
Abstract
High-density genetic linkage maps of crop species are particularly useful in detecting qualitative and quantitative trait loci (QTLs) for agronomically important traits and in improving the power of classical approaches to identify candidate genes. The aim of this study was to develop a high-density genetic linkage map in a durum wheat recombinant inbred lines population derived from two elite wheat cultivars and to identify and characterize QTLs for yellow pigment content (YPC). A dense map was constructed by genotyping with the wheat 90K iSelect array and consisted of 5,670 loci, comprising 5,019 single nucleotide polymorphism (SNP), 467 DArT, 182 SSR markers and eight genes distributed in 35 linkage groups. Data for yellow pigment content were obtained from four replicated trials conducted at two locations in southern Italy for 2 years. A total of seven QTLs on different chromosome regions (1B, 2A, 2B, 5A, 5B, 7A and 7B) were identified, three of which were consistent in three or four environments and across environments. The genome scan for QTLs and the SNP homology prediction against annotated proteins in wheat and Brachypodium genomes identified two candidate genes of the carotenoid biosynthesis pathway (aldehyde oxidase, AO1, and diphosphomevalonate decarboxylase, DMAPD) significantly associated with YPC. This study provides a basis for further genetic QTL dissection and tools for marker-assisted breeding programs, because SNP markers and some carotenoid candidate genes were found to be tightly linked to major QTLs for YPC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Durum wheat [Triticum turgidum L. ssp. durum (Desf.) Husnot] is a cereal species widely cultivated in various regions of the world, including the Mediterranean regions, Canada, USA and Mexico. With about 17 million hectares under cultivation, the global annual grain yield of durum wheat is estimated to be around 35.5 million tonnes (Source: USDA July 2013 Xldata/world/all wheat/wheat/prod) with wide variation caused by drought and heat stresses. Durum amber grain is mainly used to make semolina for alimentary pasta in Europe and North America, and to produce couscous and flatbreads in North Africa and the Middle East.
Due to the importance of common and durum wheat as a major crop, the wheat genome has been extensively studied, although the complex nature including allopolyploidy, the high chromosome number, the large genome size relative to model plants (Arabidopsis, rice, Brachypodium) and the high percentage of repetitive regions have been major barriers to genome sequencing. In 2005, a group of plant scientists, breeders and growers established the International Wheat Genome Sequencing Consortium (IWGSC, http://www.wheatgenome.org) to overcome the sequencing challenges by a strategy based on the physical mapping and sequencing of individual flow-sorted chromosomes of Chinese Spring, an international wheat reference variety. Recently, parallel massive sequencing strategies, through next-generation sequencing (NGS) technologies, have generated the first draft sequence of the bread wheat genome (Mayer et al. 2014) and of the wheat A genome progenitor Triticum urartu (Ling et al. 2013) and D genome progenitor Aegilops tauschii (Jia et al. 2013). The impact of these new techniques and resources on wheat genetics is revolutionary. Indeed, the development of high-density genetic linkage maps is particularly important for bridging information between genome sequence efforts and breeding programs (Lewin et al. 2009). Genetic maps of crop species are useful in detecting qualitative and quantitative trait loci (QTLs) for agronomically important traits, identification and localization of candidate genes, map-based cloning of genes of interest, anchoring BAC contigs to genetic maps, linkage disequilibrium and diversity studies, and for marker-assisted selection and predictive molecular breeding (for a review see Gupta et al. 2008).
The first genetic maps in common (Gale et al. 1995) and durum wheat (Blanco et al. 1998) were developed mainly based on restriction fragment length polymorphisms (RFLP), and later by more polymorphic and cheaper markers, such as amplified fragment length polymorphisms (AFLP), simple sequence repeats (SSRs) and diversity arrays technology (DArT). An updated account on mapping of wheat individual chromosomes and the whole genome is available at GrainGenes (http://wheat.pw.usda.gov/). SSR-based consensus maps have been developed by Somers et al. (2004) in bread wheat and by Marone et al. (2012) and Maccaferri et al. (2014) in durum wheat; SNP-based consensus maps are currently replacing or updating such maps. Many biparental cross populations were recently used to achieve SNP-based consensus maps in bread wheat (Cavanagh et al. 2013; Wang et al. 2014) and durum wheat (Maccaferri et al. 2014). However, the large genome size and the low level of polymorphism prevented the development of high-resolution genetic maps. Recent progresses of high-throughput next-generation DNA sequencing (NGS) have overcome these difficulties, and high-density single nucleotide polymorphism (SNP)-based linkage maps with >5,000 markers have been constructed for some agronomically important crop species, such as barley (Close et al. 2009), maize (Yan et al. 2009), soybean (Hyten et al. 2010) and oilseed rape (Delourme et al. 2013).
SNP markers are co-dominant, abundant and evenly distributed across the genomes, and can be generated in a high-throughput and cost-effective manner. These properties make SNPs an ideal marker system (Gupta et al. 2008). With the more recent NGS technologies, SNP detection and genotyping were combined in a single experiment, such as sequencing of restriction site-associated genomic DNA (RAD) (Baird et al. 2008), genotyping-by-sequencing (GBS) (Elshire et al. 2011; Poland et al. 2012) and sequence-based genotyping (SBG) (Truong et al. 2012). Recently, SBG was applied to wheat, and over 90,000 SNP markers were incorporated in linkage maps (Trebbi et al. 2011; Poland et al. 2012; van Poecke et al. 2013; Cavanagh et al. 2013; Saintenac et al. 2011; Wang et al. 2014). The implementation of SNP markers in high-density linkage maps and the availability of sequences to annotated protein datasets of sequenced model species allow high-resolution QTL analysis, fine-mapping and identification of candidate genes associated with important agronomic traits in order to track directly the segments through marker-assisted selection (MAS) (Saintenac et al. 2013; van Poecke et al. 2013).
Yellow pigment content (YPC) is an important quality trait in durum wheat semolina for both commercial (bright yellow color pasta) and nutritional (major content of micronutrients) relevance. The yellow color is derived from the accumulation of carotenoid pigments in wheat pericarp and endosperm. Carotenoids represent a large class of lipid-soluble isoprenoids (C40) with extensive polyene chains that may contain up to 15 conjugated double bounds (Moise et al. 2013).
