transferred to individual 8 cm × 8 cm plastic pots containing Fafard 2P Mix.
Analyses of surviving N. tabacum × N. africana plants
All surviving plants were first screened for expression of 35S:mgfp5-ER using a handheld near ultraviolet light in a darkened room. A subset of 182 surviving plants comprised of both gfp-expressing and non-gfp-expressing plants were also (1) genotyped using PCR to determine the presence/absence of 35S:mgfp5-ER, (2) phenotyped for the yellow burley character, (3) genotyped for the TMV-resistance gene, N, located on chromosome H of the N. tabacum genome according to Lewis et al. (2005), (4) genotyped with at least one microsatellite marker from each of the 24 N. tabacum linkage groups, and (5) quantified for nuclear DNA content using flow cytometry.
Microsatellite marker genotyping
A subset of the surviving plants from the N. tabacum × N. africana 35S:mgfp5-ER interspecific cross, as well as three bulk samples of tissue collected from plants which did not develop beyond the cotyledonary stage, were genotyped with at least one microsatellite locus per N. tabacum linkage group. Genotyping at a higher number of loci (13) for N. tabacum chromosome H (see below for chromosome H identification) was conducted because of the implied role of this chromosome in the lethality response for this cross (Burk et al. 1979). A total of 280 microsatellite primer pairs published in Bindler et al. (2011) were initially screened to identify those that clearly and easily amplified polymorphic bands between the parental lines, TN 90LC and transgenic N. africana line GH12-195-3. Thirty-nine microsatellite primer pairs were ultimately selected for genotyping of surviving plants based on the goal of representing each N. tabacum linkage group and a desire to have an increased number of markers for N. tabacum chromosome H. Although we knew a priori the genomic location of the amplified markers for the N. tabacum genome, we currently have no knowledge of the relative locations of the markers amplified for the N. africana genome because linkage maps for this species do not exist.
To identify the genetic linkage group of Bindler et al. (2011) corresponding to chromosome H of the N. tabacum genome, we hybridized N. tabacum cultivar Hicks as a pollen parent with a genetic stock of Red Russian monosomic for chromosome H (Red Russian Haplo H). Seventy-eight primer pairs amplifying polymorphic markers from loci representing each of the 24 N. tabacum linkage groups were used to genotype 94 random progeny from the Red Russian Haplo H × Hicks cross. Red Russian genotypes monosomic for chromosome H transmit the unpaired chromosome through the egg at a frequency of approximately 29.6 % (Clausen and Cameron 1944). If a marker were associated with a chromosome other than chromosome H, each marker allele would be present for each of the 94 progeny. If a marker were associated with chromosome H, however, segregation for the Red Russian allele would occur in these progeny. After identification of a candidate linkage group corresponding to chromosome H, an additional ten markers from that linkage group were also used to genotype the set of 94 progeny from the Red Russian Haplo H × Hicks cross to verify their association with chromosome H.
Detection of PCR products was conducted using the labeling method of Schuelke (2000). PCRs were performed in 15 µl volumes containing 25 ng of template DNA (5 ng μL−1), 1.5 μL of 10 × PCR buffer, 1.5 μL of 20 mM MgSO4, 1 μL of 5 M betaine, 1.2 μL of 2.5 mM dNTPs, 0.15 μL of 1 μM forward primer stock, 0.75 μL of 1 μL reverse primer stock, 0.75 μL of 1 μM M13 primer stock (IRD-700 or IRD-800), 0.2 μL of Taq DNA polymerase (5 U μL−1) (New England Biolabs, Ipswich, MA), and 2.95 μL of dH2O.
PCR conditions consisted of a denaturation step at 94 °C for 5 min; 30 cycles of 94 °C for 30 s, (Tm of primer) °C for 45 s, 72 °C for 45 s; followed by 7 cycles of 94 °C for 30 s, (Tm – 3) °C for 45 s, 72 °C for 45 s; and a final extension step at 72 °C for 5 min. Amplification products were separated using 8 % polyacrylamide gels and a LI-COR 4300 DNA Analysis System (LI-COR Biosciences, Lincoln, Nebraska) under the following conditions: 1500 V, 40 mA, 40 W, and 45 °C for 2.5 h. IRDye 700- or 800-labeled molecular weight standards (50–350 bp) were loaded on each gel to facilitate allele sizing using AFLP Quantar 1.0 software (KeyGene Products B.V., Wageningen, the Netherlands).
