
Abstract
Cross-coupling reactions are powerful synthetic tools for the formation of remarkable building blocks of many naturally occurring molecules, polymers and biologically active compounds. These reactions have brought potent transformations in chemical and pharmaceutical disciplines. In this review, we have focused on the use of cross-coupling reactions such as Suzuki, Negishi, Heck, Sonogashira and Stille in the total synthesis of some natural products of recent years (2016–2020). A short introduction of mentioned cross-coupling reactions along with highlighted aspects of natural products has been stated in separate sections. Additionally, few examples of natural products via incorporation of more than one type of cross-coupling reaction have also been added to demonstrate the importance of these reactions in organic synthesis.
Graphic abstract

Similar content being viewed by others

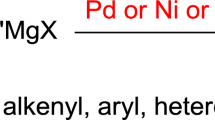
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Reactions assembling the carbon–carbon bonds in organic compounds constitute a pillar of strength to organic synthesis. Particularly, the designing of natural products framework demands a rapid and selective construction of carbon skeleton [1, 2]. Cross-coupling reactions have gathered much attention of researchers from the past few decades because of simple, general and mild synthetic approaches involving the use of highly active ligands and transition metal complexes. These protocols are involved in the accomplishment of simple to complex carbon skeletons such as functional materials, synthetic fragments, bioactive compounds, natural products and agrochemicals [3, 4]. One of the prominent cross-coupling reactions is palladium catalyzed C–C bond forming reactions that give interesting properties to the final chains of molecules [5,6,7,8]. In 2010, Nobel Prize award for palladium-catalyzed cross-coupling reactions [4] had fastened its roots in chemical synthesis and their enabling characteristics make them a center of interest in multiple domains.
Tamura and Kochi [9] reported the first iron-catalyzed cross-coupling reaction and then as the time passed, many new strategies including Suzuki [10], Heck [11], Sonogashira [12], Negishi [13], Stille [14], Cadiot–Chodkiewicz [15], Castro–Stephens [16], Hiyama [17], Fukuyama [18], Liebeskind–Srogl [19] and Kumada reactions [20], etc., were developed for framing the structures of organic compounds and the scope of cross-coupling reactions became a key topic for the investigators. Keeping in view the recent advances and extensive applications of cross-coupling reactions, several reviews on novel synthetic methodologies have been reported [21,22,23].
Owing to unique chemical features and biological potential, many of the natural products are used as core part of valuable drugs [24]. Herein, we have emphasized the role of cross-coupling reactions in the total synthesis of some natural products. Natural products are briefly introduced and categorized into alkaloids, terpenoids, steroids, polyketides, macrolides, lignans, alkamides and lactones classes.
Review of literature
Suzuki cross-coupling reaction
Palladium-catalyzed Suzuki cross-coupling reaction (Suzuki–Miyaura coupling) is a widely known method to couple organoboranes/boronic acids with organic halides/triflates under basic conditions to form C–C bonds [25]. Non-toxicity and stability, accessibility of organoboron compounds/boronic acids, their incorporation into organometallic compounds and simple workup techniques add additional features to the armory of Suzuki cross-coupling. This cross-coupling endures many functional groups and the stereochemistry of coupling reagents is retained in the resulting biaryls [26]. It is one of the well-built tool to furnish biaryl moiety which displays vast applications in pharmaceuticals and material science [27]. The first natural product synthesis via Suzuki coupling was disclosed by Rossi [28] in 1981 which involved the preparation of sex pheromone 1 in 21.6% overall yield (Fig. 1).

Structure of sex pheromone 1
Synthesis of alkaloids
Pentabromopseudilin 2 is a polyhalogenated bicyclic alkaloid [29] which was firstly isolated from Pseudomonas bromoutilis (1996) and later from Alteromonas luteoviolaceus, Chromobacteria and Pseudoalteromonas sp (Fig. 2). Biological activities such as antifungal, anti-tumour and antibiotic are associated with this polybrominated natural product. It acts as inhibitor against human lipoxygenases, myosin and exhibits the potential to act against MRSA with IC50 value of 0.1 µM [30].

Structure of pentabromopseudilin 2
Kum et al. [31] carried out the synthesis of an anti-MRSA and myosin-inhibiting natural product 2 with 38% overall yield through two steps by using Suzuki coupling protocol (Scheme 1). Synthesis was started from the coupling of phenol 3 with N-methyliminodiacetic acid (MIDA) boronate 4 and conditions were optimized to achieve the best results. The use of 10 mol% Buchwald 2nd generation precatalyst (Sphos Pd G2) along with 4 M KOH (base) in tetrahydrofuran (THF)/H2O led to the formation of compound 5 which was treated with pyridinium tribromide 6 to shape the core of pentabromopseudilin 2 in 60% yield. The synthesized compounds 2 and 5 were evaluated for biological potential against Staphylococcus epidermidis; however among them, compound 2 showed clear zones of inhibition at 50 and 5 µM.

Synthesis of pentabromopseudilin 2
Marine environment provides a number of valuable natural products with potential of anticancer, antibiotic and therapeutic activities. Among them, actinobacteria are of vital importance as these are the sources of market valued chemical substances. Lynamicin D 7 is a chlorinated bisindole pyrrole alkaloid which has been obtained from marine actinomycetes such as SCSIO 03,032 and NPS12745 (Fig. 3). This compound has the capability to act against drug-resistant pathogens (Staphylococcus aureus and Enterococcus faecium) [32, 33].

Structure of lynamicin D 7
The first total synthesis of lynamicin D 7 was reported by Sigala et al. [34] which involved Suzuki coupling of dicarboxylate 9 with boronate 11 (Scheme 2). In this regard, pyrrole 8 was subjected to POCl3 (63% yield) and CH3COONa and resulting product was oxidized (using KMnO4) followed by Fischer esterification (with CH3OH, H2SO4) and bromination (using Br2, 95%) provided indole-based dibromo dicarboxylate derivative 9. While boronate 11 was obtained from indole 10 by employing iodination followed by protection with Boc group (94% yield, 2 steps) and then treatment with pinacolborane in the presence of PdCl2(dppf) (84% yield). Suzuki coupling of synthesized fragments 9 and 11 in the presence of Pd(OAc)2, triphenylphosphine (PPh3), Na2CO3 in THF/H2O mixture at 60 °C produced bisindole pyrrole 12 in good yield (76%) which on treatment with trifluoroacetic acid (TFA) afforded the acquired product 7 in an excellent yield (97%). Biological activity of lynamicin D 7 was also studied which revealed that it had a minor effect on cell viability, but it could regulate splicing of pre-mRNAs as it alters the levels of kinase (SRPK1).

Synthesis of lynamicin D 7
Carbazole alkaloids bearing pyrrole ring constitute a major class of natural products and their source of extraction includes various plant species including bacteria and fungi. From the last few decades, extensive work has been carried out on carbazole compounds due to their versatile chemical characteristics and biological potentials such as anti-HIV, anti-malarial, anti-tumour, anti-TB, etc. [35] Some of the biologically important carbazole alkaloids 13–24 are listed in Fig. 4.

Structures of carbazole alkaloids 13–24
A convenient strategy for the total synthesis of carbazole alkaloids 13–24 was reported by Bhatthula et al. [36] with moderate to excellent yields (27–96%). The synthetic route involved Suzuki cross-coupling and Cadogan reductive cyclization as vital steps. Synthesis of mukonine 13 was started from benzoic acid 25 which underwent esterification (with CH3I, Cs2CO3, 99%) followed by addition of bis(pinacolato diboron) to give compound 26 in 66% yield. Coupling of compound 26 with o-nitrobenzene 27 using Pd(PPh3)4 and K2CO3 in refluxing toluene in 5 h gave the coupled product 28 in 88% yield. Later, compound 28 in the presence of PPh3 and o-dichlorobenzene (o-DCB) experienced Cadogan reductive cyclization to furnish the desired natural product 13 (66%) along with its regioisomer 13a (27%). Reduction of compound 13 with diisobutylaluminium hydride (DIBAL-H) and LiAlH4 afforded koenoline 15 and murrayafoline A 17, respectively, while oxidation of compound 15 with MnO2 gave murrayanine 16 in 81% yield. Saponification of compound 13 (using aq. KOH) produced mukoeic acid 14 in 96% yield. Carbazole 13a provided regioisomers 14a, 15a and 16a under saponification, reduction and oxidation conditions, respectively (Scheme 3). Synthesis of glycoborine 18 and clauszoline K 19 was achieved in 93% and 75% yield using compound 31/32 and 2-cholro-4-methyl-1-nitrobenzene 33 as Suzuki coupling partners (Scheme 4). Treatment of compound 41 (obtained from 4-bromotoluene 39) with o-nitrobenzene 27 in the presence of Pd(PPh3)4 gave biphenyl 43 in 86% yield which was cyclized (using PPh3, o-DCB) to obtain 2-methyl-9H-carbazole 24 (94%). Moreover, the use of 3-bromophenol 37 as starting material and 2-cholro-4-methyl-1-nitrobenzene 33 (for Suzuki coupling) framed the structures of glycozolicine 20 (38%), glycozoline 21 (38%), mukolidine 22 (85%) and mokuline 23 (91%) (Scheme 5).

Synthesis of carbazole alkaloids 13–17

Synthesis of carbazole alkaloids 18 and 19

Synthesis of carbazole alkaloids 20–24
Isocrytolepine 44 (obtained from Cryptolepis sanguinolenta) is an indoloquinoline alkaloid reported independently by Pousset et al. [37] and Sharaf et al. [38] in 1995 (Fig. 5). It consists of tetracyclic skeleton based on angularly fused indolo[3,2-c]quinoline ring with wide range of biological activities such as antiplasmodial, antimicotic, antihyperglycemic, antimuscarinic and antibacterial, etc. [39].

Structure of isocrytolepine 44
Håheim et al. [40] presented the synthesis of tetracyclic core 48 of isocrytolepine 44 (Scheme 6). Suzuki coupling was employed between 3-bromoquinoline 45 and boronic acid 46 in the presence of PdCl2(dppf) (5 mol%), K2CO3 in EtOH/H2O at 60 °C for 20 h to give compound 47 in 84% yield. The latter compound 47 was cyclized using PdCl2(dppf) (20 mol%) as catalyst and 1,3-bis(2,4,6-trimethylphenyl)-imidazolium (IMes) (5 mol%) as ligand to produce regiospecific compound 48 in good yield (73%).