As many important agronomic traits, YPC in wheat is a complex character controlled by several genes with additive effects. The amber color of the grain is caused by the accumulation of lutein as a major component, and zeaxanthin, α-carotene, β-carotene and β-cryptoxanthin as minor components. Quantitative trait loci (QTLs) for YPC have been located on chromosomes 3A and 7A by Parker et al. (1998), 3B, 4B, 5B and 7A by Mares and Campbell (2001), 1B, 4A, 4B, 6A, 7A and 7B by Zhang et al. (2008), 1B and 7A by Zhang et al. (2009) and 2A, 3B, 5A and 7A by Blanco et al. (2011). The homoeologous group 7 chromosomes account for up to 50 % of the phenotypic variation (Parker et al. 1998; Eluoafi et al. 2001; Kuchel et al. 2006), whereas the QTLs on chromosomes 5B and 3B were responsible for 12–20 % phenotypic variation (Mares and Campbell 2001; Blanco et al. 2011). Identification of associated markers or functional carotenoid genes mapped in QTL regions would facilitate the selection of improved durum varieties. Indeed, the lack of molecular information about the carotenoid structural genes and their wheat map position has limited the study of carotenoid pigments and the regulation mechanism of accumulation in the wheat grain. As far as we know, only the genes for phytoene synthase (PSY) mapped on homoeologous group 5 and group 7 chromosomes (Pozniak et al. 2007; He et al. 2008; Dibari et al. 2012), lycopene ε-cyclase (ε-LCY) mapped on group 3 chromosomes (Howitt et al. 2009; Crawford and Francki 2013) and carotenoid β-hydroxylase (HYD) mapped on group 2 and group 4 chromosomes (Qin et al. 2012) have been cloned and characterized in wheat.
In this study, we reported a high-density SNP-based genetic linkage map developed in a RIL population derived from two elite durum wheat cultivars characterized by a different carotenoid pigment content. The objectives within this context were to describe general results on (1) physical mapping of SNP markers on individual wheat chromosomes of the A, B and D genomes; (2) development of a high-resolution SSR-, DArT- and SNP-based linkage map in durum wheat; (3) identification and characterization of QTLs for high carotenoid pigment content; (4) identification of carotenoid candidate genes and SNP markers tightly linked to major QTLs for the synthesis and degradation of yellow pigment content to be used in marker-assisted breeding programs.
Materials and methods
Plant materials
A recombinant inbred line (RIL) mapping population, previously developed from the cross Svevo × Ciccio, was genotyped using the recently developed wheat SNP iSelect array comprising of approximately 90,000 gene-associated SNPs (Wang et al. 2014) to saturate the genetic map described in Gadaleta et al. (2009). The elite durum wheat cultivars Svevo and Ciccio were different for qualitative and quantitative traits, such as grain yield components, grain protein content, grain yellow pigments and adaptive traits.
Nulli-tetrasomic lines (NTs) (Sears 1954, 1966) of Chinese Spring were used to assign SNP markers to each chromosome.
Yellow pigment content
Parents and RILs were evaluated for YPC in replicated trials in southern Italy at two locations (Valenzano and Gaudiano) in 2006 and 2007. A randomized complete block design with three replications and plots consisting of 1-m rows, 30 cm apart, with 80 germinating seeds per plot, was used in the field experiments. Yellow pigment content (YPC) was evaluated according to the AACC Approved Method 14–50 (AACC, 2000) with slight modifications as described by Fares et al. (1991): 1 g of each sample was extracted with 5 mL of water-saturated n-butyl alcohol on an orbital incubator for 3 h at 260 oscillations per minute. To minimize pigment degradation by oxidative enzymes, grain samples were ground in a laboratory mill with a 1 mm sieve and the resulting whole flour stored at 4 °C for a maximum of 24 h before analysis. One gram of each sample was extracted with 5 mL of water-saturated n-butyl alcohol on an orbital incubator for 3 h at 260 oscillations per minute. Samples were centrifuged for 7 min at 4,600 rpm, and absorbance of n-butyl alcohol extracts was measured by a UV-spectrophotometer at 435.8 nm. YPC was correlated to grain protein content and grain yield components (grain yield per spike, number of kernels per spike, 1,000 kernel weight) evaluated on the same mapping population and reported by Blanco et al. (2012).
SNP genotyping
A total of 50 ng/μL of genomic DNA of each RIL, parental lines (Svevo and Ciccio) and Chinese Spring nulli-tetrasomic lines was analyzed with the wheat 90K iSelect array (Wang et al. 2014). Genotyping was performed at TraitGenetics GmbH (http://www.traitgenetics.de) following the manufacturer’s recommendations as described in Akhunov et al. (2009). The genotyping assays were carried out to the Illumina iScan reader and performed using GenomeStudio software version 2011.1 (Illumina).
Candidate genes for carotenoid content
Wheat carotenoid candidate genes (CGs) underlying QTLs for YPC were identified using an in silico strategy. The sequences of SNP markers linked to QTL regions for YPC were BLASTX analyzed (e-value threshold ≤E−10 and identity percentage higher than 80 %) using homology prediction in NCBI nonredundant protein database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) of wheat and Brachypodium genomes. Information regarding the CGs expression was obtained using the expression profiling data publically available at Plant Expression Database (PLEXdb; http://www.plexdb.org).
Segregation analysis and map construction
The segregation ratio for each SNP marker was tested by Chi-square analysis for deviation from the expected 1:1 ratio, and those showing segregation distortion at P ≤ 0.001 and with more than 10 % missing data (reported as failed in the original SNP data file) were excluded from further analyses. Ten RILs with more than 10 % missing data were also excluded. SNPs with a null allele at one parent and segregating presence/absence in the mapping population were eliminated for the inability to discriminate null alleles from failed data. Linkage analysis between markers and determination of the linear order of loci was performed by JoinMap 4.0 (Van Ooijen 2006) using the regression mapping algorithm. Grouping was performed using the independence LOD parameter with groups presenting a LOD > 6. The Haldane mapping function was used to calculate map distances (Haldane 1919). Previously, physically mapped gSSR and EST-SSR markers (Gadaleta et al. 2009) and SNP data from the nulli-tetrasomic analysis were used as anchor loci and for assigning linkage groups to specific chromosomes or chromosome bins. Linkage groups were named according to the wheat chromosome nomenclature followed by a number.
Statistical analysis and QTL detection
The software MSTAT-C was used as the standard procedure for the analysis of variance for YPC. Genetic variance and broad-sense heritability were obtained using the variance component estimates. Pearson phenotypic correlation coefficients were calculated between YPC and grain protein content and grain yield components. QTL mapping was carried out by the composite interval mapping (CIM) method (Zeng 1994) using the software QGene 4.0 (Joehanes and Nelson 2008). A scanning interval of 1 cM between markers and putative QTL with a window size of 10 cM was used to detect QTL. The number of marker cofactors for background control was set by forward regression with a maximum of five controlling markers. A QTL was considered significant when one or more markers were associated with YPC at LOD > 3. Suggestive QTL at the sub-threshold 2.0 < LOD < 3.0 values were reported for further investigation. For additive QTL effects, positive and negative signs of the estimates indicate, respectively, the contribution of Svevo and Ciccio toward higher trait value. The proportion of phenotypic variance explained by a single QTL was determined by the square of the partial correlation coefficient (R 2). Graphical representation of linkage groups and QTL was carried out using MapChart 2.2 software (Voorrips 2002).