Flow cytometry
A subset of surviving plants from the N. tabacum × N. africana cross were analyzed for nuclear DNA content by flow cytometry using nuclei from N. tabacum cv. TN 90LC and Glycine max Merr. cv. NC-Raleigh as internal controls. DNA extraction and propidium iodide staining were performed with a CyStain PI Absolute P kit (Partec North America, Inc, Swedesboro, NJ). Each sample was prepared by adding 600 μL of cold nuclei extraction buffer to a 5-cm petri dish containing the standards and sample tissue. The sample leaf tissue (approximately 2.5 cm2) and internal standards were co-chopped with a doubled-edged razor blade for approximately 60 s, incubated in the extraction buffer for approximately 90 s, and then filtered through a CellTrics 30-μm filter (Partec North America, Inc, Swedesboro, NJ) into a 2-mL microcentrifuge tube. Samples were stored on ice and centrifuged for 30 s at 13,200 rpm to pellet the nuclei. The supernatant was then poured off, and the nuclei were resuspended in 600 μL of cold staining buffer, 200 μL of extraction buffer, 6 μL CyStain propidium iodide, and 3 μL RNase (3.33 mg/mL) (Partec North America, Inc, Swedesboro, NJ). The samples were incubated in the dark at room temperature for 45–60 min. After incubation, the samples were filtered through a CellTrics 30-μm filter into 5-mL BD Falcon polystyrene tubes (BD Biosciences, Franklin Lakes, NJ) and stored on ice in the dark until analyses on a BD LSR II flow cytometer (Becton–Dickinson Biosciences, San Jose, CA) fitted with a 15-mW argon laser (excitation at 488 nm) and propidium iodide fluorescence (FLA-2) detector. The flow rate was approximately 300 particles per second. Signals from subcellular debris were gated out, and the position of the G1 histogram peaks was measured using BD FACSDiva software (Becton–Dickinson Biosciences, San Jose, CA). Histograms were based on 20,000 scanned events that were not gated out. DNA content (pg nuclei−1) was calculated as (sample histogram peak/standard histogram peak) × DNA content of the standard. When interspecific aneuploid hybrid sample histogram peaks overlapped with the diploid standard, subsequent samples of those individuals were analyzed using the soybean internal control only.
Cytogenetic analysis
Somatic chromosome numbers were determined for plants of interest by counting mitotic metaphase chromosomes in corolla tissues of the flower buds. Collection, pretreatment, and fixation of corolla tissues were carried out as described by Burns (1964). Chromosome squashes were conducted using the enzymatic digestion and ‘drop’ method as described by Andres and Kuraparthy (2013). Cells were examined using an Olympus BX53 Dark Fluorescence microscope (Olympus Corporation, Tokyo, Japan) connected to a Prior Lumen 200 light source (Prior Scientific, Cambridge, UK). Chromosomes counts were made for at least 10–15 cells for each slide. Images were captured using a Hamamatsu ORCA-03 camera (Hamamatsu Photonics, Hamamatsu, Japan) and processed using cell sense dimension imaging software (Olympus Corporation, Tokyo, Japan).
Results
Generation of genetic materials
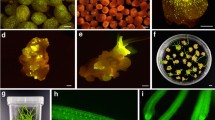
A N. africana line homozygous for a single 35S:mgfp5-ER transgene insertion and stably expressing the GFP signal (Supplementary Figure 1) was isolated and designated as GH12-195-3. This line was hybridized as the pollen parent with burley tobacco cultivar, TN 90LC, and approximately 65,990 seeds from these crosses were germinated. An estimated 99.62 % of these seeds failed to develop past the cotyledonary stage (Fig. 1). The total number of surviving plants was 251 (0.381 %), of which 76 expressed GFP and 175 did not (Fig. 1). The level of GFP expression in GFP-expressing N. tabacum × N. africana progeny appeared to be lower than that of the transgenic N. africana line per se (Supplementary Figure 1; Fig. 1), but expression or lack thereof in the midveins and stems (Fig. 1) permitted efficient discrimination between the two phenotypic classes. PCR was used to test for the presence/absence of 35S:mgfp5-ER in all surviving plants, and complete correspondence between genotypic and phenotypic results was observed.