Synthesis of tetracyclic motif 48 of isocrytolepine 44
α-Cyclopiazonic acid 49 is a member of prenylated alkaloids possessing the potential to inhibit calcium-ATPase enzyme in sarcoendoplasmic reticulum due to its toxic nature (Fig. 6). For the first time, it was obtained from fungus Penicillium cyclopium [41].

Structure of α-cyclopiazonic acid 49
A shortest synthetic pathway for the preparation of α-cyclopiazonic acid 49 involving seven steps was reported by Shi et al. [42] (Scheme 7). Synthesis was commenced from palladium-catalyzed coupling of 4-bromoindole 50 with t-prenylboronate 51 using Pd(PPh3)4, NaOH in toluene/H2O at 90 °C to form regioisomer 52 in 91% yield. Vilsmeier–Haack formylation (with POCl3, dimethylformamide (DMF), aq. KOH, 75%) and N-tosylation (with TsCl, 53%) produced aldehyde 53 which on reaction with compound 54 in the presence of LiHMDS yielded indole derivative 55 (78%). It was then subjected to [3 + 2] annulation by employing Tf2NH (50% yield) to achieve diastereomeric mixture of three tetracycles. Only small amount of one diastereomer was separated and was treated with excess of Mg (50% yield) to give mixture of diastereomeric amines (56a:b:c = 1:0.3:0.7) from which the amine 56a was successfully separated. It was converted to the target product 49 in 59% yield on treatment with diketene 57 in dichloromethane (DCM) and t-BuOK.

Synthesis of α-cyclopiazonic acid 49
Synthesis of polyketides
Anthracyclines account for a medically prime class of aromatic polyketides and demonstrate anti-cancer potential. Their structure contains aglycone chromophore attached with one or more deoxysugars. Nogalamycin 58 (extracted from Streptomyces nogalater) possesses antibacterial and anticancer potential, whereas its semisynthetic derivative, menogaril 59 exhibits anticancer properties. These compounds are naturally occurring polyketides (Fig. 7) [43].

Structures of nogalamycin 58 and menogaril 59
The synthesis of anthracyclines 58 and 59 was achieved by Peng and VanNieuwenhze [44] via implementation of Suzuki cross-coupling protocol for the preparation of the DFT-ring system 65 (Scheme 8). In this regard, enol triflate 60 and D-ring precursor 61 in the presence of PdCl2(dppf) and KOH in toluene at room temperature produced coupled product 62 in 69% yield. Then, compound 62 on subsequent primary alcoholic group protection (with TBSCl, 88%) followed by Staudinger reduction (using PPh3, H2O/THF) provided amine which was further protected with 2-naphthylsulfonyl group. In the following step, addition of formic acid selectively cleaved the primary TBS group to produce compound 63 (88% over 3 steps) which on treatment with PySO3 followed by the addition of HC(OCH3)3 accomplished dimethyl acetal 64 in an excellent yield (95% over 2 steps). Epoxidation of alkene 64 was carried out with m-chloroperoxybenzoic acid (m-CPBA, 57%) which was reduced (using LiAlH4, 59%) and cyclized in acidic conditions (3 M HCl, acetic acid) to afford DFT-ring system 65 in 77% yield.

Synthesis of DFT- ring 65 of nogalamycin 58 and menogaril 59
Synthesis of terpenoids
The presence of tricarbocyclic core is a peculiarity of naturally occurring diterpenoid hamigerans. Furthermore, hamigerans possess three or four stereogenic centers arranged in adjacent positions around cis bicyclic core [45]. (−)-Hamigeran B 66 with magnificent anti-viral property was first isolated from Hamigera tarangaensis (a sponge) in 2000 (Fig. 8) [46].

Structure of (−)-hamigeran B 66
Total synthesis of (−)-hamigeran B 66 was disclosed by Kuwata et al. [47] over 17 steps via Suzuki cross-coupling of (R)-triflate 68 (obtained from a cyclic ketone 67) with compound 69 using PdCl2(dppf)·DCM, K2CO3 in dimethyl sulfoxide (DMSO) at 80 °C (Scheme 9). The coupling product (R)-70 (68%) was obtained as a mixture of two rotamers which was divulged through 1H NMR interpretations. Further, the treatment of (R)-70 with SmI2 for reductive coupling of two aldehydes resulted in two diastereomers 71a (51%) and 71b (49%). The stereochemistry of the target product 66 was maintained during the synthetic route and was obtained over several steps from diastereomer 71a.

Synthesis of (−)-hamigeran B 66
Magnolia officinalis var. biloba is a commonly used Chinese medicine. Its active ingredient known as terpenoid quinone is also named as magterpenoid C 72. It is famous for the remedy of phlegm, dyspepsia and abdominal distension (Fig. 9) [48].

Structure of magterpenoid C 72
The first total synthesis of magterpenoid C 72 by using Suzuki coupling and silica-gel accelerated [4 + 2] cycloaddition protocols was explained by Kumar et al. [49] (Scheme 10). Hydroquinone and 4-allyl anisole gave 2-bromo-1,4-dimethoxy-benzene 73 and compound 74, respectively, which were subjected to Suzuki coupling in the presence of Pd(PPh3)4, Na2CO3 in refluxing dimethoxyethane (DME)/H2O for 6 h to give intermediate 75 in 60% yield. In the next step, oxidative demethylation of compound 75 was done using phenyliodine bis(trifluoroacetate) (PIFA) in acetonitrile/water mixture to access quinone 76 (62%). Quinone 76 was subjected to reduction (with NaBH4, 99%) followed by the addition of AlCl3 for demethylation (50% yield) and NaIO4 to furnish the desired quinone 77 in 95% yield. Final framework of terpenoid 72 (67% over 2 steps) was achieved in a regioselective manner by the combination of quinone 77 with β-myrcene 78 in the presence of SiO2-gel and MnO2 for [4 + 2] cycloaddition and subsequent aromatization.

Synthesis of magterpenoid C 72
Miscellaneous
Insects constitute a major portion of animals on earth. Although a number of peptides and proteins have been isolated from various insect species from past few years however, their biologically activity has been less investigated. Aspongopus chinensis Dallas, found in China, is used as traditional medicine as well as food. Investigation revealed that it exhibits pronounced biological activities such as anticancer, analgesic and angiogenesis, etc. Proliferation of neural stem cells is also associated with this insect species. Aspongpyrazine A 79 is a pyrazine obtained from insect A. chinensis (Fig. 10) [50].

Structure of aspongpyrazine A 79
Bendre et al. [51] reported an efficient, simple and environmental friendly biological route for the preparation of palladium nanoparticles (PdNPs) using palladium chloride and oxytoxin (a harmone). These PdNPs were then applied in the Suzuki cross-coupling for the synthesis of aspongpyrazine A 79 with 86.4% overall yield (Scheme 11). Readily available 2-bromo-6-methylpyrazine 80 was subjected to Suzuki coupling with boronic acid 81 over PdNPs and K2CO3 in DMF/H2O at 100 °C to obtain compound 82 in 90% yield. The latter compound 82 underwent deprotection with 47% HBr and aliquat© 336 (catalyst) to afford final skeleton of aspongpyrazine A 79 in 96% yield.

Synthesis of aspongpyrazine A 79
Kehokorin A 83 and kehokorin B 84 contain dibenzofuran core in their structures and have been extracted from field-collected sample of fruit bodies of Trichiaceae (Trichia favoginea var. persimilis) (Fig. 11). Biological evaluation of these compounds showed that kehokorin A 83 possesses cytotoxicity potential against HeLa cell line which was attributed to the presence of rhamnose unit [52].

Structures of kehokorin A 83 and kehokorin B 84
Fujiwara et al. [53] implemented Suzuki cross-coupling approach twice in the total synthesis of kehokorin A 83 and kehokorin B 84. In their work, phenol 85 was transformed into bromo-phenol derivative 86 (95%) on treatment with N-bromosuccinimide (NBS) in the presence diisopropylamine (i-Pr2NH) followed by the addition of TMSCHN2 (trimethylsilyldiazomethane) for methylation of phenolic hydroxyl group. In the next step, compound 86 was coupled with boronic acid 87 to access biaryl compound 88 in 94% yield under the conditions of Pd2(dba)3, SPhos and K3PO4 in toluene at 105 °C which over three steps provided dibenzofuran core 89. Compound 89 produced was further transformed into compound 90 which again underwent Suzuki coupling with boronic acid 91 under similar conditions to give p-terphenyl dibenzofuran 92 in 55% yield (Scheme 12). The benzyl group of the dibenzofuran 92 was cleaved by using H2, Pd/C to obtain compound 93 which on treatment (with K2CO3, BuOH, 1,4-dioxane) furnished kehokorin B 84 (70%), whereas on incorporation of imidate 94 (using TMSOTf) followed by the addition of K2CO3, BuOH, 1,4-dioxane and H2, Pd/C for hydrogenolysis produced kehokorin A 83 (48%) (Scheme 13).

Synthesis of p-terphenyl dibenzofuran 92

Completion of the total synthesis of kehokorin A 83 and kehokorin B 84
11,12-Dihydro-3-hydroxyretinol 95 has been collected from Laurencia nipponica (red alga) at Muroran, Hokkaido in Japan. It possesses moderate antifouling property in 5–10 µg/cm2 against the larvae of Amphibalanus amphitrite (Fig. 12) [54].

Structure of (R)-11,12-dihydro-3-hydroxyretinol 95
Rivas et al. [55] reported the stereocontrolled synthesis of (R)-11,12-dihydro-3-hydroxyretinol 95 with 73% yield via Suzuki cross-coupling of enantiopure alkenyl iodide 99 with pinacolboranedienoate 98 (Scheme 14). Compound 98 was obtained when methyl geranoate 96 with 2-propenylboronate 97 was coupled with second generation Grubbsˈ catalyst in 48% yield. In the following step, Suzuki coupling in the presence of 7 mol% Pd(PPh3)4 and 10% aq. TlOH in THF at 25 °C in 2 h gave all-trans-tetraenoate 100 (71%) which on reduction with DIBAL-H yielded the desired product 95.

Synthesis of (R)-11,12-dihydro-3-hydroxyretinol 95
Bradykinin binding inhibitor, a metabolite termed as L-755,807 101 (non-peptide natural product) was isolated from Microsphaeropsis sp. It acted as 3H-bradykinin binding inhibitor with IC50 of 71 µM (Fig. 13) [56].