Results
Chromosome assignment of SNP markers
Out of 81,587 SNP arrays from the 90K array, 13,900 (17.0 %) totally failed and 4,872 (6.0 %) failed on three or more nulli-tetrasomic (NT) lines; 34,727 (42.6 %) were monomorphic; therefore, a total of 53,499 arrays were excluded from further analyses (Table S1). The remaining 28,088 arrays were grouped according to the number of failed NTs (one or two) and to the number of NTs showing a SNP in comparison with Chinese Spring. A total of 7,601 (9.3 %) arrays failed on only one NT and the SNPs were assigned to the missing chromosome in the NT; an additional 814 (1.0 %) arrays failed on two different NT lines and then were assigned to the two missing chromosomes. A total of 8,350 (10.2 %) arrays showed a NT with one SNP and then were assigned to the missing chromosome in that specific NTs, while 302 (0.4 %) showed two SNPs in two different NTs and were assigned to two different chromosomes. Further, 1,399 (1.7 %) and 204 (0.3 %) showed one NT line with a SNP and one or two failed NTs and were tentatively assigned to two or three chromosomes, respectively. A total of 7,270 (8.9 %) assays showed a NT line with an ambiguous nucleotide (M, K, R, Y, YUPAC codes), and the SNPs were assigned to the missing chromosome in that specific NT lines, while 789 (1.0 %) and 1,359 (1.7 %) showed a NT line with an ambiguous nucleotide and a failed NT or with a SNP and were assigned to two chromosomes.
The accuracy of SNP assignment to the A, B or D wheat chromosomes was checked on a sample of 1,859 SNP markers that were genetically mapped on the durum wheat RIL population Svevo × Ciccio (see paragraph below). Out of 551 physically mapped SNPs according to the failed NT line, 482 (87.5 %) showed the same position in the genetic and the physical maps, while the accuracy decreased to 69.1 % when the chromosome assignment was given according to the two failed NT lines. The accuracy rate was high when the markers were assigned to the A, B or D genome chromosomes according to the NT line showing a SNP compared to Chinese Spring; indeed, only 2.8, 3.7 and 0.0 % of contrasting localization between genetical and physical mapping was observed when the chromosome assignment was made according to the NT line with a SNP and 0, 1 or 2 failed NT lines, respectively. A total of 46 out of 47 checked SNP markers showed a correct chromosome assignment to one of the two NTs showing a SNP. The accuracy was 82.0 % when the chromosome assignment was made according to the NT line with an ambiguous nucleotide (M, K, R, Y).
The assignment of 28,387 SNP markers to the A, B and D genome chromosomes is reported in Table S2 with the indication F (failed), N (nucleotide), M, K, R, Y (ambiguous nucleotide) to take into account the accuracy rate according to the different criteria used for the chromosome assignment. The SNPs tentatively assigned to two chromosomes according to the two failed NTs were not reported because of their low accuracy rate. A summary of the distribution of 24,451 SNP loci on the 21 wheat chromosomes is given in Table S3, taking into account that the nullisomics for chromosomes 4D and 7D were not available and that the ditelosomic 4BS was used instead of the nullisomic 4B. SNPs assigned to two or three different chromosomes were not included. A higher number of SNPs were mapped on the B genome (43.0 %) than on the A (36.5 %) and D (20.5 %) genomes. Among the seven homoeologous groups, a maximum of 22.6 % SNPs were mapped on the group 2 chromosomes, and a minimum of 9.9 % on groups 4 and 7 chromosomes. The highest number of SNPs was mapped on chromosome 2B (2,342 loci = 9.6 %) and only 433 loci (1.8 %) on the chromosome 6D.
The durum linkage map
The Svevo × Ciccio RIL population was genotyped with the wheat 90K SNP iSelect assay (Wang et al. 2014). Out of 81,587 assays, 15,701 (19.2 %) failed completely and 69,216 (84.8 %) were monomorphic in the mapping population. A total of 6,796 (8.9 %) were polymorphic; however, 1,078 had more than 10 % missing data and 28 showed strong segregation distortion (P > 0.001) and were excluded from further analyses. A total of 421 (6.2 %) SNPs were dominant (pattern with one failed allele) and were excluded from mapping due to the inability to discriminate the null allele from the failed assay. The remaining 5,297 SNPs could be used for mapping and were combined with the 527 DArT, 70 EST-SSR, 112 SSR, two morphological markers (glume color, Bla1, and spike glaucousness, Ws), two seed storage protein loci (Gli-A2 and Gli-B2) included in the map described by Gadaleta et al. (2009) and two glutamine synthetase plastidic genes (GS2-A2 and GS2-B2) reported by Gadaleta et al. (2011). The complete linkage map is reported in supplementary materials (Table S4) and a summary in Table 1; Fig. 1.


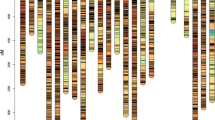
Schematic representation of the durum wheat linkage map and closest markers to QTLs for yellow pigments. Each linkage group has been represented by the first and the last marker, following a progressive and implied numeric order (see details in the supplementary Table 2S). The number of markers in each linkage group is reported in italic above the linkage group bar. All loci are indicated as follows: gSSR markers by “X,” EST-SSR markers by the international accession number, DArT markers through “wPt or tPt” code and SNP markers with “IWB” abbreviation. The genetic map is aligned with the deletion bin map, and the colored lines show the correspondent genetic/physical position of one SSR marker per bin. The physical mapping is referred on data of SSR markers (Gadaleta et al. 2009) and SNP markers (Gadaleta et al. 2014). The closest marker to QTLs for YPC is indicated in red; on the left side, the red arrows report the environments in which the QTLs were detected (V06, Valenzano 2006, G06, Gaudiano 06, V07, Valenzano 2006, G06, Gaudiano 07). Centromeres are indicated by a black circle. (Color figure online)
Out of 6,012 loci, 5,670 assembled in 35 linkage groups by using a LOD ≥ 6, while 352 remained unlinked or assembled in small linkage groups (LGs) without any chromosomal anchor locus. LGs were assigned to chromosome bins by the physically and genetically mapped gSSR and EST-SSR markers previously reported by Gadaleta et al. (2009) and by the physically mapped SNPs previously described in the present work. The anchor loci for chromosomes 5A and 5B were also represented by SNP markers physically mapped by Gadaleta et al. (2014). Two chromosomes were assembled in a single LG; most chromosomes were assembled in 2–3 LGs, while chromosomes 3A and 5B were each represented by four LGs.