Surviving GFP-expressing and non-GFP-expressing individuals from interspecific hybridization between N. tabacum × N. africana
Analysis of non-GFP-expressing plants
A subset of 182 surviving individuals (75 GFP-expressing and 107 non-GFP-expressing plants) were randomly selected for further analysis using flow cytometry and microsatellite marker genotyping to generate information on the genetic composition of these individuals. Of the 107 non-GFP-expressing plants, 103 had a nuclear DNA content of approximately half of that of TN 90LC (9.94 pg nuclei−1) with a range of 4.298–5.632 pg nuclei−1 (Fig. 2). One plant (#4) had approximately the same estimated DNA content (9.271 pg nuclei−1) as TN 90LC and exhibited strong phenotypic resemblance to this cultivar. This individual may have been the result of inadvertent self-pollination, a rare apomictic event, or spontaneous chromosome doubling of a haploid. Three non-GFP-expressing plants (#42, #80, and #104) had nuclear DNA contents that were intermediate between that of haploid and diploid DNA contents for N. tabacum (Fig. 2). These three plants had abnormal phenotypes that were very atypical of those generally observed for N. tabacum × N. africana progeny.

Nuclear DNA contents for N. tabacum cv. TN 90LC, GFP-expressing N. africana, and 182 individuals derived from hybridization between the two
The 107 non-GFP-expressing plants were also genotyped using at least one microsatellite marker per N. tabacum linkage group and also genotyped for the presence of the TMV-resistance gene, N (a total of 40 markers) (Supplementary Table 1). Although we knew the genomic location of the N. tabacum alleles (Bindler et al. 2011), we had no prior information on the genomic positions of the N. africana alleles. Only the N. tabacum alleles, and no N. africana alleles, were amplified for 101 of the 107 non-GFP-expressing plants. This is in contrast to the observation of all N. tabacum and N. africana marker alleles being amplified from DNA extracted from cotyledonary tissue of dying plants. For the three plants with intermediate DNA contents, all N. tabacum alleles were amplified, but variable numbers of N. africana alleles were also amplified (24 for plant #42, 12 for plant #80, and 21 for plant #104) (Supplementary Table 1). The mitotic chromosome numbers for these plants were determined to be 36–37, 30, and 39, respectively (Fig. 3). A small number of N. africana markers (2–6) were also amplified for three plants (#6, #107, and #108) with haploid nuclear DNA contents. These three plants were of abnormal phenotype relative to other haploid plants and had chromosome numbers of 27, 27, and 26, respectively (Fig. 4). All non-GFP-expressing plants were positive for the presence of N located on chromosome H. All of these plants also exhibited the yellow burley phenotype except the three individuals with intermediate DNA contents (#42, #80, and #104) and one plant with haploid DNA content and that carried two N. africana markers (plant #6).

Chromosome counts for three non-GFP-expressing plants with intermediate DNA contents. Plants # 42, 80, and 104 have 36–37, 30, and 39 chromosomes, respectively

Chromosome counts for three non-GFP-expressing plants with approximate haploid DNA contents. Plants # 6, 107, and 108 have 27, 27, and 26 chromosomes, respectively
Analysis of GFP-expressing plants
Nuclear DNA contents for GFP-expressing plants were much more variable than those for the non-GFP-expressing plants and ranged from 7.819 to 11.653 pg nuclei−1 (Fig. 2), excluding the DNA content for an obvious paternal N. africana haploid (plant #149; DNA content = 5.486 pg nuclei−1). No GFP-expressing plant exhibited the yellow burley characteristic.
Microsatellite marker genotyping of GFP-expressing progeny indicated that the majority of these plants, except one (the N. africana paternal haploid), possessed most of the N. tabacum and N. africana marker alleles. No GFP-expressing plant carried all of the N. africana and N. tabacum alleles, however, and the number of N. africana and N. tabacum alleles that were present was variable (Supplementary Table 1). Linkage group 11 of Bindler et al. (2011) was found to correspond to N. tabacum chromosome H, as markers from this linkage group (and only this linkage group) were found to exhibit segregation in progeny of the Red Russian Haplo H × Hicks cross (Supplementary Figure 2). Based upon genotypes at 13 well-spaced microsatellite loci on linkage group 11, 28 GFP-expressing plants were estimated to possess an intact N. tabacum chromosome H, while 12 plants were missing chromosome H, and 35 plants carried a fragmented chromosome H at various break points along the chromosome (Table 1). In cases where chromosome H was fragmented, N. tabacum markers from positions 0.0–72.8 cM were generally present, while those more distant were absent (Table 1). For the 28 plants possessing an intact N. tabacum chromosome H, the N. africana marker amplified by PT30342 was deleted in all plants except one (plant #184).