Structure of L-755,807 101
A convergent synthetic route for the total synthesis of L-755–807 101 and its stereoisomers via Suzuki coupling and E-selective HWE (Horner-Wadsworth-Emmons) protocol was presented by Tanaka et al. [57]. (R)(−)-3-Hydroxy-2-methylpropionate 102 gave (R,R)-vinyl bromide 103 over several steps. Then, (R,R)-103 was treated with boronic acid 104 by loading Pd(PPh3)4, TlOEt in THF/H2O to accomplish (R,R)-triene alcohol 105 in 65% yield which on oxidation (with MnO2) provided (R,R)-triene aldehyde 106 in 79% yield (Scheme 15). Moreover, amide 107 was converted to phosphonate 108 on treatment with n-BuLi and dimethoxy methyl phosphonate which was further assembled with already synthesized fragment 106 under HWE reaction conditions (t-BuOK in 1 M THF) to form compound 109 in 65% yield. Deprotection of TES group of compound 109 using 3HF·TEA followed by the addition of AZADOL for oxidation afforded the target skeleton of L-755,807 101 in 63% yield (Scheme 16). Additionally, structure–activity relationship (SAR) for L- 755,807 101 and its isomers was studied for the first time and it was observed that the prepared compounds showed potent Aβ aggregation inhibitory activity (IC50 values 5–21 µM).

Synthesis of (R,R)-triene aldehyde 106

Completion of synthesis of L-755,807 101
Anithiactins A-C (110–112) are modified phenylthiazole derivatives and have been extracted from Streptomyces sp. These compounds displayed moderate acetylcholinesterase inhibitory activity with no cytotoxicity [58]. Later on, thiasporine A 113 was extracted from Actinomycetospora chlora in addition to anithiactins A and C (Fig. 14) [59].

Structures of anithiactin A 110, anithiactin B 111, anithiactin C 112 and thiasporine A 113
Vaaland et al. [60] designed the synthetic route for four title natural products 110–113 by employing Suzuki coupling as main step (Scheme 17). The starting substrates i.e. boronic acid hydrochloride 46 and thiazole bearing carboxylate 114 were coupled in the presence of 15 mol% Pd2dba3, 43 mol% XPhos and CsF in refluxing dioxane for 17 h to form a common intermediate 115 in 64% yield. Furthermore, protocols such as hydrolysis (using aq. NaOH, CH3OH) and aminolysis (using 4 M NH3, CH3OH) were implemented to obtain the structures of desired naturally occurring thiazole derivatives 110–113 in moderate to excellent yields (57–95%).

Synthesis of thiazole derivatives 110–113
Fumimycin 116 (obtained from Aspergillus fumisynnematus) is an unusual metabolite bearing interesting alanine motif connected to a phenyl moiety at the α-carbon (Fig. 15). It showed peptide deformylase inhibitory potential with IC50 of 4.1 µM in addition to antibacterial activity [61].

Structure of (±)-fumimycin 116
Zaghouani et al. [62] gave a concise total synthesis of (±)-fumimycin 116 in 11.6% overall yield based on 7 steps by employing Suzuki coupling approach (Scheme 18). For this purpose, pyruvic acid 117 and phenol 118 underwent esterification using N,N'-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) followed by intramolecular aza-Friedel–Crafts cyclization (using Cbz-NH2, 15 mol% TfOH) and regioselective chlorination (with N-chlorosuccinimide (NCS), 5 mol% PalauˈChlor) to give chlorobenzofuranone 119 in 76% yield. Decorated styrene 121 with complete E-selectivity in 73% yield was obtained by coupling of chlorobenzofuranone 119 with boronate 120 by using Pd(OAc)2 (catalyst), SPhos (ligand), K3PO4 (base) in THF/H2O at 60 °C. The synthesis of natural product 116 was achieved in 42% yield (over 3 steps) when styrene derivative 121 underwent Cbz-deprotection (with Pd(OAc)2/Et3SiH, trimethylamine (TEA)) followed by fumaryl coupling (using fumaryl chloride) and hydrolysis (with silica/H2O).

Synthesis of (±)-fumimycin 116
Sonogashira cross-coupling reaction
Numerous natural products contain alkynes as a fundamental component in their structure and Sonogashira cross-coupling is a vital approach to incorporate alkynes in the skeleton of a wide range of natural products [63]. This reaction includes coupling of a terminal alkyne with vinyl/aryl halide in the presence of palladium catalyst and copper additive as a co-catalyst. Under certain circumstances, copper additives may be harmful to reaction results for some substrates. To solve such kind of problems, copper-free Sonogashira coupling has also been discovered [64, 65]. Furthermore, Sonogahira coupling is employed for the building of bioactive compounds and electronic materials [66].
Synthesis of alkaloids
(±)-Aspergilline A 122 (extracted from Aspergillus versicolor) is a cyclopiazonic acid derived alkaloid with a rigid and highly oxygenated hexacyclic skeleton which is based on indole, tetrahydrofuran and tetramic acid moieties (Fig. 16). This natural product shows inhibitory activity against tobacco mosaic virus and moderate cytotoxicity in various human cell lines [67].

Structure of (±)-aspergilline A 122
Nakhla and Wood [68] described the total synthesis of (±)-aspergilline A 122 in 16 steps by employing Sonogashira coupling in addition to various protocols such as oxidation, cyclization, [3 + 2] cycloaddition and Aldol reaction (Scheme 19). The synthesis was commenced from N-methylation of readily available bromoisatin 123 to obtain N-methylated product 124 (using CH3I and K2CO3) in good yield (94%) which on coupling with propargyl amine 125 in the presence of 4 mol% Pd(PPh3)4, 8 mol% CuI, Cs2CO3 and i-Pr2NEt (Hünig’s base) in toluene at 85 °C gave isatin 126 in 72% yield. Aldol substrate 128 was prepared in 69% yield from acid chloride 127 by premixing it with i-Pr2NEt followed by the addition of isatin 126, Raney Ni and DMP. The compound 128 was further converted into (±)-aspergilline A 122 over several steps.

Synthesis of (±)-aspergilline A 122
Cyclopiamide A 129 (obtained from Penicillium cyclopium) and speradine E 130 (obtained from Aspergillus oryzae) are N-methyl-2-oxindoles [69] having structural resemblance to alkaloid 122. However, speradine E 130 consists of an additional β-dicarbonyl motif than cyclopiamide A 129 (Fig. 17).

Structures of cyclopiamide A 129 and speradine E 130
The preparation of cyclopiamide A 129 and speradine E 130 was described by Nakhla et al. [70] via a unified approach (Scheme 20). Isatin 126 (obtained from Sonogashira coupling) was advanced to pyrrolinone 132 (45%) by treating allyl malonyl chloride 131 with i-Pr2NEt followed by the addition of isatin 126. The compound 132 was treated with sodium hydride to get tetracycle 133 in 25% yield which in the presence of 1.2 mol% Pd(PPh3)4 and morpholine underwent decarboalkoxylation and dehydrative aromatization and the addition of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) furnished cyclopiamide A 129 in 93% yield. The framework of speradine E 130 was obtained in 54% yield from the reaction of methyl malonyl fluoride 134 with alkaloid 129.

Synthesis of cyclopiamide A 129 and speradine E 130
(+)-Anatoxin-a 135 and (+)-homoanatoxin-a 136 are natural alkaloids possessing distinctive structural features and strong biological properties that have made them a theme of considerable research work in synthetic and pharmacological studies (Fig. 18). These are extremely neurotoxic compounds generated during surface-water blooms by benthic and planktonic cyanobacteria. These compounds may prompt asphyxiation, respiratory paralysis and ultimately death as they function as neuromuscular blocking agents [71,72,73,74].

Structures of (+)-anatoxin-a 135 and (+)-homoanatoxin-a 136
Addante-Moya et al. [75] reported an easy and systematic synthetic methodology for the construction of natural and unnatural enantiomers of anatoxin-a 135 and homoanatoxin-a 136 by utilizing ring opening reaction, cyclization, Sonogashira coupling and chemo- and regioselective hydration as key steps. 9-Oxabicyclo[6.1.0]non-4-ene 137 and chiral α-methylbenzylamine 138 reacted under microwave irradiation in methanol to give a mixture of diastereoisomeric amino-alcohols (-)-139 and (+)-140 in 34% and 34.2% yields, respectively (Scheme 21). Azabicyclo ketone (-)-141 required for the accomplishment of natural enantiomers (+)-anatoxin-a 135 and (+)-homoanatoxin-a 136 was prepared from (-)-139 over several steps. Enol triflate (+)-142 produced from azabicyclo ketone (-)-141 (using potassium bis(trimethylsilyl)amide (KHMDS) and Cominsˈ reagent) was coupled with trimethylsilyl enyne (for anatoxin-a) and propyne (for homoanatoxin-a) by loading PdCl2(PPh2)2, CuI, TEA in DMF at room temperature to give corresponding compounds (-)-143 (86%) and (-)-144 (75%), respectively. Next, trimethylsilyl group of compound (-)-143 was unmasked using K2CO3 in methanol followed by triple bond hydration (using HgO, boron trifluoride etherate, trichloroacetic acid). The addition of trifluoroacetic acid cleaved Boc group and afforded trifluoroacetate salt of (+)-135 in 99% yield. The final structure of homoanatoxin-a (+)-136 was also achieved in 99% yield by using the similar conditions for the triple bond hydration and Boc deprotection (Scheme 22).

Preparation of enol triflate (+)-142

Synthesis of (+)-anatoxin-a 135 and (+)-homoanatoxin-a 136
Synthesis of terpenoids
Cleviolide 145 is a first monoterpene isolated from Senecio clevelandii by Bohlmann et al. [76] It is reported as a precursor of two monoterpenes named cis-dihydrocleviolide (obtained from S. clevelandii) and trans-dihydrocleviolide (obtained from Psathyrella scobinacea) (Fig. 19) [77].