A total of 3,360 (59.3 %) markers were localized on the B genome with a total length of 704.2 cM, while 2,308 (40.7 %) mapped on the A genome (total length 869.6 cM). Approximately 20 % more markers mapped on the B genome indicating that the B genome was more polymorphic than the A genome. The whole map covered 1,773.8 cM with an average chromosome length of 126.7 cM. The length of individual chromosomes varied from 68.0 cM (chromosome 3A) to 181.9 cM (5A), whereas the number of loci per chromosome ranged from 220 (4A) to 690 (2B). Chromosomes 3A and 6B showed the highest number of markers per cM (4.8–5.1), and chromosome 5A had the lowest marker density (1.7). Linkage analysis showed a very high number of DArT and SNP markers that co-segregated and therefore mapped on the same locus. A percentage of 83.1 % of the 5,019 mapped SNPs and 45.7 % of the 467 mapped DArTs showed redundancy (identical and co-migrating loci mapped within 0.1 cM). Chromosomes 1A, 2B, 5B and 7A exhibited the maximum redundancy for SNP markers (86.8–88.9 %) and chromosome 3B for DArT markers (78.1 %). The SNP markers were generally well distributed throughout the genome, although some chromosomes exhibited a higher density. The overall SNP density was 2.8 markers/cM, with a maximum of 4.7 for chromosome 3A and a minimum of 1.4 for chromosome 4A. Four major SNP clusters (>40 loci/0.1 cM) were identified on chromosomes 1A, 4B, 6A and 7A. The percentage of mapped SNPs that showed segregation distortion at P ≥ 0.05 was estimated at 8.2 %. Most of the skewed loci clustered in regions of the chromosomes 1A, 1B, 2A, 3A, 4A, 5B, 6A, 6B and 7B.
Detection of QTLs for yellow pigment content
Yellow pigment content (YPC) was evaluated in four replicated trials conducted in two locations in southern Italy (Valenzano and Gaudiano) in 2006 and 2007. The analysis of variance revealed highly significant differences (P < 0.01) among genotypes in each of the four environments; environments, genotypes and the environment × genotype interaction were significant in the combined analysis across environments (data not reported). Mean, range, genetic variance and heritability estimates of YPC in the RIL population Svevo × Ciccio and the parents in each environment are reported in Table 2. The parental lines had significantly different YPC values in each environment, and Svevo always had a higher value (7.67–9.44 µg/g) than Ciccio (5.85–7.36 µg/g). The means of the RIL population were near the mid-parental values (6.89–8.52 µg/g), but transgressive segregation in both directions was observed in each environment, indicating that positive alleles for YPC were present in both parents. The pattern of variability was typical of a quantitative trait, and the broad-sense heritability estimates (genotype mean basis) ranged from 0.83 to 0.94 indicating the stability of the trait and that the phenotypic expression was mainly due to genotypic effect.
Data on grain yield components (grain yield per spike, 1,000 kernel weight and kernel number per spike) and protein content, previously evaluated in the same RIL population (Blanco et al. 2012), were used to investigate their relationships with YPC. Correlation coefficients in individual environments are shown in Table S5. Grain yield per spike and 1,000 kernel weight both demonstrated a slightly negative correlation with yellow pigment concentration, but it was significant only between YPC and grain yield per spike at Gaudiano 2007 (P < 0.01), and between YPC and 1,000 kernel weight at Valenzano 2007 (P < 0.01) and Gaudiano 2007 (P < 0.001). No correlation was found between YPC and kernel number per spike and protein content.
Identification of QTLs associated with yellow pigment content
QTLs for yellow pigment content were determined based on the genetic map reported in Gadaleta et al. (2009) and integrated with DArT and SNP markers as described in the current work. The composite interval mapping (CIM) method as proposed by Zeng (1994) was employed, and QTLs with LOD values >3.0 were considered; suggestive QTLs at the sub-threshold 2.5 < LOD < 3.0 were reported only for further investigation. Putative QTLs for YPC in individual environments and across environments are listed in Table 3, and map positions of the closest marker are reported in Fig. 1. CIM identified seven QTLs on chromosomes 1B, 2A, 2B, 5A, 5B, 7A and 7B accounted for a large proportion of the total phenotypic variation for YPC scored in four environments (Table 3). The amount of phenotypic variation explained by individual QTLs (R 2) ranged from 12.6 to 60.4 %. The major QTL detected on chromosome arm 7AL (LG 7A_2, closest marker IWB59875) was significant in all four environments and explained 16.1–60.4 % of YPC variation. The QTL detected on chromosome arm 1BL (LG 1B_2, closest marker IWB73028) was consistent in three environments and explained 13.6–27.2 % of phenotypic variation. Three QTLs detected on chromosome arms 2AS (LG 2A_1), 2BL and 5BL (LG 5B_2) were consistent in two environments and explained 13.5–18.0 % of YPC variation. QTLs on 1BL, 5BL and 7AL were consistent also across environments with a coefficient of determination of 19.3, 21.8 and 51.6 %, respectively (Fig. 2). The remaining two QTLs on 5A_1 and 7B_2 were significant only in single environments with LOD scores of 3.1 and 7.8, respectively. Both parents contributed positive alleles (elevated YPC) with Svevo contributing the positive alleles at the LGs 1B_2, 2B, 5A_1 and 5B_2 7B_2 QTLs, and Ciccio at the QTLs on LGs 2A_1 and 7A_2. The allelic effect changed greatly in the different environments, ranging from nonsignificant values to 1.03 pigment content unit (QTL on 7A_2 at Valenzano 2007).

LOD score scan on linkage groups 5B_2 and 7A_2 for QTLs associated with yellow pigment concentration (YPC) across four environments. The position and the name of molecular markers are shown on the chromosomes along the horizontal axis. The LOD score scan was obtained by CIM using five cofactors. Cofactor markers on linkage groups 5B_2 and 7A_2 are shown highlighted
Candidate genes related to carotenoid QTLs
The wheat 90K array developed by Wang et al. (2014) comprises approximately 90,000 gene-associated SNPs that provides dense coverage of the wheat genome. In order to identify potential candidate genes (CGs) involved in the major QTLs for carotenoid pigment content detected in the Svevo × Ciccio segregating population, the sequences of the SNP markers located in the regions underlying the QTLs were analyzed for annotated proteins involved in the carotenoid pathway in the wheat and Brachypodium genomes databases. We detected an aldehyde oxidase (AO) and a diphosphomevalonate decarboxylase (DMAPD) located in the major QTLs on the chromosome arms 7AL and 5BL, respectively. In particular, the DNA sequence of the IWB59875 SNP marker closest to the 7AL QTL showed correspondence with the aldehyde oxidase AO1 sequence in the Brachypodium genome, whereas in the wheat genome was still uncharacterized. The candidate gene diphosphomevalonate decarboxylase (DMAPD), identified by the gene-associated SNP marker IWB76257, mapped within the 5BL QTL region at 0.84 cM from the closest DArT marker tPt-1253.
Discussion
Single nucleotide polymorphism (SNP) markers are particularly useful in the development and saturation of genetic maps and detection of QTLs for agronomic traits. In the current study, we used the wheat 90K chip developed by Wang et al. (2014) to generate a high-density linkage map in a RIL population derived from two durum elite cultivars, Svevo and Ciccio, widely grown in Italy.