Discussion
The primary objective of this research was to determine whether GFP expression in N. africana could serve as a dominant marker to permit efficient separation of haploids and interspecific hybrids that survive from the N. tabacum × N. africana cross. Although the level of GFP expression appeared to be less in gfp/--- hemizygous interspecific hybrids as compared to the stable homozygous transgenic N. africana line, results indicated the system to be useful for this purpose. Interspecific hybrids expressed GFP, while maternal haploids did not. Although a small number of non-GFP-expressing plants were found to carry variable numbers of N. africana chromosomes, these would not have been selected for use in a practical plant breeding program because of their abnormal plant types. A transgenic purple seedling marker has also been used to identify spontaneous maternal haploids from N. tabacum × N. tabacum crosses (Lewis and Rose 2011). There is a benefit to using the N. africana system, however, as the rate of haploidy in N. tabacum × N. africana crosses has been found to be approximately seven times greater than that for N. tabacum × N. tabacum crosses (Lewis and Rose 2011). Although the frequency of haploidy is low in either system, the high fecundity of N. tabacum (~3000 seeds cross−1) permits the generation of about three haploids per hand pollination, which are exceptionally easy to make for this species.
We also generated new information on the genetic composition of surviving plants from the N. tabacum × N. africana interspecific cross. Most previous studies on such progeny have been based solely upon cytological observations (Burk et al. 1979; Gerstel et al. 1979; Kramer and Reed 1988), although some DNA marker research has also been recently presented (Tezuka et al. 2012). According to Burk et al. (1979), the observed haploid frequency of approximately 1 or 2 haploids per 1000 seed resembled the rate of spontaneous parthenogenesis, leading to the suggestion that haploid production was the result of this asexual form of reproduction that occurs when an embryo develops in the absence of fertilization of the egg (Burk et al. 1979; Wernsman 1992). Indeed, it is possible that the primary mechanism of haploid production in the N. tabacum × N. africana system is parthenogenesis.
Interspecific hybridization between some species can also give rise to haploids through chromosome elimination, however. Chromosome elimination is a phenomenon where the chromosomes of one species are selectively eliminated during the initial mitotic divisions of a newly formed embryo. Complete uniparental chromosome elimination has been documented in hybrids from interspecific crosses such as Hordeum vulgare × Hordeum bulbosum (Subrahmanyam and Kasha 1973), Avena sativa × Zea mays (Riera Lizarazu et al. 1996), Triticum aestivum × Zea mays (Laurie and Bennett 1989), and Triticum aestivum × Pennisetum glaucum (Gernand et al. 2005). Using flow cytometry and microsatellite marker genotyping, we identified N. tabacum × N. africana progeny with nuclear DNA contents and chromosome numbers that were intermediate between those of haploid N. tabacum and of the majority of interspecific F1 hybrids. Similarly, Kramer and Reed (1988) reported unusual chromosome numbers between 25 and 45 in progeny from this cross. These authors did not discriminate chromosomes from the two parental genomes, however, and did not speculate on how these individuals might have been derived. In the current research, we also identified three individuals (out of a total of 107 surviving non-GFP-expressing plants) with near-haploid nuclear DNA content, but that also carried N. africana DNA markers and exhibited n = 24 + 2 or n = 24 + 3 chromosomes. The actual number of plants possessing N. africana introgression may actually have been greater because, with the exception of linkage group 11, we only genotyped haploid plants with 1 or 2 microsatellite markers per N. tabacum linkage group. These findings strongly suggest at least a partial role for chromosome elimination in the development of maternal haploids from the N. tabacum × N. africana cross. Further research involving observations of early embryonic mitotic divisions would be needed to determine the relative importance of parthenogenesis versus chromosome elimination in the production of haploids from this cross. It is interesting to note that the rate of maternal haploidy in the N. tabacum × N. africana system has been found to be approximately seven times greater than that in N. tabacum × N. tabacum crosses (Lewis and Rose 2011). An appropriate investigation of this question may be difficult, however, as chromosome elimination likely involves less than 1 % of all seedlings from the cross, as evidenced by the fact greater than 99 % of seedlings die at the cotyledonary stage with no DNA marker-based evidence of chromosome loss in these individuals. If this is truly the case, it would be interesting to gain insight on the set of genetic or physiological circumstances that become necessary to stimulate chromosome elimination at such a low frequency.