Structure of cleviolide 145
The three-step preparation of natural acetylenic monoterpene cleviolide 145 was described by Cheval et al. [78] in 40% overall yield via Sonogashira coupling (Scheme 23). Nosylate 146 bearing 2,5-dihydrofuran-2-one is a component of various natural products and has been utilized for the synthesis of cleviolide 145. Nosylate 146 and 4-methylpent-yn-3-ol 147 were reacted in the presence of PdCl2(PPh3)2 as catalyst, CuI as co-catalyst and i-Pr2NEt as base in acetonitrile to produce compound 148 in 67% yield. Treatment of compound 148 with diphosphorous pentaoxide in benzene gave the desired natural product cleviolide 145 in good yield (81%). Vinyl nosylates could be efficient coupling partners for Sonogashira coupling approach either in the presence of Cu or Ag salts and p-nitro substituted nosylate allowed this cross-coupling to be done at room temperature.

Synthesis of cleviolide 145
Over the years, natural products have been considered as valuable compounds in synthetic as well as therapeutic industry owing to the diversity in their chemical structures and biological capabilities. They are obtained from various species. In this regard, daphnane 149 and tigliane 150 have been extracted from thymelaeaceae and euphorbiaceae (Fig. 20). Daphnane and tigliane are structurally complex naturally occurring diterpenes comprising of [5–7-6] tri-cyclic carbon framework and these natural compounds are of great importance due to their antimalarial, antimicrobial, anti-HIV and neurotrophic properties [79, 80].

Structures of daphnane 149 and tigliane 150
Dai et al. [81] reported a concise synthesis of common tricyclic carbon core 151 (Fig. 21) of daphnane and tigliane via Sonogashira coupling followed by gold catalyzed furan preparation and furan/allene [4 + 3] cycloaddition (Scheme 24). Synthesis was commenced from the reaction of aldehyde 153 with compound 152 in the presence of CuI and i-Pr2NH followed by the addition of bromo moiety 154 and sodium hydride to produce fragment 155 in 70% yield. In the following step, allene 157 was obtained by the combination of compound 155 with fragment 156 (obtained from cyclopentanone) in the presence of palladium catalyst [PdCl2(PPh3)2], CuI and i-Pr2NH at 70 °C. The gold catalysis (with 10% PPh3AuCl/AgOTf) followed by [4 + 3] cycloaddition (with 13% tBuXPhosAuCl/AgSbF6) of substrate 157 produced the target skeleton of diterpenes 158a and 158b in acceptable ratio (1.8:1). It was concluded that gold-catalyzed [4 + 3] cycloaddition could be used for the accomplishment of complex oxa-bridged polycyclic frameworks of natural products in a stereoselective way.

Common tricyclic core 151 of daphnane 149 and tigliane 150

Synthesis of common tricyclic carbon core of daphnane and tigliane
Fenical and co-workers in 2010 isolated secondary metabolites, nitropyrrolins A-E from CNQ-509 strain. Nitropyrrolin A 159, nitropyrrolin B 160 and nitropyrrolin D 161 (hybrid isoprenoid natural products) showed cytotoxic activity toward human colon cancer cells with IC50 values 31.1 µM, 31.0 µM and 5.7 µM, respectively (Fig. 22) [82, 83].

Structures of nitropyrrolins A, B, D (159–161)
Ding et al. [84] outlined a synthetic methodology for the synthesis of nitropyrrolins A, B, D via Sonogashira coupling approach for substituted pyrrole formation. This approach further involves carboxylative cyclization, sulfonylcarbamate preparation and deprotection protocol for the formation of hydroxyl ketone. The synthesis was commenced with the coupling of N-Boc protected iodide 162 with alkyne 163 using PdCl2(PPh3)2, CuI, TEA in DMF to furnish pyrrole substrate 164 in 90% yield. Pyrrole 164 was subjected to carboxylative cyclization by using silver carbonate, PPh3 in DCM to afford desired carbonate which was then treated with p-TsNH2 and K2CO3 in DMF followed by refluxing in pyridine/CH3OH afforded the required hydroxyl ketone 165 (60% yield). Next step involved the diastereoselective reduction of ketone 165 through Corey–Bakshi–Shibata reduction approach (using (R)-Me-CBS and BH3·DMS) to give diols 166a and 166b with good diastereoselectivity (6:1). The final structure of natural product 159 was achieved in 92% yield by deprotection of Boz group of diol 166a with aqueous K2CO3 in methanol (Scheme 25). Similarly, the skeleton of nitropyrrolin B 160 was constructed in 60% overall yield from N-Boz protected iodide 162 by employing Sonogashira coupling with ent-163 in the presence of PdCl2(PPh3)2 and diastereoselective reduction (with CBS) of ent-165 to furnish ent-166a which was subjected to one-pot epoxidation and mesylation (using MsCl, TEA) followed by deprotection of N-Boz group under mild conditions yielded nitropyrrolin B 160 in 92% yield. It was further transformed to nitropyrrolin D 161 as previously reported by Morimoto and coworkers [82] (Scheme 26).

Synthesis of nitropyrrolin A 159

Synthesis of nitropyrrolin B and D (160, 161)
Heronapyrrole B 167 is a secondary metabolite obtained from CMB-M0423 strain of Streptomyces sp. by Capon research group [85, 86]. This natural product shows antibacterial activity against Gram-positive bacteria (S. aureus, B. subtilis) (Fig. 23) [87].

Structure of heronapyrrole B 167
The synthesis of nitropyrrolin A 159 and heronapyrrole B 167 through Sonogashira coupling was reported by Ding et al. [88] (Scheme 27). Heronapyrrole B 167 was accomplished in 90% yield by treating compound 166 (prepared from iodide 162) with AD-mix-α, MeSO2NH2 in tert-butanol/water mixture followed by deprotection of Boz group with aq. K2CO3 in methanol.

Synthesis of heronapyrrole B 167
Synthesis of steroids
Among all the categories of natural products, steroids have played a vital role in evoking latest ideas in total synthesis. A significant amount of knowledge has been added to organic chemistry in the context of tracking suggestions for the assembly of fragments to form steroids. Evidently, this curiosity continues to present day. Aplykurodine obtained from mollusks represents a class of most degraded steroids and (±)-aplykurodinone-1 169 (extracted from Synphonota geographica) belongs to this family of natural products. Its skeleton consists of a cis-fused ring with epimeric C8, an unsaturated side chain [89] and six contiguous stereocenters (Fig. 24) [90].

Structure of (±)-aplykurodinone-1 169
Tao et al. [91] reported a highly efficient formal synthesis of (±)-aplykurodinone-1 169 via one pot hetero-Pauson–Khand reaction (h-PKR), desilylation and Sonogashira coupling as key steps (Scheme 28). h-PKR has rare applications in the formation of natural products; however it is known as a convenient method to shape butenolides and α,β-unsaturated lactams. The synthesis was commenced with the iodination of enone 170 (using iodine, pyridine in DCM) to afford vinyl halide 171 in good yield (85%). Then, vinyl iodide 171 underwent Sonogashira coupling with TMS-acetylene in the presence of PdCl2(PPh3)2, CuI, TEA as base in THF at room temperature to give 2-alkynyl-2-cyclopentanone 172 (90% yield). The compound 172 was treated with 2-iodobenzoic acid (IBX) in DMSO/THF for TBS deprotection followed by the addition of Mo(CO)6, CO in toluene/DMF to perform one pot cycloaddition and desilylation to produce tricyclic compound 173 in 60% yield. The formal synthesis of (±)-aplykurodinone-1 169 was achieved by converting tricyclic compound into required enone 174 (20% overall yield) over several steps. The final framework of natural product 169 with high stereoselectivity was established through Micheal addition [92] of enone 174.

Synthesis of aplykurodinone-1 169
Synthesis of polyketides
Bysspectin A 175 is a polyketide-derived octaketide dimer isolated from endophytic fungus (Byssochlamys spectabilis) and consists of a unique carbon skeleton based on two hydrophobic ketone chains and 2-phenylbenzofuran motif (Fig. 25). It acts as an inhibitor against hCE2 (human carboxylesterase) owing to its hydrophobic nature [93].

Structure of bysspectin A 175
Yang et al. [94] designed the synthetic strategy for bysspectin A 175 for the first time and utilized copper catalyzed domino Sonogashira cyclization to obtain the targeted product with 7.7% overall yield (Scheme 29). In order to complete the synthesis, compound 177 was obtained from 176 [95] and was further converted into compound 178 in 92% yield via Sonogashira coupling using trimethylsilyl acetylene, PdCl2(PPh3)2, CuI, TEA in THF at 50 °C. Phenolic hydroxyl group of compound 178 was protected (using SEMCl, i-Pr2NEt) followed by the addition of octylmagnesium bromide, Dess–Martin periodinane (DMP) and potassium carbonate to form terminal alkyne 179 in good yield (89%). Then, domino Sonogashira cyclization was done between terminal alkyne 179 and fragment 180 in the presence of [Cu(phen)(PPh3)2]NO3, Cs2CO3 in toluene to obtain benzofuran ring in 83% yield. Deprotection of SEM group with tetra-n-butylammonium fluoride (TBAF) afforded the required product 175 in 65% yield.

Synthesis of bysspectin A 175
Synthesis of macrolides
Salarins are marine macrolides extracted from Madagascan sponge (Facaplysinopsis sp.) comprising of 17-membered lactone set with a trisubstituted oxazole or a trisacylamine core. Salarin C 181 (a nitrogenous macrolide) isolated by Kashman and coworkers belongs to this class and acts as antiproliferative agent (Fig. 26) [96, 97].

Structure of salarin C 181
Schӓckermann and Lindel [98] reported the synthesis of eastern portion 189 of salarin C 181 by employing halogen dance reaction to join the trisubstituted oxazole ring. In the first step, methylation of oxazole 182 was done with CH3I to furnish trisubstituted oxazole 183 in 90% yield which in the next step was coupled with propargyl alcohol in the presence of PdCl2(PPh3)2, CuI, TEA in acetonitrile to give compound 184 in 83% yield. Hydrosilylation of compound 184 with Et3SiH [Cp*Ru(MeCN)3]PF6] and desilylation with TBAF was performed to get allylic alcohol. Trisubstituted n-butanesulfonyloxazole 185 was prepared from allylic alcohol by O-silylation (TBSCl, imidazole) followed by oxidation (H2O2, (NH4)6Mo7O24·4H2O) (Scheme 30). Then, trisubstituted n-butanesulfonyloxazole 185 was coupled with (Z)-iodoalkene 187 (prepared from TMS-protected alkyne 186) in the presence of n-BuLi followed by the addition of p-TsOH in methanol and DMP in DCM to form unsaturated aldehyde in good yield (93%). Phosphonate 188 was used for the Still Gennari olefination of aldehyde (using KHMDS in THF) to accomplish the partial segment 189 (bisalkenyloxazole) of salarin C 181 in 88% yield (Scheme 31). Additionally, photooxidation of bisalkenyloxazole was carried out by using singlet oxygen which supported Kashman’s hypothesis of conversion of salarin C to salarin A.