The map includes eight genes, 5019 SNP, 467 DArT and 182 SSR markers used to anchor the linkage groups (LGs) to the wheat chromosome bins. Although a good coverage of the genomes was obtained, a number of gaps over 20 cM for LGs belonging to the same chromosome limited the connections of all LGs in 14 chromosomes, as expected in the durum wheat genome. Indeed, the Svevo × Ciccio map consists of 35 LGs, and in part, this is the result of mapping gene-associated SNPs in a large genome of 13 Gb characterized by gene-rich regions interspersed within a vast amount of repeated DNA (>80 %) (Erayman et al. 2004). The fragmentation of some genomic regions is also due to the stringent parameters (LOD score >6 and distance <20 cM) used in the markers grouping in order to take into account the chromosome bin position of the SSR markers previously mapped by deletion lines (Gadaleta et al. 2009). Moreover, in some instances, we preferred to have two distinct LGs rather than one merged LG because of the physical position of the LGs on not adjacent chromosome bins. So the LG 6B_2, positioned on the bin 6BS5-0.76-1.05 by the physically mapped SSR markers Gli-B2, Xgwm508, TC80528, Xbarc68, Xwmc398 and BJ213673b (Sourdille et al. 2004; Gadaleta et al. 2009; Kumar et al. 2013), and the LG 6B_3, positioned on the bin C-6BL3-0.36 by the anchor markers Xgwm88, Xgwm58 and TC101037 (Sourdille et al. 2004; Gadaleta et al. 2009), could be merged in a single LG at LOD = 18, thus including the bin C-6BS5-0.76 estimated to be long 228.25 Gb without any marker (Kumar et al. 2013). Although the link of the two LGs is likely due to lack or reduced recombination in the centromeric region, such genetic map could be of reduced or no benefit for some marker applications such as positional cloning or marker-assisted selection.
As expected, the Svevo × Ciccio linkage map presents a higher marker density than those previously developed by Gadaleta et al. (2009) and Menzo et al. (2013) related to the same RIL population. The total length of the map was 1,773.8 cM, with a mean of 3.2 markers/cM and ranges from 1.7 for chromosomes 5A to 5.1 markers/cM for chromosome 3A. The relatively low marker density of the Svevo × Ciccio map is likely due to the low level of polymorphism of the two commercial cultivars used to develop the RIL population.
The average number of mapped markers per chromosome was 404.9, with a range from 220 markers on chromosome 4A and 690 on chromosome 2B. However, 83.1 % of the SNP mapped showed redundancy, i.e., identical or co-migrating loci mapped within 0.1 cM. More markers were mapped on genome B (3,361) than in genome A (2,309) (Table 1). Lack of genome coverage was observed for chromosomes 1A, 1B, 3B, 4A, 4B, 6A, 6B and 7A in centromeric regions, as reported in several wheat maps (Sourdille et al. 2003; Somers et al. 2004; Torada et al. 2006; Maccaferri et al. 2014). Moreover, the short arm of chromosome 4A is not covered at all, which is consistent with other published wheat maps (Gadaleta et al. 2009; Marone et al. 2012; Cavanagh et al. 2013). This low coverage of some Svevo × Ciccio map regions could be due to the lack of polymorphism between the two parental lines, and/or to the small difference in recombination frequencies of molecular markers as shown in other maps (Somers et al. 2004; van Poecke et al. 2013), and/or to evolutionary events that induced limitations in sequence variations. As demonstrated by Ma et al. (2013), translocation and inversion events of chromosome 4A with 5A and 7B chromosomes had re-located the arm 4AS, justifying the lack of mapped markers and consequently of genetic maps related to 4AS.
QTL analysis and identification of candidate genes
Despite the economic and nutritional importance of carotenoids, the wheat genetic improvement for high YPC by conventional breeding has been only practiced in the last two decades. Although many QTLs controlling YPC were previously localized on wheat chromosomes in various segregant populations (Crawford and Francki 2013; Kuchel et al. 2006; Mares and Campbell 2001; Parker et al. 1998; Pozniak et al. 2007; Zhang et al. 2008), the complete QTL characterization and introgression into commercial varieties still represent an important task.
The present study was designed to detect QTLs for YPC in two elite durum wheat cultivars widely grown in Italy. We identified seven QTLs in the Svevo × Ciccio population mapped on chromosome arms 1BL, 2AS, 2BL, 5AS, 5BL, 7AL and 7BL. The detected QTLs explained 12.6–60.4 % of the YPC phenotypic variation. In comparison with previous studies (Blanco et al. 2011), the enhanced number of markers (5,670) gave a major dissection of chromosome segments involved in the carotenoid expression trait as confirmed by the size reduction of single QTL regions.
The presence of a major QTL for YPC on 7AL was repeatedly detected in common and durum wheat (Parker et al. 1998; Mares and Campbell 2001; Patil et al. 2008; Zhang et al. 2008; Zhang and Dubcovsky 2008; Howitt et al. 2009; Blanco et al. 2011), and shown to be associated with the phytoene synthase Psy-A1 locus localized on the bin 7AL18-0.90-1.00. A major QTL for YPC (16.1–60.4 % phenotypic variation) showing stability across all examined environments was detected in the Svevo × Ciccio population on chromosome arm 7AL (Table 3). However, composite interval mapping analysis showed the peak on the marker IWB59875 tightly linked to the physically mapped SSR markers BJ262177e and Xcfd6 on the bin 7AL21-0.74-0.86 (Fig. 2). Zhang and Dubcovsky (2008) and Blanco et al. (2011) detected two separated peaks on the adjacent bins 7AL21-0.74-0.86 and 7AL18-0.90-1.00, and supported the presence of two different QTLs for YPC on 7AL. The bin position of the major QTL detected in the current study on 7AL21-0.74-0.86 and the position of the common marker Xgwm1061a on 7AL18-0.90-1.00 tightly linked to Psy-A1 (Psy-A1 was not polymorphic between Svevo and Ciccio) indicated that the 7AL QTL was allelic to the second QTL detected in the Kofa × UC1113 and Latino × Primadur mapping population on the bin 7AL21-0.74-0.86 by Zhang and Dubcovsky (2008) and Blanco et al. (2011), respectively. The QTL on bin 7AL18-0.90-1.00 was functionally associated with phytoene synthase (Psy-A1) (Zhang and Dubcovsky 2008), a key enzyme involved in the first committed step of carotenoid pathway, while the QTL on bin 7AL21-0.74-0.86 was considered highly associated with YPC but not well characterized and documented.