The precise molecular basis for uniparental chromosome elimination is yet to be understood in any specific cross, but several mechanisms have been proposed to be involved. These include asynchronous mitotic processes (Gupta 1969; Gernand et al. 2005), uniparental inactivation of centromeric genes or proteins (Finch 1983; Schwarzacher et al. 1987; Kim et al. 2002; Mochida et al. 2004; Sanei et al. 2011), formation of multipolar spindles (Subrahmanyam and Kasha 1973), failure of parental chromosomes to form normal spindle attachments and to migrate to spindle poles during cell division (Bennett et al. 1976; Laurie and Bennett 1989), spatial separation of genomes during interphase (Finch 1983; Linde-Laursen and von Bothmer 1999), uniparental nondisjunction of anaphase chromosomes (Ishii et al. 2010), formation of micronuclei during mitosis in early embryonic development (Kasha and Kao 1970; Gernand et al. 2005, 2006), and degradation of alien chromosomes by host-specific nucleases (Davies 1974). These possibilities are not meant to be mutually exclusive, and chromosome elimination following interspecific hybridization in plants may involve several complex mechanisms that are not fully understood to date. Partial elimination of chromosomes from one parent in interspecific crosses (Laurie and Bennett 1989; Riera Lizarazu et al. 1996; Kynast et al. 2001; Gernand et al. 2005; Li et al. 2009) and introgression of chromatin from the paternal parental species into haploid plants have also been previously reported (Wilkinson et al. 1995; Chen et al. 1998; Li et al. 2009). Evidence of chromosome elimination (Zhang et al. 2008) and incorporation of DNA from the pollen parent into maternal haploids derived from intraspecific Z. mays × Z. mays crosses has also been described (Li et al. 2009; Zhao et al. 2013).
The initiation and timing of chromosome elimination may affect the potential for introgression of paternal DNA into haploid plants through intergenomic translocation or genome rearrangement following hybridization, with later initiation increasing the possibility of such occurrences (Gernand et al. 2005; Li et al. 2009). Asynchronous DNA replication for chromosomes of parental species or other cell cycle perturbations might play roles in the transfer of chromosome segments from one species to a chromosome of the other species during chromosome elimination, as inhibition of DNA replication can induce DNA double-strand breaks, genome rearrangements, and deletions (Michel 2000; Ravi and Chan 2010). The direct transfer of N. africana chromosomes or chromosome segments to the N. tabacum genome could have practical utility if commercially useful characteristics could be transferred from this species. For example, N. africana has been well documented to exhibit high levels of resistance to Potato Virus Y (Lucas et al. 1980).
We also investigated the genetics of an interspecific hybrid lethality mechanism in this research. Hybrid lethality is a post-zygotic form of reproductive isolation in plants and is a phenomenon that occurs in a number of interspecific crosses in Nicotiana, particularly between N. tabacum and species of section Suaveolentes (of which N. africana is a member) (Tezuka 2012). This reaction was first studied by Burk et al. (1979) and Gerstel et al. (1979) who crossed all possible 24 N. tabacum monosomics with N. africana. All crosses resulted in the typical high rate of seedling lethality, except that involving N. tabacum monosomic for chromosome H. The rate of seedling survival for this cross (68.6 %) was approximately the same as that for the observed ovular transmission rate (70.4 %) for the H monosome (Clausen and Cameron 1944), leading to the suggestion that one or more genes on N. tabacum chromosome H are involved in the expression of lethality in progeny from this cross. A recent publication (Tezuka et al. 2012) contradicts these results and suggests chromosome Q and associated DNA marker linkage group 11 (Bindler et al. 2011) to carry a gene or genes contributing to this reaction. Our data agree with the finding that a genetic factor on linkage group 11 of Bindler et al. (2011) is involved in the lethality reaction, but clearly associates this linkage group with N. tabacum chromosome H, thus agreeing with the original results of Burk et al. (1979) and Gerstel et al. (1979). It is well documented that the TMV-resistance gene, N, resides on N. tabacum chromosome H in modern tobacco cultivars (Gerstel 1945; Gerstel and Burk 1960; Lewis et al. 2005). Further confirmation of the role of chromosome H in the interspecific lethality reaction is indicated by data showing N (contributed by modern tobacco cultivar TN 90LC used in our study) to co-segregate with markers on linkage group 11 in surviving interspecific hybrids with fragmented chromosome H (Table 1). This is consistent with knowledge of this disease-resistance gene’s relative position to linkage group 11 markers on the beginning of this linkage group (Lewis 2003). In addition, it has been well reported that the breeding line Holmes Samsoun carries a N. glutinosa chromosome substituted for N. tabacum chromosome H (Gerstel 1945; Gerstel and Burk 1960; Lewis et al. 2005). Crosses between the Holmes Samsoun substitution line and N. africana result in surviving F1 progeny (Lewis 2003), further implicating the role of chromosome H on the N. tabacum × N. africana hybrid lethality reaction.