Synthesis of trisubstituted n-butanesulfonyloxazole 185

Synthesis of partial section 189 of salarin C
Synthesis of alkamides
Alkamides are secondary metabolites obtained from various plant families and constitute a well-defined class of natural compounds in which peptide bonding connects unsaturated fatty acids with different amino acids. Tingling and pungent effects are the characteristic features of this class. Spilanthol (affinin) 190 is a familiar bioactive alkamide which has been extracted from plants of Asteraceae family and it also produces numbing, tingling, pungency and mouth-watering effects. According to scientific research, it exhibits a variety of biological activities such as antimicrobial, antimutagenic and insecticidal, etc. (Fig. 27) [99].

Structure of spilanthol 190
Alonso et al. [100] reported the synthesis of spilanthol 190 in five steps via Sonogashira coupling, Z-selective alkyne semi-reduction and HWE as main steps (Scheme 32). During the connection of double bonds, control of alkene geometry was maintained in the synthetic route. The first step in synthetic strategy involved the Sonogashira coupling of alcohol 191 with 1-bromo-1-propene in the presence of Pd(PPh3)4, PPh3, CuI, diisopropanolamine (DIPA) in DMF to afford the compound 192 (53%) which was further transformed to a diene 193 (59% yield) by using Zn/Cu/Ag in the presence of TMSCl through Z-selective alkyne semi-reduction. Later, alcohol 193 underwent Swern oxidation followed by the addition of phosphonoacetamide 194 to crude aldehyde to accomplish the synthesis of 190 in 63% yield. Moreover, it was found that spilanthol could be an advantageous contemporary anesthetic in medical field.

Synthesis of spilanthol 190
Negishi cross-coupling reaction
After the revelation of palladium catalyzed Negishi coupling of aryl/vinyl chlorides by Fu and co-workers [101], a significant success has been made in developing new catalytic methodologies related to this approach. It involves the use of organozinc reagent as a coupling partner and immensely utilized to compose natural products [102]. It implies mild reaction conditions with advantage of low toxicity of readily available organozinc substrates and their fast transmetalation to palladium. In some cases, other metal catalyst systems containing nickel, iron, copper or cobalt are also used instead of palladium-based catalyst [103, 104].
Synthesis of macrocycles
Argyrins are macrocyclic natural products which have been isolated from myxobacteria in 2002 by Sasse et al. These are known as heptapeptides based on thiazole heterocycle and tryptophan amino acid. Biological interpretation showed that these compounds act as cytotoxic and immunosuppressant agents, etc. (Fig. 28) [105, 106].

Structure of argyrin C 195
For the first time, Stempel et al. [107] carried out an efficient and short synthesis of AzuAla1 argyrin C 204 in a stereocontrolled manner by incorporating Negishi coupling reaction (Scheme 33). By keeping in consideration to track and detect bioactive compounds by using non-invasive probes, β-(1-azulenyl)-ʟ alanine (termed as AzuAla1, unnatural deep blue amino acid) was prepared with fluorescent/photophysical features and then utilized it for the preparation of natural product 204. Iodoalanine 197 (obtained from alanine 196) was converted into organozinc reagent by using zinc and 20 mol% iodine that underwent Negishi coupling with C3-iodinated compound 199 (prepared from indole 198) in the presence of 2.5 mol% Pd2(dba)3 as catalyst and 5 mol% SPhos as ligand at 35 °C to give compound 200 in 88% yield. Compound 200 was saponified (sodium hydroxide, methanol) with subsequent benzenesulfonyl group cleavage and addition of glycine methyl ester to produce dipeptide 201 in good yield (79%). Next, the dipeptide 201 was fused with fragment 202 to give tripeptide unit 203 (81% yield) which over several steps gave Azuala1 argyrin C 204. Nevertheless, it was the first synthetic approach to introduce fluorophore group such as AzuAla1 in complex natural product.

Synthesis of AzuAla1 argyrin C 204
Synthesis of lignans
Lignan natural products; (±)-dimethylretrodendrin 205, (±)-dimethylmetairesinol 206, (±)-kusunokinin 207, (±)-bursehernin 208, (±)-yatein 209, (±)-collinusin 210 bearing dibenzylbutyrolactone and aryltetralin cores are associated with anti-tumour, anti-viral, fungicidal, antibiotic and anti-HIV activities (Fig. 29) [108, 109].

Structures of lignan natural products (205–210)
KC et al. [110] reported regioselective dicarbofunctionalization of unactivated olefins via Ni-catalyzed tandem cyclization/Negishi cross-coupling and implemented this methodology to construct the skeletons of six lignan natural products (205–210) in good yields (65–86%) and diastereoselectivities (dr, 19:1–40:1). For this purpose, organozinc reagent 213 and substrate 211 were coupled in the presence of NiBr2, terpy 212 in N-methyl-2-pyrrolidone (NMP) at 50 °C followed by Jones oxidation to produce lactone 214 in 62% yield. Lactone 214 on treatment with fragments 215, 216 and 217 after subjection to lithium diisopropylamide (LDA) gave natural products 205 (73%), 206 (71%) and 207 (86%), respectively (Scheme 34). The same procedure was repeated for the accomplishment of remaining lignans 208–210 by employing different organozinc reagents 218 and 222 (Scheme 35).

Synthesis of (±)-dimethylretrodendrin 205, (±)-dimethylmetairesinol 206, (±)-kusunokinin 207

Synthesis of (±)-bursehernin 208, (±)-yatein 209, (±)-collinusin 210
Synthesis of polyketides
Seragamide A 224 was isolated from Suberites japonicas (Okinawan sponge) as a cytotoxic metabolite having peptide motif. It plays a vital role in the polymerization of G-actin and stabilizes F-actin filaments (Fig. 30) [111].

Structure of seragamide A 224
Lang and Lindel [112] disclosed the synthesis of polyketide Sect. 230 of seragamide A 224 via Negishi coupling reaction. tert-Butyl ester 225 (obtained from enantiomerically pure (R)-propylene oxide) was subjected to reduction (DIBAL-H) followed by oxidation and Corey-Fuchs reaction (CBr4, PPh3, TEA) to access dibromoalkene 226 (81%). Then, the compound 226 was treated with LDA followed by methylation (using CH3I) and hydrozirconation/iodination (with Cp2ZrHCl and I2) to get (E)-olefin 227 in 84% yield with perfect stereo- and regioselectivity. Olefin 227 was coupled with organozinc homoenolate 228 (prepared from β-bromopropionic acid ester) in the presence of PdCl2(dppf) x DCM at room temperature to produce compound 229 in 75% yield which on subjection with ceric ammonium nitrate (CAN) afforded polyketide portion 230 (82%) (Scheme 36). Further, tripeptide Sect. 231 of seragamide 224 was synthesized by using D-tyrosine as starting material that was converted to tripeptide 231 over several steps which in the presence of TFA/DCM followed by the addition of BEP, i-Pr2NEt in DCM was fused with polyketide unit 232 (from 230) to give pepetide-polyketide 233 (60%) with higher diastereoselectivity (dr, 9:1) than tripeptide fragment 231 (dr, 4:1) (Scheme 37).

Synthesis of polyketide section 230 of seragamide A 224

Synthesis of tripeptide-polyketide 233 of seragamide A 224
Synthesis of terpenoids
Stachyflin 234 possessing cis-fused rings and an ethereal bond is a terpenoid and shows anti-influenza A virus activity with EC50 value of 0.003 µM. Its biological potential and structural features have made it a fascinating molecule in synthetic chemistry (Fig. 31) [113].

Structure of stachyflin 234
Haut et al. [114] and Wildermuth et al. [115] independently reported a total synthesis of (+)-stachyflin 234 through sp2-sp3 Negishi cross-coupling reaction of isoindolinone 236 with dehydrodecalin 237 in the presence of Pd-SPhos G2 catalyst, SPhos ligand and dimethylacetamide (DMA) base in THF to furnish compound 238 in 56% yield (Scheme 38). Cleavage of MOM-ether group (with HCl), cationic cyclization (with BF3·OEt2) and hydrogenation (with H2, Pd/C) gave cis-fused decalin 239 (62% over 3 steps) which on treatment with PIFA in benzene and demethylation by potassium n-dodecanthiolate provided required (+)-stachyflin 234 (43% over 2 steps).

Synthesis of stachyflin 234
Miscellaneous
In 1990, first naturally occurring paracyclophanes were extracted from blue-green algae and contained 22-membered cyclic infrastructure exhibiting cytotoxic potential against tumor cell lines (KB and LoVo) with IC50 value 2–10 µg/mL. (−)-Cylindrocyclophane F 240 belongs to these types of natural products (Fig. 32) [116,117,118].

Structure of (−)-cylindrocyclophane F 240
Berthold and Breit [119] reported a short and convergent route for the preparation of (−)-cylindrocyclophane F 240 using Pd-catalyzed Negishi coupling and cross olefin metathesis protocol (Scheme 39). Compound 241 (obtained from ʟ-(+)-lactic acid precursor) underwent iodine lithium exchange (t-BuLi, Et2O) followed by transmetalation (ZnCl2, THF) and Negishi cross-coupling reaction with triflate 242. As a result, diene 243 was obtained in 96% yield with 5 mol% PdCl2(dppf) loading in THF. Next step involved olefin cross metathesis using Grubbs II followed by hydrogenation (H2, Pd/C, AcOEt) and deprotection (BBr3, DCM) to obtain targeted (−)-cylindrocyclophane F 240.

Synthesis of (−)-cylindrocyclophane F 240
Heck cross-coupling reaction
Heck cross-coupling reaction includes the coupling of aryl iodide/bromide/chloride, triflates, mesylates, tosylates and diazonium salts with olefins to accomplish aryl-substituted alkenes. This methodology has been extended to the coupling of alkenyl compounds with olefins [120]. This interesting strategy finds its applications in the formation of medicinally important analogues, polymers and natural products. Some known examples are the synthesis of retinoid x receptor antagonist and diazepinylbenzoic acid via Heck coupling [121].
Synthesis of alkaloids
Ergot alkaloids are a source of indole alkaloids exhibiting a wide spectrum of biological activities such as antiprolactin and anti-Parkinson’s activity. These are obtained from Claviceps pupurea (fungus) which grows on rye and other grains. (+)-Lysergol 244 is an indole alkaloid belonging to this attractive family of natural products (Fig. 33) [122].