The bioinformatics analysis of the mapped SNP markers in the QTL regions detected in the SC population indicated that the closest SNP marker IWB59875 associated with the major 7AL QTL for YPC (Table 3; Fig. 2) corresponded to the candidate gene aldehyde oxidase (AO1) that belongs to the AO gene family (AO; EC 1.2.3.1). The aldehyde oxidase enzymes (AO1, AO2 and AO3 in Arabidopsis) catalyze the conversion of abscisic aldehyde (an epoxy-carotenoid) in abscisic acid (ABA) and are expressed in leaves, roots and seeds (Seo et al. 1998, 2000a, b; Xiong et al. 2001). AO proteins were also detected in pea (Zdunek-Zastocka 2008), barley (Omarov et al. 2003), maize (Qin et al. 2013) and recently in common wheat (Gallé et al. 2013), but in all plants they might play different roles (Omarov et al. 2003). AO2 and AO3 were found to be more expressed in roots suggesting their function in ABA accumulation in direct response of stresses (Gallé et al. 2013). The direct relationship of the AO1 gene-associated SNP (IWB59875) with the detected QTL on 7AL suggested its possible implication in the grain carotenoid accumulation. Indeed, the comparative matching analysis of the AO1 gene-associated SNP IWB59875 to wheat 61 k microarray platform, carried out using the Plant Expression Dataset (PLEXdb) BLAST, showed a high expression level of the AO1 gene (TaAffx.97075.1.S1_at) in wheat seeds. However, since AO genes are involved in a number of biological processes, a more detailed experimental analysis should be carried out to support the role of AO1 in the carotenoid accumulation in wheat grain.
The 5B QTL (closest marker tPt-1253), mapped on bin 5BL16-0.79-1.00, was found to have a significant effect in two out of four environments and across environments and explained 16.9–21.8 % of the phenotypic variation. A QTL at the same chromosome position was previously detected by Mares and Campbell (2001) in common wheat. The bioinformatics analysis of the SNP markers mapped on the 5B QTL region identified the diphosphomevalonate decarboxylase (DMAPD) gene-associated SNP (IWB76257) at 0.84 cM from the closest DArT marker tPt-1253. DMAPD is responsible for the reversible isomerization of isopentenyl diphosphate (IPP) into its allelic isomer DMAPP, both precursors of carotenoids (Kumari et al. 2013). The important role of DMAPD gene in the carotenoid pathway was already reported in plants (Hirschberg 2001; Moise et al. 2013).
Minor variations were observed for QTLs on chromosomes 2A, 2B, 3A, 5A and 7B. They seem to be more sensitive to the environment since they are expressed in only one or two sites. The 3A QTL on the short chromosome arm was detected in previous studies (Mares and Campbell 2001), while the 1B QTL appears to be at the same position as a minor QTL detected by Zhang et al. (2008). Also, the 7B QTL located within the same chromosomal segment as reported by Eluoafi et al. (2001), Pozniak et al. (2007) and Zhang et al. (2008).
Conclusion
In the present study, we reported a high-density SNP-based genetic map that comprises 5,019 SNP, 467 DArT and 182 SSR markers with a substantial increment of genome coverage in comparison with previous published maps on the same Svevo × Ciccio population. This high-resolution map and data on the yellow pigment content of the RIL population evaluated in four environments provided a first deep knowledge and characterization of chromosome segments involved in the expression of YPC. The seven detected QTLs for YPC show an evident environmental influence with a predominant polygenic nature control, represented by heritability estimates higher than 0.9 in three sites. The major QTLs on chromosome arms 7AL and 5BL were found to be coincident or in close proximity with candidate genes for carotenoid accumulation in the grain. These results provide valuable information for marker-assisted selection (MAS) for yellow pigment content to expedite the selection of durum varieties with improved pasta quality. In addition, the genetic data obtained in this study will support further studies on QTL dissection for agronomically important traits differentiating the two parental lines.
References
Akhunov E, Nicolet C, Dvorak J (2009) Single nucleotide polymorphism genotyping in polyploid wheat with the Illumina GoldenGate assay. Theor Appl Genet 119:507–517
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376
Blanco A, Bellomo MP, Cenci A, De Giovanni C, D’Ovidio R, Iacono E, Laddomada B, Pagnotta MA, Porceddu E, Sciancalepore A, Simeone R, Tanzarella OA (1998) A genetic linkage map of durum wheat. Theor Appl Genet 97:721–728
Blanco A, Colasuonno P, Gadaleta A, Mangini G, Schiavulli A, Simeone R, Digesù AM, De Vita P, Mastrangelo AM, Cattivelli L (2011) Quantitative trait loci for yellow pigment concentration and individual carotenoid compounds in durum wheat. J Cereal Sci 54:255–264
Blanco A, Mangini G, Giancaspro A, Giove S, Colasuonno P, Simeone R, Signorile A, De Vita P, Mastrangelo A, Cattivelli L, Gadaleta A (2012) Relationships between grain protein content and grain yield components through QTL analyses in a RIL population derived from two elite durum wheat cultivars. Mol Breeding 30:69–92
Cavanagh CR, Chao S, Wang S, Huang BE, Stephen S, Kiani S, Forrest K, Saintenac C, Brown-Guedira GL, Akhunova A, See D, Bai G, Pumphrey M, Tomar L, Wong D, Kong S, Reynolds M, da Silva ML, Bockelman H, Talbert L, Anderson JA, Dreisigacker S, Baenziger S, Carter A, Korzun V, Morrell PL, Dubcovsky J, Morell MK, Sorrells ME, Hayden MJ, Akhunov E (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci USA 110:8057–8062
Close TJ, Bhat PR, Lonardi S, Wu Y, Rostoks N, Ramsay L, Druka A, Stein N, Svensson JT, Wanamaker S, Bozdag S, Roose ML, Moscou MJ, Chao S, Varshney RK, Szűcs P, Sato K, Hayes PM, Matthews DE, Kleinhofs A, Muehlbauer GJ, DeYoung J, Marshall DF, Madishetty K, Fenton RD, Condamine P, Graner A, Waugh R (2009) Development and implementation of high-throughput SNP genotyping in barley. BMC Genom 10:582
Crawford AC, Francki MG (2013) Lycopene-e-cyclase (e-LCY3A) is functionally associated with QTL for flour b* colour on chromosome 3A in wheat (Triticum aestivum L.). Mol Breed 31:737–741