The observation that chromosome H (linkage group 11) appeared to be broken in many cases of N. tabacum × N. africana F1 hybrid survival was somewhat surprising. Break points at various lengths along chromosome H allowed for the production of graphical genotypes for surviving plants and permitted tentative placement of a gene or gene(s) involved in the lethality reaction near the end of the chromosome, near marker PT52778. The reason for the high frequency of observed cases of chromosome fracture is not known, but one might speculate that some form of genomic instability exists after fertilization events between N. tabacum and N. africana, resulting in increased potential for DNA breakage and genome rearrangement. Finally, the lethality reaction from the N. tabacum × N. africana cross is believed to require genetic factors from both N. tabacum and N. africana, where a loss of either results in survival of the hybrid individual. Our data show that 28 F1 hybrid individuals possessed all tested N. tabacum microsatellite alleles, but still survived. The current data do not provide clear evidence for the genomic location of the hypothetical N. africana genetic factor involved in the lethality response, but the fact that N. africana marker allele PT30342 was absent in all but one of 28 individuals with an apparently intact N. tabacum chromosome H suggests that this marker may be closely associated with a N. africana genetic contributor to the lethality reaction. Interpretation of this N. africana marker data is made difficult because we do not know the relative genomic positions of the N. africana marker alleles, although a fair degree of marker synteny between some Nicotiana species has been reported (Wu et al. 2009).
References
Adachi T (2001) Introduction and concept of breeding barriers. In: Adachi T, Imanishi S, Hoffmann F (eds) How to overcome breeding barriers by means of plant biotechnology: a case study in tomato. Osaka Municipal Universities Press, Osaka, pp 1–4
An G, Watson BD, Cheng CC (1986) Transformation of tobacco, tomato, potato, and Arabidopsis thaliana using a binary Ti vector system. Plant Physiol 891:301–305
Andres RJ, Kuraparthy V (2013) Development of an improved method of mitotic metaphase chromosome preparation compatible for fluorescence in situ hybridization in cotton. J Cotton Sci 17:149–156
Bennett MD, Finch RA, Barclay IR (1976) The time rate and mechanism of chromosome elimination in Hordeum hybrids. Chromosoma 54:175–200
Bindler G, Pileske J, Bakaher N, Gunduz I, Ivanov N, van der Hoeven R, Ganal M, Donini P (2011) A high density genetic map of tobacco (Nicotiana tabacum L.) obtained from large scale microsatellite marker development. Theor Appl Genet 23:219–230
Bourgin J-P, Nitsch JP (1967) Production of haploid Nicotiana from excised stamens. Ann Physiol Veg 9:377–382
Burk LG (1962) Haploids in genetically marked progenies of tobacco. J Hered 53:222–225
Burk LG, Gerstel DU, Wernsman EA (1979) Maternal haploids of Nicotiana tabacum L. from seed. Science 206:585
Burns JA (1964) A technique for making preparations of mitotic chromosomes from Nicotiana flowers. Tob Sci 8:1–2
Chen CX, Zhu LH, Sun JS (1998) Molecular evidence on maize specific DNA fragment transferred into wheat through sexual hybridization. Sci China Ser C-Life Sci 41:126–132
Chiu WL, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter in plants. Curr Biol 6:325–330
Clausen RE, Cameron DR (1944) Inheritance in Nicotiana tabacum. XVIII. Monosomic analysis. Genetics 29:447–477
Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY (1995) Understanding, improving and using green fluorescent proteins. Trends Biochem Sci 20:448–455
Davies DR (1974) Chromosome elimination in inter-specific hybrids. J Hered 32:267–270
de Nettancourt D, Stokes GW (1960) Haploidy in tobacco. J Hered 51:102–104
Finch RA (1983) Tissue-specific elimination of alternative whole parental genomes in one barley hybrid. Chromosoma 88:386–393
Flowers RA, Stokes GW, Smiley JH (1967) Identification of tobacco haploids by stomatal size. Tob Sci 11:72–74