Structure of (+)-lysergol 244
Milde et al. [123] used anti-carbopalladation/Heck reaction to achieve the enantioselective synthesis of (+)-lysergol 244 in 12 steps and with 13% overall yield (Scheme 40). 2-Bromoindole 245 was used as starting precursor to obtain alcohol 246 over several steps. Racemic alcohol 246 was converted to required domino precursor 249 on treatment with DMP followed by enantioselective reduction (using Noyori’s catalyst 247) and Mitsunobu reaction (using sulfonamide 248). Next step involved anti-carbopalladation/Heck reaction by the addition of 10 mol% [PdCl2(PhCN)2] as catalyst, 20 mol% XPhos as ligand in DMA at 120 °C for 2 h to form two rings of target product 250 in a stereospecific manner in 80% yield along with side product 251 (17%). Proceeding from compound 250, the final structure of (+)-lysergol 244 was accomplished over several steps.

Synthesis of (+)-lysergol 244
Lycorine-type alkaloids are derived from plants and show antimitotic, antiviral and antineoplastic activities, etc. (±)-γ-Lycorane 252 is not associated with any significant pharmaceutical property; however, it has become a popular molecule in the aspect of describing the potential of new synthetic techniques for the combination of fragments in lycorine-type alkaloids (Fig. 34) [124].

Structure of (±)-γ-lycorane 252
Monaco et al. [125] reported the synthesis of (±)-γ-lycorane 252 via Heck cyclization reaction (Scheme 41). Firstly, compound 254 was prepared in 87% yield by treating piperonylamine 253 with bromine in acetic acid followed by the addition of cyclohexanone, diacetoxyacetyl chloride and boron trifluoride diethyl etherate. Next, compound 254 was cyclized in the presence of PdCl2(PPh3)2 in DMF to afford pentacyclic motif 255 in 74% yield. The required product 252 (85%) was formed by the hydrogenation of double bonds of compound 255 by using H-Cube hydrogenation flow reactor, 10% palladium on charcoal in ethanol/ethyl acetate mixture and then followed by its reduction with LiAlH4.

Synthesis of (±)-γ-lycorane 252
Tuberculosis (TB) is an infectious disease and needs to be treated with effective drugs. Advent of new drugs with low toxicity, high potency, good interaction and novel mechanism of action remains a challenge for the cure of tuberculosis. However, natural resources serve best in producing molecules with unique chemical and biological attributes. 3,4-Diarylpyrrole alkaloids are suitable example of captivating bioactive metabolites obtained from marine species. Denigrin A 256 and denigrin B 257 (extracted from Dendrilla nigra) with potent antitubercular activity belongs to this category of natural products (Fig. 35) [126].

Structures of denigrin A 256 and denigrin B 257
Karak et al. [127] reported the first synthesis of denigrin A and B (256, 257) with 62% and 31% overall yields based on three and five steps, respectively, via Heck reaction as a key step (Scheme 42). The starting substrates; maleic anhydride 258 and diaryliodonium tosylate 259 were coupled through Heck reaction (Pd(OAc)2, sodium acetate in acetonitrile) to produce monoarylated 260 and diarylated compound 261 (69%, 4:96). To obtain denigrin A 256 (97%), compound 261 was reacted with p-methoxyphenethylamine 263 in acetic acid followed by the addition of BBr3 in DCM. On the other hand, treatment of diarylated compound 261 with LiAlH4 and p-methoxybenzaldehyde provided Z-isomer 262 as sole product in 89% yield. Later, compound 262 was reacted with fragment 263 in acetic acid to yield Z-benzylidene-diarylpyrrol-2(5H)-one 264Z and 264E through one-pot reaction. The cleavage of methyl ether groups of 264Z with BBr3 afforded denigrin B 257 in 89% yield.

Synthesis of denigrin A and B (256, 257)
Synthesis of lactones
Zeaenol 265 and 7-epi-zeaenol 266 extracted from a filamentous fungi belongs to resorcyclic acid lactones. Such compounds are known for cytotoxic potential having anticancer potential against human tumor cell line with same IC50 act as NF-ҡB inhibitor. (Fig. 36) [128].

Structures of zeaenol 265 and 7-epi-zeaenol 266
Doda et al. [129] disclosed the asymmetric synthesis of zeaenol 265 and 7-epi-zeaenol 266 in 7 and 9 steps with 32% and 21% overall yield, respectively, by employing Heck reaction as fundamental step (Scheme 43). In this regard, trans allyl alcohol 267 obtained from D-mannitol was protected with MOM group to give compound 268 which underwent coupling with aromatic triflate 269 in the presence of Pd(OAc)2, PPh3, Bu4NBr, K2CO3 in DMF at 80 °C for 5 h to provide trans olefin 270 in 88% yield over 2 steps. Deprotection of compound 270 with camphorsulfonic acid (CSA) and macrolactonization (using sodium hydride) gave macrolactone 271 in good yield (85%) which on treatment with TiCl4 produced desired 7-epi-zeaenol 266 in 88% yield. Moreover, subjection of macrolactone 271 with TMSCl, 4-nitrobenzoic acid, potassium carbonate and TiCl4 furnished zeaenol 265 in 83% yield.

Synthesis of resorcyclic acid lactones 265 and 266
Miscellaneous
Abscisic acid 272 isolated in 1960s is a plant hormone. It plays an important role in growth of plants from different aspects including seed development, germination and alteration to abiotic environmental stress (Fig. 37) [130].

Structure of abscisic acid 272
Dumonteil et al. [131] reported an environment friendly synthesis of abscisic acid 272 via Heck reaction in ligand and solvent free conditions with 54% yield (Scheme 44). In order to proceed, diketone 273 was reacted with (S,S)-hydrobenzoin and pyridinium p-toluenesulfonate (PPTS) in cyclohexane followed by the loading of vinylmagnesium bromide in THF to give compound 274 in quantitative yield. Heck coupling of compound 274 with Z-enoate 275 in the presence of 5 mol% Pd(OAc)2 and silver carbonate at 50 °C for 17 h delivered (E/Z)-diene 276 in 96% yield which on saponification using NaOH and TBACl followed by acidic treatment (with HCl) yielded the S-enantiomeric enriched 272 (59% over 2 steps).

Synthesis of abscisic acid 272
Stille cross-coupling reaction
Stille cross-coupling reaction represents the coupling of organostannanes with organic halides/triflates usually in the presence of palladium catalyst to connect carbon–carbon bonds in a variety of compounds. It finds enormous use in the preparation of precursors of drugs, for instance valsartan and imatinib C and in the formulation of electronic materials such as transistors and photovoltaic cells [132, 133].
Synthesis of lactones
Glabramycin B 277 was isolated through an antisense screening method from fermentation broth of Neosartorya glabra by Singh and co-workers in 2009 (Fig. 38). It exhibited antibacterial activity [134].

Structure of glabramycin B 277
Yamamoto et al. [135] reported an enantioselective synthesis of glabramycin B 277 through Stille coupling as one of the main step and corrected its relative configuration (Scheme 45). Compound 278 was used as starting material to produce tricyclic core 279 over several steps and was converted to vinyl triflate 280 on hydrogenation using H2, Rh/Al2O3 in ethyl acetoacetate followed by the addition of KHMDS and Tf2O in DME. Later, vinyl triflate 280 and trienylstannane 281 were coupled in the presence of Pd-PEPPSI-IPr 282, CsF in THF at 60 °C and cleavage of MOM group with TMSBr produced compound 283 in good yield (75% over 3 steps). The final skeleton of glabramycin B 277 was obtained by oxidation (with DMP) and deprotection (with tris(dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F)).

Synthesis of glabramycin B 277
Heliolactone 284, a non-sesquiterpene lactone stimulates the germination of root parasitic weeds which causes harmful effects on many crops (Fig. 39). It has been investigated that production of heliolactone increased in the presence of water while lowered in the presence of phosphorous and nitrogen supplements [136].

Structure of heliolactone 284
Woo and McErlean [137] reported enantioselective total synthesis of heliolactone 284 by using Stille coupling and confirmed the stereochemical nature (Scheme 46). α-Ionone 285 and chiral iodide 287 gave enantiomers 286 and 288, respectively, which were coupled in the presence of Pd2dba3, AsPh3, CuI in NMP to obtain target product 284 in 49% yield.

Synthesis of heliolactone 284
Synthesis of terpenoids
Plant of East Asia of genus Isodon is used as traditional medicine for the treatment of different infections including respiratory, cancer and inflammation problems in China. Mainly, its aerial parts such as stems and leaves are used for this purpose and some species of Isodon bears swollen rhizomes which also exhibit medicinal value. Hispidanin A-D are asymmetric dimeric diterpenoids which have been obtained from rizhomes of Isodon (Fig. 40) [138].

Structure of hispidanin A 289
Deng et al. [139] reported the asymmetric total synthesis of hispidanin A 289 via Stille coupling approach (Scheme 47). Compound 290 produced 291 over 3 steps, masking of phenol of tricyclic compound 291 (using MOMCl) and coupling with substrate 292 in the presence of palladium catalyst PdCl2(PhCN)2, CuI, Ph3As in NMP at 120 °C furnished compound 293 in 75% yield (2 steps). The compound 293 upon deprotection (with Amberlyst-15) followed by base promoted lactonization (with K2CO3) gave dienophile 294. Diels–Alder cycloaddition was conducted between fragment 294 and diene 295 (obtained from an epoxide) in toluene followed by addition of NaBH4 and MgClO4, acetic anhydride to produce diterpenoid 289 in 75% yield.

Synthesis of hispidanin A 289
Synthesis of macrolides
The resorcyclic macrolides account for important family of natural products containing β-resorcylate motif and macrocyclic lactone core. trans-Resorcylide 296 with a distinctive structure acts as plant growth inhibitor and is an influential template from synthetic point of view (Fig. 41) [140, 141].