Delourme R, Falentin C, Fomeju BF, Boillot M, Lassalle G, André I, Duarte J, Gauthier V, Lucante N, Marty A, Pauchon M, Pichon JP, Ribière N, Trotoux G, Blanchard P, Rivière N, Martinant JP, Pauquet J (2013) High-density SNP-based genetic map development and linkage disequilibrium assessment in Brassica napus L. BMC Genom 14:120
Dibari B, Murat F, Chosson A, Gautier V, Poncet C, Lecomte P, Mercier I, Bergès H, Pont C, Blanco A, Salse J (2012) Deciphering the genomic structure, function and evolution of carotenogenesis related phytoene synthases in grasses. BMC Genom 13:221
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Eluoafi I, Nachit MM, Martin LM (2001) Identification of a microsatellite on chromosome 7B showing a strong linkage with yellow pigment in durum wheat (Triticum turgidum L. var. durum). Hereditas 135:255–261
Erayman M, Sandhu D, Sidhu D, Dilbirligi M, Baenziger PS, Gill KS (2004) Demarcating the gene-rich regions of the wheat genome. Nucl Acids Res 32:123546–123565
Fares C, Platani C, Tamma G, Leccese F (1991) Microtest per la valutazione del colore in genotipi di frumento duro. Molini d’Italia 12:19–21
Gadaleta A, Giancaspro A, Giove SL, Zacheo S, Mangini G, Simeone R, Signorile A, Blanco A (2009) Genetic and physical mapping of new EST-derived SSRs on the A and B genome chromosomes of wheat. Theor Appl Genet 118:1015–1025
Gadaleta A, Nigro D, Giancaspro A, Blanco A (2011) The glutamine synthetase (GS2) genes in relation to grain protein content of durum wheat. Funct Integr Genomics 11:665–670
Gadaleta A, Giancaspro A, Nigro D, Giove SL, Zacheo SA, Incerti O, Simeone R, Piarulli L, Colasuonno P, Blanco A (2014) Development of a high-density genetic and physical map of wheat chromosome 5A for the detection of candidate genes in QTL analysis. Mol Breed (in press)
Gale MD, Atkinson MD, Chinoy CN, Harcoout RL, Jia J, Li QY, Devos KM (1995) Genetic maps of hexaploid wheat. In: Li ZS, Xin ZY (eds) Proceedings of 8th international wheat genet symposium, China Agricultural Scientech Press, Beijing, pp 29–40
Gallé A, Csiszár J, Benyó D, Laskay G, Leviczky T, Erdei L, Tari I (2013) Isohydric and anisohydric strategies of wheat genotypes under osmotic stress: biosynthesis and function of ABA in stress responses. J Plant Phys 170:1389–1399
Gupta PK, Mir RR, Mohan A, Kumar J (2008) Wheat genomics, present status and future prospects. Int J Plant Genomics 2008:1–36. doi:10.1155/2008/896451
Haldane JBS (1919) The combination of linkage values, and the calculation of distance between linked factors. J Genet 8:299–309
He XY, Zhang YL, He ZH, Wu YP, Xiao YG, Ma CX, Xia XC (2008) Characterization of phytoene synthase 1 gene (Psy1) located on common wheat chromosome 7A and development of a functional marker. Theor Appl Genet 116:213–221
Hirschberg J (2001) Carotenoid biosynthesis in flowering plants. Curr Opin Plant Biol 4:210–218
Howitt CA, Cavanagh CR, Bowerman AF, Cazzonelli C, Rampling L, Mimica JL, Pogson BJ (2009) Alternative splicing, activation of cryptic exons and amino acid substitutions in carotenoid biosynthetic genes are associated with lutein accumulation in wheat endosperm. Funct Integr Genomics 9:363–376
Hyten DL, Choi IY, Song Q, Specht JE, Carter TE Jr, Shoemaker RC, Hwang EY, Matukumalli LK, Cregan PB (2010) A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping. Crop Sci 50:960–968
Jia J, Zhao S, Kong X, Li Y, Zhao G, He W, Appels R, Pfeifer M, Tao Y, Zhang X et al (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95
Joehanes R, Nelson JC (2008) QGene 4.0, an extensible Java QTL-analysis platform. Bioinformatics 24:2788–2789
Kuchel H, Langridge P, Mosionek L, Williams K, Jefferies SP (2006) The genetic control of milling yield, dough rheology and baking quality of wheat. Theor Appl Genet 112:1487–1495
Kumar S, Goyal A, Mohan A, Balyan HS, Gupta PK (2013) An integrated physical map of simple sequence repeats in bread wheat. Aust J Crop Sci 7:460–468
Kumari S, Priya P, Misra G, Yadav G (2013) Structural and biochemical perspectives in plant isoprenoid biosynthesis. Phytochem Rev 12:255–291
Lewin HA, Larkin DM, Pontius J, O’Brien SJ (2009) Every genome sequence needs a good map. Genome Res 19:1925–1928
Ling HQ, Zhao S, Liu D, Wang J, Sun H, Zhang C, Fan H, Li D, Dong L, Tao Y, Gao C, Wu H, Li Y, Cui Y, Guo X, Zheng S, Wang B, Yu K, Liang Q, Yang W, Lou X, Chen J, Feng M, Jian J, Zhang X, Luo G, Jiang Y, Liu J, Wang Z, Sha Y, Zhang B, Wu H, Tang D, Shen Q, Xue P, Zou S, Wang X, Liu X, Wang F, Yang Y, An X, Dong Z, Zhang K, Zhang X, Luo MC, Dvorak J, Tong Y, Wang J, Yang H, Li Z, Wang D, Zhang A, Wang J (2013) Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496:87–90
Ma J, Stiller J, Berkman PJ, Wei Y, Rogers J, Feuillet C, Dolezel J, Mayer KF, Eversole K, Zheng YL, Liu C (2013) Sequence-based analysis of translocations and inversions in bread wheat (Triticum aestivum L.). PLoS ONE. doi:10.1371/journal.pone.0079329
Maccaferri M, Ricci A, Salvi S, Milner S, Noli E, Martelli P, Casadio R, Akhunov E, Ammar K, Blanco A, Desiderio F, Distelfeld A, Dubcovsky J, Fahima T, Faris J, Korol A, Massi A, Mastrangelo A, Morgante M, Pozniak C, Xu S, Tuberosa R (2014) A high-density, SNP-based consensus map of tetraploid wheat as a bridge to integrate durum and bread wheat genomics and breeding. Plant Biotechnol J (in press)
Mares D, Campbell A (2001) Mapping components of flour and noodle color in Australian wheat. Aust J Agric Res 52:1297–1309
Marone D, Panio G, Ficco DBM, Russo MA, De Vita P, Papa R, Rubiales D, Cattivelli L, Mastrangelo AM (2012) Characterization of wheat DArT markers: genetic and functional features. Mol Genet Genomics 287:741–753
Mayer KX, Rogers J, Dole J, Pozniak C, Eversole K, Feuillet C, Gill B, Friebe B, Lukaszewski J, Sourdille P et al (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788. doi:10.1126/science.1251788
Menzo V, Giancaspro A, Giove S, Nigro D, Zacheo S, Colasuonno P, Marcotuli I, Incerti O, Blanco A, Gadaleta A (2013) TRAP molecular markers as a system for saturation of the genetic map of durum wheat. Euphytica. doi:10.1007/s10681-013-0891-5
Moise AR, Al-Babili S, Wurtzel ET (2013) Mechanistic aspects of carotenoid biosynthesis. Chem Rev. doi:10.1021/cr400106y
Omarov R, Dräger D, Tischner R, Lips H (2003) Aldehyde oxidase isoforms and subunit composition in roots of barley as affected by ammonium and nitrate. Physiol Plant 117:337–342