Gernand D, Rutten T, Varshney A, Rubtsova M, Prodanovic S, Brüß C, Kumlehn J, Matzk F, Houben A (2005) Uniparental chromosome elimination at mitosis and interphase in wheat and pearl millet crosses involves micronucleus formation, progressive heterochromatinization, and DNA fragmentation. Plant Cell 17:2431–2438
Gernand D, Rutten T, Pickering R, Houben A (2006) Elimination of chromosomes in Hordeum vulgare × H. bulbosum crosses at mitosis and interphase involves micronucleus formation and progressive heterochromatization. Cytogenet Genome Res 114:169–174
Gerstel DU (1945) Inheritance in Nicotiana tabacum. XIX. Identification of the tabacum chromosome replaced by one from N. glutinosa in mosaic-resistant Holmes Samsoun tobacco. Genetics 30:448–454
Gerstel DU, Burk LG (1960) Controlled introgression in Nicotiana: a cytological study. Tob Sci 4:147–150
Gerstel DU, Burns JA, Burk LG (1979) Interspecific hybridizations with an African tobacco, Nicotiana africana Merxm. J Hered 70:342–344
Gupta SB (1969) Duration of mitotic cycle and regulation of DNA replication in Nicotiana plumbaginifolia and a hybrid derivative of N. tabacum showing chromosome instability. Can J Genet Cytol 11:133–142
Haseloff J, Siemering KR, Prasher D, Hodge S (1997) Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc Natl Acad Sci USA 94:2122–2127
Heim R, Prasher DC, Tsien RY (1994) Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci USA 91:12501–12504
Ishii T, Ueda T, Tanaka H, Tsujimoto H (2010) Chromosome elimination by wide hybridization between Triticeae or oat plant and pearl millet: pearl millet chromosome dynamics in hybrid embryo cells. Chromosome Res 18:821–831
Kasha KJ, Kao KN (1970) High frequency haploid production in barley (Hordeum vulgare L.). Nature 225:874–876
Kim NS, Armstrong KC, Fedak G, Ho K, Park NI (2002) A microsatellite sequence from the rice blast fungus (Magnaporthe grisea) distinguishes between the centromeres of Hordeum vulgare and H. bulbosum in hybrid plants. Genome 45:165–174
Kramer MG, Reed SM (1988) An evaluation of maternal nullihaploidy for Nicotiana tabacum L. nullisomic production. I. An interspecific hybridization approach. J Hered 79:24–27
Kynast RG, Riera-Lizarazu O, Vales MI, Okagaki RJ, Maquieira SB, Chen G, Ananiev EV, Odland WE, Russell CD, Stec AO, Livingston SM, Zaia HA, Rines HW, Philips RL (2001) A complete set of maize individual chromosome additions to the oat genome. Plant Physiol 125:1216–1227
Laurie DA, Bennett MD (1989) The timing of chromosome elimination in hexaploid wheat × maize crosses. Genome 32:953–961
Lewis RS (2003) Unpublished information. North Carolina State University
Lewis RS, Rose C (2011) Identification of tobacco haploids on the basis of transgenic overexpression of PAP1 from Arabidopsis thaliana. Crop Sci 51:1491–1497
RS, Milla SR, Levin JS (2005) Molecular and genetic characterization of Nicotiana glutinosa L. chromosome segments in tobacco mosaic virus-resistant tobacco accessions. Crop Sci 45:2355–2362
Li L, Xu X, Jin W, Chen S (2009) Morphological and molecular evidences for DNA introgression in haploid induction via a high oil inducer CAUHOI in maize. Planta 230:367–376
Linde-Laursen I, von Bothmer R (1999) Orderly arrangement of the chromosomes within barley genomes of chromosome-eliminating Hordeum lechleri × barley hybrids. Genome 42:225–236
Lucas GB, Gooding GV Jr, Sasser JN, Gerstel DU (1980) Reaction of Nicotiana africana to black shank, Granville wilt, root knot, tobacco mosaic virus, and potato virus Y. Tob Sci 24:141–142
Michel B (2000) Replication fork arrest and DNA recombination. Trends Biochem Sci 25:173–178
Mino M, Maekawa K, Ogawa K, Yamagishi H, Inoue M (2002) Cell death processes during expression of hybrid lethality in interspecific F1 hybrid between Nicotiana gossei Domin and Nicotiana tabacum. Plant Physiol 130:1776–1787
Mochida K, Tsujimoto H, Sasakuma T (2004) Confocal analysis of chromosome behavior in wheat × maize zygotes. Genome 47:199–205
Nitsch JP, Nitsch C (1969) Haploid plants from pollen grains. Science 163:85–87