Structure of trans-resorcylide 296
Luo et al. [142] prepared trans-resorcylide 296 via Stille carbonylation protocol (Scheme 48). In this regard, bromide 297 reacted with magnesium metal, CuI and epoxide 298 for regioselective ring opening to form alcohol which was transformed to cis-vinylstannane 299 (66%). It involved the treatment with cesium carbonate, subsequent formation of terminal alkyne (using n-BuLi, n-Bu3SnCl) and stereoselective reduction of alkyne bond (using Cp2ZrHCl). Compound 301 was shaped from starting material 300 over 4 steps. Fragments 299 and 301 gave Stille carbonylative precursor 302 in 52% yield with TEA in DCM. In the following step, 302 was reacted in the presence of 10 mol% Pd(PPh3)4, 20 mol% P(2-furyl)3 and CO in dioxane to furnished macrocyclic product 303 in 36% yield. The addition of BCl3 produced the required structure of trans-resorcyclide 296 in an excellent yield (97%).

Synthesis of trans-resorcylide 296
Miscellaneous
Mycophenolic acid 304 isolated from Penicillium fungus is associated with a variety of biological properties including antifungal, antibacterial, antiviral, anticancer and acts as immunosuppressive agent, etc. The presence of arene bearing six substituents make it an interesting molecule in synthetic chemistry (Fig. 42). Halle et al. [143] reported a short synthesis of mycophenolc acid 304 (quantitative yield) via Stille coupling of compound 306 (prepared from ethyl allenoate 305) with stannane 308 (obtained from acetate 307) in the presence of Pd(PPh3)4, PPh3 in DMF at 100 °C followed by hydrolysis using LiOH/H2O mixture (Scheme 49).

Structure of mycophenolic acid 304

Synthesis of mycophenolic acid 304
Schisandrene 310 (Suzuki and Stille coupling) [144], hemigeran C 311 and hemigeran D 312 (Negishi and Heck coupling) [145], patellazole B 313, raputimonoindole A 314 (Suzuki and Heck coupling) [146, 147] and diptoindonesin G 315 (Suzuki and Sonogashira coupling) [148] are examples of natural products incorporating more than one type of cross-coupling reactions to connect their synthons (Fig. 43).