Parker GD, Chalmers KJ, Rathjen AJ, Langridge P (1998) Mapping loci associated with flour colour in wheat (Triticum aestivum L.). Theor Appl Genet 97:238–245
Patil RM, Oak MD, Tamhankar SA, Sourdille P, Rao VS (2008) Mapping and validation of a major QTL for yellow pigment content on 7AL in durum wheat (Triticum turgidum L. ssp. durum). Mol Breed 21:485–496
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253
Pozniak CJ, Knox RE, Clarke FR, Clarke JM (2007) Identification of QTL and association of a phytoene synthase gene with endosperm colour in durum wheat. Theor Appl Genet 114:525–537
Qin X, Zhang W, Dubcovsky J (2012) Cloning and comparative analysis of carotenoid β-hydroxylase genes provides new insights into carotenoid metabolism in tetraploid (Triticum turgidum ssp durum) and hexaploid (Triticum aestivum) wheat grains. Plant Mol Biol. doi:10.1007/s11103-012-9972-4
Qin S, Zhang Z, Ning T, Ren S, Su L, Li Z (2013) Abscisic acid and aldehyde oxidase activity in maize ear leaf and grain relative to post-flowering photosynthetic capacity and grain-filling rate under different water/nitrogen treatments. Plant Physiol Biochem 70:69–80
Saintenac C, Jiang D, Akhunov ED (2011) Targeted analysis of nucleotide and copy number variation by exon capture in allotetraploid wheat genome. Genome Biol 12:R88
Saintenac C, Jiang D, Wang S, Akhunov ED (2013) Sequence-based mapping of the polyploid wheat genome. G3 (Bethesda) 3:1105–1114
Sears ER (1954) The aneuploids of common wheat. Univ Mol Res Bull No 572
Sears ER (1966) Nullisomic-tetrasomic combinations in hexaploid wheat. In: Riley R, Lewis KR (eds) Chromosome manipulations and plant genetics. Oliver and Boyd Ltd., Edinburgh
Seo M, Akaba S, Oritani T, Delarue M, Bellini C, Caboche M, Koshiba T (1998) Higher activity of an aldehyde oxidase in the auxin-overproducing superroot1 mutant of Arabidopsis thaliana. Plant Physiol 116:687–693
Seo M, Koiwai H, Akaba S, Komano T, Oritani T, Kamiya Y, Koshiba T (2000a) Abscisic aldehyde oxidase in leaves of Arabidopsis thaliana. Plant J 23:481–488
Seo M, Peeters AJM, Koiwai H, Oritani T, Marion-Poll A, Zeevaart JAD, Koornneef M, Kamiya Y, Koshiba T (2000b) The Arabidopsis aldehyde oxidase 3 (AAO3) gene product catalyzes the final step in abscisic acid biosynthesis in leaves. Proc Natl Acad Sci USA 97:12908–12913
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114
Sourdille P, Cadalen T, Guyomarc’h H, Snape JW, Perratant MR, Charmet G, Boeuf C, Bernard S, Bernard M (2003) An update of the Courtot × Chinese Spring intervarietal molecular marker linkage map for the QTL detection of agronomic traits in wheat. Theor Appl Genet 106:530–538
Sourdille P, Singh S, Cadalen T, Brown-Guedira GL, Gay G, Qi Bikram S, Gill L, Dufour P, Murigneux A, Bernard M (2004) Microsatellite-based deletion bin system for the establishment of genetic-physical map relationships in wheat (Triticum aestivum L.). Funct Integr Genomics 4:15–25
Torada A, Koike M, Mochida K, Ogihara Y (2006) SSR-based linkage map with new markers using an intraspecific population of common wheat. Theor Appl Genet 112:1042–1051
Trebbi D, Maccaferri M, de Heer P, Sørensen A, Giuliani S, Salvi S, Sanguineti MC, Massi A, van der Vossen EAG, Tuberosa R (2011) High-throughput SNP discovery and genotyping in durum wheat (Triticum durum Desf.). Theor Appl Genet 123:555–569
Truong HT, Ramos AM, Yalcin F, de Ruiter M, van der Poel HJA, Huvenaars KHJ, Hogers RCJ, van Enckevort LJG, Janssen A, van Orsouw NJ, van Eijk MJT (2012) Sequence-based genotyping for marker discovery and co-dominant scoring in germplasm and populations. PLoS ONE 7:e37565
Van Ooijen JW (2006) JoinMap 4.0, software for the calculation of genetic linkage maps in experimental populations. Kyazma B. V., Wageningen
Van Poecke RMP, Maccaferri M, Tang J, Truong HT, Janssen A, van Orsouw NJ, Salvi S, Sanguineti MC, Tuberosa R, van der Vossen EAG (2013) Sequence-based SNP genotyping in durum wheat. Plant Biotechnol J 11:809–817
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93(1):77–78
Wang S, Wong D, Forrest K, Allen A, Chao S, Huang BE, Maccaferri M, Salvi S, Milner SG, Cattivelli L, Mastrangelo AM, Stephen S, Barker G, Wieseke R, Plieske J, International Wheat Genome Sequencing Consortium, Lillemo M, Mather D, Appels R, Dulferos R, Brown-Guedira G, Korol A, Akhunova AR, Feuillet C, Salse J, Morgante M, Pozniak C, Luo MC, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E (2014) Characterization of polyploid wheat genomic diversity using the high-density 90,000 SNP array. Plant Biotech J. doi:10.1111/pbi.12183
Xiong L, Ishitani M, Lee H, Zhu JK (2001) The Arabidopsis LOS5/ABA3 locus encodes a molybdenum cofactor sulfurase and modulates cold and osmotic stress responsive gene expression. Plant Cell 13:2063–2083
Yan J, Yang X, Shah T, Sanchez-Villeda H, Li J, Warburton M, Zhou Y, Crouch JH, Xu Y (2009) High-throughput SNP genotyping with the GoldenGate assay in maize. Mol Breed 25:441–451
Zdunek-Zastocka E (2008) Molecular cloning, characterization and expression analysis of three aldehyde oxidase genes from Pisum sativum L. Plant Physiol Biochem 46:19–28
Zeng Z (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang W, Dubcovsky J (2008) Association between allelic variation at the phytoene synthase 1 gene and yellow pigment content in the wheat grain. Theor Appl Genet 116:635–645
Zhang W, Chao S, Manthey F, Chicaiza O, Brevis JC, Echenique V, Dubcovsky J (2008) QTL analysis of pasta quality using a composite microsatellite and SNP map of durum wheat. Theor Appl Genet 117:1361–1377
Zhang YL, Wu YP, Xiao YG, He ZH, Zhang Y, Yan J, Zhang Y, Xia XC, Ma CX (2009) QTL mapping for flour color components, yellow pigment content and polyphenol oxidase activity in common wheat (Triticum aestivum L.). Euphytica 165:435–444
Acknowledgments
The authors acknowledge the financial support of Italian MIUR Projects “PON-ISCOCEM” and “PRIN 2010-11.”
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Colasuonno, P., Gadaleta, A., Giancaspro, A. et al. Development of a high-density SNP-based linkage map and detection of yellow pigment content QTLs in durum wheat. Mol Breeding 34, 1563–1578 (2014). https://doi.org/10.1007/s11032-014-0183-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-014-0183-3