Pelletier G, Ferault M, Goujaud J, Vedel F, Caboche M (1987) The use of rootless mutants for the screening of spontaneous androgenetic and gynogenetic haploids in Nicotiana tabacum: evidence for the direct transfer of cytoplasm. Theor Appl Genet 75:13–15
Ravi M, Chan SWL (2010) Haploid plants produced by centromere-mediated genome elimination. Nature 464:615–618
Reed SM (1993) Use of stomatal size to distinguish between haploid and dihaploid tobacco plants. Tob Sci 37:84–86
Riera Lizarazu O, Rines HW, Phillips RL (1996) Cytological and molecular characterization of oat × maize partial hybrids. Theor Appl Genet 93:123–135
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sanei M, Pickering R, Kumke K, Nasuda S, Houben A (2011) Loss of centromeric histone H3 (CENH3) from centromeres precedes uniparental chromosome elimination in interspecific barley hybrids. Proc Natl Acad Sci USA 108:498–505
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Schwarzacher Robinson T, Finch RA, Smith JB, Bennett MD (1987) Genotypic control of centromere positions of parental genomes in Hordeum × Secale hybrid metaphases. J Cell Sci 87:291–304
Siemering KR, Golbik R, Sever R, Haseloff J (1996) Mutations that suppress the thermosensitivity of green fluorescent protein. Curr Biol 6:1653–1663
Stines BJ, Mann TG (1960) Diploidization in Nicotiana tabacum: a study of the yellow burley character. J Hered 51:222–227
Subrahmanyam NC, Kasha KJ (1973) Selective chromosomal elimination during haploid formation in barley following interspecific hybridization. Chromosoma 42:111–125
Tezuka T (2012) Hybrid lethality in the genus Nicotiana. In: Mworia JK (ed) Botany. InTech, Rijeka, pp 191–210
Tezuka T, Matsuo C, Lizuka T, Oda M, Marubashi W (2012) Identification of Nicotiana tabacum linkage group corresponding to the Q chromosome gene(s) involved in hybrid lethality. PLoS One 7:e37822
Tsien RY (1998) The green fluorescent protein. Annu Rev Biochem 67:509–544
Wernsman EA (1992) Varied roles for the haploid sporophyte in plant improvement. In: Stalker HT, Murphy JP (eds) Plant breeding in the 1990s. CAB Int, Wallingforde, pp 461–481
Wernsman EA, Matzinger DF, Rufty RC (1989) Androgenetic vs. gynogenetic doubled haploids of tobacco. Crop Sci 29:1151–1155
Wilkinson MJ, Bennett ST, Clulow SA, Allainguillaume J, Harding K, Bennett MD (1995) Evidence for somatic translocation during potato dihaploid induction. Heredity 74:146–151
Wu F, Eanetta NT, Xu Y, Plieske J, Ganal M, Pozzi C, Bakaher N, Tanksley SD (2009) COSII genetics maps of two diploid Nicotiana species provide a detailed picture of synteny with tomato and insights into chromosome evolution in tetraploid N. tabacum. Theor Appl Genet 120:809–827
Zhang ZL, Qiu FZ, Liu YZ, Ma KJ, Li ZY, Zu SZ (2008) Chromosome elimination and in vivo haploid production induced by Stock 6-derived inducer line of maize (Zea mays L.). Plant Cell Rep 27:1851–1860
Zhao Z, Xu X, Xie H, Chen S, Jin W (2013) Fertilization and uniparental chromosome elimination during crosses with maize haploid inducers. Plant Physiol 163:721–731
Acknowledgments
We thank the laboratory of Dr. George Allen at N.C. State University for providing us with the 35S:mgfp5-ER construct. We also gratefully acknowledge the financial support of our research program by Philip Morris International and Altria Client Services.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Author contributions
WGH performed the research, helped to analyze the data, and helped draft the manuscript; VK conducted cytological analyses; SPK performed plant transformations; RSL designed the research, helped analyze the data, and wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hancock, W.G., Kuraparthy, V., Kernodle, S.P. et al. Identification of maternal haploids of Nicotiana tabacum aided by transgenic expression of green fluorescent protein: evidence for chromosome elimination in the N. tabacum × N. africana interspecific cross. Mol Breeding 35, 179 (2015). https://doi.org/10.1007/s11032-015-0372-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0372-8