Structures of natural products (310–315) illustrating the role of more than one type of cross-coupling in the synthesis
Conclusion
In summary, cross-coupling reactions have made immense progress in shaping structurally diverse natural products. We have explored the synthesis of natural products via Suzuki, Negishi, Heck, Sonogashira and Stille cross-coupling reactions to recognize the benefits of these strategies in synthetic as well as pharmaceutical sectors. The interesting features of these reactions have played role in the production of diastereoselective, stereoselective, regioselective and enantioselective fragments of naturally occurring molecules with moderate to good yields. Mostly palladium-based catalysts including Pd(PPh3)4, Pd(OAc)2, PdCl2(PPh2)2, PdCl2(dppf) and Pd2(dba)3 with various combination of ligands, bases and solvents have been used in this respect. Furthermore, the important biological activities of natural compounds such as anticancer, antiviral, antituberculosis, hCE2 inhibitor, ATPase inhibitor, etc. add supremacy to the entire constructed framework. However, this review would serve as a parameter to attract the attention of chemists towards the applications of cross-coupling reactions to make more adorable advancements with no shortcomings.
References
Denmark SE, Liu JHC (2010) Silicon-based cross-coupling reactions in the total synthesis of natural products. Angew Chem Int Ed 49:2978–2986. https://doi.org/10.1002/anie.200905657
Slagt VF, de Vries AH, De Vries JG, Kellogg RM (2010) Practical aspects of carbon−carbon cross-coupling reactions using heteroarenes. Org Process Res Dev 14:30–47. https://doi.org/10.1021/op900221v
Kapdi AR, Prajapati D (2014) Regioselective palladium-catalysed cross-coupling reactions: a powerful synthetic tool. RSC Adv 4:41245–41259. https://doi.org/10.1039/c4ra07895k
Li H, Seechurn CCCJ, Colacot TJ (2012) Development of performed Pd catalysts for cross-coupling reactions, beyond the 2010 Nobel prize. ACS Catal 2:1147–1164. https://doi.org/10.1021/cs300082f
Nicolaou KC, Bulger PG, Sarlah D (2005) Palladium-catalyzed cross-coupling reactions in total synthesis. Angew Chem Int Ed 44:4442–4489. https://doi.org/10.1002/anie.200500368
Siamaki AR, Abd El Rahman SK, Abdelsayed V, El-Shall MS, Gupton BF (2011) Microwave-assisted synthesis of palladium nanoparticles supported on graphene: A highly active and recyclable catalyst for carbon–carbon cross-coupling reactions. J Catal 279:1–11. https://doi.org/10.1016/j.jcat.2010.12.003
Chen X, Engle KM, Wang DH, Yu JQ (2009) Palladium (II)-catalyzed C-H activation/C–C cross-coupling reactions: versatility and practicality. Angew Chem Int Ed 48:5094–5115. https://doi.org/10.1002/anie.200806273
Bruno NC, Tudge MT, Buchwald SL (2013) Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem Sci 4:916–920. https://doi.org/10.1039/c2sc20903a
Tamura M, Kochi JK (1971) Vinylation of Grignard reagents. Catalysis by iron. J Am Chem Soc 93:1487–1489. https://doi.org/10.1021/ja00735a030
Kubo T, Scheutz GM, Latty TS, Sumerlin BS (2019) Synthesis of functional and boronic acid-containing aliphatic polyesters via Suzuki coupling. Chem Commun 55:5655–5658. https://doi.org/10.1039/c9cc01975h
Gurak JA Jr, Engle KM (2018) Practical Intermolecular hydroarylation of diverse alkenes via reductive heck coupling. ACS Catal 8:8987–8992. https://doi.org/10.1021/acscatal.8b02717
Touj N, Yaşar S, Özdemir N, Hamdi N, Özdemir İ (2018) Sonogashira cross-coupling reaction catalysed by mixed NHC-Pd-PPh3 complexes under copper free conditions. J Organomet Chem 860:59–71. https://doi.org/10.1016/j.jorganchem.2018.01.017
Price GA, Hassan A, Chandrasoma N, Bogdan AR, Djuric SW, Organ MG (2017) A Supported catalyst for challenging Negishi coupling reactions in flow. Angew Chem Int Ed 56:13347–13350. https://doi.org/10.1002/ange.201708598
Holz J, Pfeffer C, Zuo H, Beierlein D, Richter G, Klemm E, Peters R (2019) In situ generated gold nanoparticles on active carbon as reusable highly efficient catalysts for a C−C stille coupling. Angew Chem Int Ed 58:10330–10334. https://doi.org/10.1002/anie.201902352
Knutson PC, Fredericks HE, Ferreira EM (2018) Synthesis of 1,3-diynes via Cadiot-Chodkiewicz coupling of volatile, in situ generated bromoalkynes. Org Lett 20:6845–6849. https://doi.org/10.1021/acs.orglett.8b02975
Besset T, Poisson T, Pannecoucke X (2014) Access to difluoromethylated alkynes through the Castro-Stephens reaction. Eur J Org Chem 2014:7220–7225. https://doi.org/10.1002/ejoc.201402937
Zhang L, Qing J, Yang P, Wu J (2008) Palladium-catalyzed Hiyama cross-coupling reactions of aryl mesylates. Org Lett 10:4971–4974. https://doi.org/10.1021/ol802049t
Cherney AH, Reisman SE (2014) Pd-catalyzed Fukuyama cross-coupling of secondary organozinc reagents for the direct synthesis of unsymmetrical ketones. Tetrahedron 70:3259–3265. https://doi.org/10.1016/j.tet.2013.11.104
Sun Q, Suzenet F, Guillaumet G (2012) Optimized Liebeskind-Srogl coupling reaction between dihydropyrimidines and tributyltin compounds. Tetrahedron Lett 53:2694–2698. https://doi.org/10.1016/j.tetlet.2012.03.067
Breitenfeld J, Ruiz J, Wodrich MD, Hu X (2013) Bimetallic oxidative addition involving radical intermediates in nickel-catalyzed alkyl–alkyl Kumada coupling reactions. J Am Chem Soc 135:12004–12012. https://doi.org/10.1021/ja4051923
Akhtar R, Zahoor AF, Parveen B, Suleman M (2019) Development of environmental friendly synthetic strategies for Sonogashira cross coupling reaction: An update. Synth Commun 49:167–192. https://doi.org/10.1080/00397911.2018.1514636
Yousaf M, Zahoor AF, Akhtar R, Ahmad M, Naheed S (2019) Development of green methodologies for Heck, Chan-Lam, Stille and Suzuki cross-coupling reactions. Mol Divers 23:1–19. https://doi.org/10.1007/s11030-019-09988-7
Munir I, Zahoor AF, Rasool N, Naqvi SAR, Zia KM, Ahmad R (2019) Synthetic applications and methodology development of Chan-Lam coupling: a review. Mol Divers 23:215–259. https://doi.org/10.1007/s11030-018-9870-z
Sarker SD, Nahar L (2012) Natural products isolation. In: Sarker SD, Nahar L (eds), 3rd edn. Springer, Humana Press, pp 1–25
Matos K, Soderquist JA (1998) Alkylboranes in the Suzuki−Miyaura coupling: Stereochemical and mechanistic studies. J Org Chem 63:461–470. https://doi.org/10.1021/jo971681s
Littke AF, Dai C, Fu GC (2000) Versatile catalysts for the Suzuki cross-coupling of arylboronic acids with aryl and vinyl halides and triflates under mild conditions. J Am Chem Soc 122:4020–4028. https://doi.org/10.1021/ja0002058
Molander GA, Rivero MR (2002) Suzuki cross-coupling reactions of potassium alkenyltrifluoroborates. Org Lett 4:107–109. https://doi.org/10.1021/ol0169729
Rossi R, Carpita A, Quirici MG (1981) Dienic sex pheromones: Stereoselective syntheses of (7E, 9Z)-7, 9-dodecadien-1-yl acetate, (E)-9, 11-dodecadien-1-yl acetate, and of (9Z), 11E)-9, 11-tetradecadien-1-yl acetate by palladium-catalyzed reactions. Tetrahedron 37:2617–2623. https://doi.org/10.1016/S0040-4020(01)98966-5
Vetter W (2012) The alkaloids: chemistry and biology. In: Knӧlker HJ (ed), Elsevier, Academic Press, pp 211–276
Schwalm CS, de Castro IB, Ferrari J, de Oliveira FL, Aparicio R, Correia CRD (2012) Synthesis of pentabromopseudilin and other arylpyrrole derivatives via Heck arylations. Tetrahedron Lett 53:1660–1663. https://doi.org/10.1016/j.tetlet.2012.01.086
Kum DY, Nazari M, McPhail KL, Cooper CS, Suyama TL (2017) Two-step total synthesis of an anti-MRSA and myosin-inhibiting marine natural product pentabromopseudilin via Suzuki-Miyaura coupling of a MIDA boronate ester. Tetrahedron Lett 58:3374–3376. https://doi.org/10.1016/j.tetlet.2017.07.057
Saurav K, Zhang W, Saha S, Zhang H, Li S, Zhang Q, Wu Z, Zhang G, Zhu Y, Verma G (2014) In silico molecular docking, preclinical evaluation of spiroindimicins AD, lynamicin A and D isolated from deep marine sea derived Streptomyces sp. SCSIO 03032. Interdiscip Sci Comput Life Sci 6:187–196. https://doi.org/10.1007/s12539-013-0200-y
McArthur KA, Mitchell SS, Tsueng G, Rheingold A, White DJ, Grodberg J, Lam KS, Potts BC (2008) Lynamicins A-E, chlorinated bisindole pyrrole antibiotics from a novel marine actinomycete. J Nat Prod 71:1732–1737. https://doi.org/10.1021/np800286d
Sigala I, Ganidis G, Thysiadis S, Zografos AL, Giannakouros T, Sarli V, Nikolakaki E (2017) Lynamicin D an antimicrobial natural product affects splicing by inducing the expression of SR protein kinase 1. Bioorg Med Chem 25:1622–1629. https://doi.org/10.1016/j.bmc.2017.01.025
Bauer I, Knölker HJ (2011) Alkaloid Synthesis, In: Knӧlker HJ (ed), Springer, Berlin, Heidelberg, pp 203–253
Kumar Goud Bhatthula B, Reddy Kanchani J, Reddy Arava V, Subha MCS (2019) Total synthesis of carbazole alkaloids. Tetrahedron 75:874–887. https://doi.org/10.1016/j.tet.2019.01.003
Pousset JL, Martin MT, Jossang A, Bodo B (1995) Isocryptolepine from Cryptolepis sanguinolenta. Phytochemistry 39:735–736. https://doi.org/10.1016/0031-9422(94)00925-J
Sharaf MH, Schiff PL Jr, Tackie AN, Phoebe CH Jr, Martin GE (1996) Two new indoloquinoline alkaloids from Cryptolepis sanguinolenta: cryptosanguinolentine and cryptotackieine. J Heterocycl Chem 33:239–243. https://doi.org/10.1002/jhet.5570330204
Parvatkar PT, Parameswaran PS, Tilve SG (2011) Isolation, biological activities and synthesis of indoloquinoline alkaloids: cryptolepine, isocryptolepine and neocryptolepine. Curr Org Chem 15:1036–1057. https://doi.org/10.1002/chin.201138237
Håheim KS, Helgeland ITU, Lindbäck E, Sydnes MO (2019) Mapping the reactivity of the quinoline ring-system–Synthesis of the tetracyclic ring-system of isocryptolepine and regioisomers. Tetrahedron 75:2949–2957. https://doi.org/10.1016/j.tet.2019.04.026
Griffiths-Jones CM, Knight DW (2011) A total synthesis of (±)-α-cyclopiazonic acid using a cationic cascade as a key step. Tetrahedron 67:8515–8528. https://doi.org/10.1016/j.tet.2011.08.094
Shi S, Yuan K, Jia Y (2019) Seven-step total synthesis of α-cyclopiazonic acid. Chin Chem Lett 31:401–403. https://doi.org/10.1016/j.cclet.2019.06.048
Siitonen V, Claesson M, Patrikainen P, Aromaa M, Mäntsälä P, Schneider G, Metsä-Ketelä M (2012) Identification of late-stage glycosylation steps in the biosynthetic pathway of the anthracycline nogalamycin. ChemBioChem 13:120–128. https://doi.org/10.1002/cbic.201100637
Peng R, VanNieuwenhze MS (2017) Construction of the DEF-ring system of nogalamycin and menogaril via an efficient Suzuki-Miyaura reaction. Tetrahedron Lett 58:2236–2239. https://doi.org/10.1016/j.tetlet.2017.04.085
Cao BC, Wu GJ, Yu F, He YP, Han FS (2018) A total synthesis of (−)-hamigeran B and (−)-4-bromohamigeran B. Org Lett 20:3687–3690. https://doi.org/10.1021/acs.orglett.8b01490
Taber DF, Tian W (2008) Synthesis of (−)-hamigeran B. J Org Chem 73:7560–7564. https://doi.org/10.1021/jo8010683
Kuwata K, Fujita R, Hanaya K, Higashibayashi S, Sugai T (2018) Formal total synthesis of (−)-hamigeran B from a chemo-enzymatically prepared building block with quaternary chiral center. Tetrahedron 74:740–745. https://doi.org/10.1016/j.tet.2017.12.054
Li C, Li CJ, Ma J, Chen FY, Li L, Wang XL, Ye F, Zhang DM (2018) Magterpenoids A-C, three polycyclic meroterpenoids with PTP1B inhibitory activity from the Bark of Magnolia officinalis var. biloba. Org Lett 20:3682–3686. https://doi.org/10.1021/acs.orglett.8b01476
Kumar D, Kumar V, Salam A, Khan T (2019) A silica-gel accelerated [4 + 2] cycloaddition-based biomimetic approach towards the first total synthesis of magterpenoid C. Tetrahedron Lett 60:151137–151141. https://doi.org/10.1016/j.tetlet.2019.151137
Di L, Shi YN, Yan YM, Jiang LP, Hou B, Wang XL, Zuo ZL, Chen YB, Yang CP, Cheng YX (2015) Nonpeptide small molecules from the insect Aspongopus chinensis and their neural stem cell proliferation stimulating properties. RSC Adv 5:70985–70991. https://doi.org/10.1039/c5ra12920f
Bendre AD, Patil VP, Terdale SS, Kodam KM, Waghmode SB (2020) A simple, efficient and green approach for the synthesis of palladium nanoparticles using Oxytocin: application for ligand free Suzuki reaction and total synthesis of aspongpyrazine A. J Organomet Chem 909:121093. https://doi.org/10.1016/j.jorganchem.2019.121093
Kaniwa K, Ohtsuki T, Yamamoto Y, Ishibashi M (2006) Kehokorins A-C, novel cytotoxic dibenzofurans isolated from the myxomycete Trichia favoginea var. persimilis. Tetrahedron Lett 47:1505–1508. https://doi.org/10.1016/j.tetlet.2006.01.012
Fujiwara K, Motousu R, Sato D, Kondo Y, Akiba U, Suzuki T, Tokiwano T (2019) Total synthesis of kehokorins A and B. Tetrahedron Lett 60:1299–1301. https://doi.org/10.1016/j.tetlet.2019.04.011
Oguri Y, Watanabe M, Ishikawa T, Kamada T, Vairappan CS, Matsuura H, Kaneko K, Ishii T, Suzuki M, Yoshimura E, Nogata Y (2017) New marine antifouling compounds from the Red Alga Laurencia sp. Mar Drugs 15:267–276. https://doi.org/10.3390/md15090267
Rivas A, Alvarez R, de Lera AR (2019) Stereocontrolled synthesis and configurational assignment of (R)-all-trans-11, 12-dihydro-3-hydroxyretinol. Tetrahedron Lett 60:150972. https://doi.org/10.1016/j.tetlet.2019.150972
Lam YT, Hensens OD, Ransom R, Giacobbe RA, Polishook J, Zink D (1996) L-755,807, A new non-peptide bradykinin binding inhibitor from an endophytic Microsphaeropsis sp. Tetrahedron 52:1481–1486. https://doi.org/10.1016/0040-4020(95)00985-X
Tanaka K III, Honma Y, Yamaguchi C, Aoki L, Saito M, Suzuki M, Arahata K, Kinoshita K, Koyama K, Kobayashi K, Kogen H (2019) Total synthesis, stereochemical assignment, and biological evaluation of L-755,807. Tetrahedron 75:1085–1097. https://doi.org/10.1016/j.tet.2019.01.020
Kim H, Yang I, Patil RS, Kang S, Lee J, Choi H, Kim MS, Nam SJ, Kang H (2014) Anithiactins A-C, modified 2-phenylthiazoles from a mudflat-derived Streptomyces sp. J Nat Prod 77:2716–2719. https://doi.org/10.1021/np500558b
Fu P, MacMillan JB (2015) Thiasporines A-C, thiazine and thiazole derivatives from a marine-derived Actinomycetospora chlora. J Nat Prod 78:548–551. https://doi.org/10.1021/np500929z
Vaaland IC, Lindbäck E, Sydnes MO (2019) Total synthesis of anithiactins AC and thiasporine A. Tetrahedron Lett 60:610–612. https://doi.org/10.1016/j.tetlet.2019.01.038
Kwon YJ, Sohn MJ, Zheng CJ, Kim WG (2007) Fumimycin: a peptide deformylase inhibitor with an unusual skeleton produced by Aspergillus fumisynnematus. Org Lett 9:2449–2451. https://doi.org/10.1021/ol0703231
Zaghouani M, Bögeholz LA, Mercier E, Wintermeyer W, Roche SP (2019) Total synthesis of (±)-fumimycin and analogues for biological evaluation as peptide deformylase inhibitors. Tetrahedron 75:3216–3230. https://doi.org/10.1016/j.tet.2019.03.037
Djakovitch L, Rollet P (2004) Sonogashira cross-coupling reactions catalysed by heterogeneous copper-free Pd-zeolites. Tetrahedron Lett 45:1367–1370. https://doi.org/10.1016/j.tetlet.2003.12.077
Li JH, Zhang XD, Xie YX (2005) Efficient and copper-free Pd (OAc) 2/DABCO-catalyzed Sonogashira cross-coupling reaction. Synthesis 2005:804–808. https://doi.org/10.1055/s-2005-861805
Li JH, Liang Y, Xie YX (2005) Efficient palladium-catalyzed homocoupling reaction and Sonogashira cross-coupling reaction of terminal alkynes under aerobic conditions. J Org Chem 70:4393–4396. https://doi.org/10.1021/jo0503310
Ljungdahl T, Bennur T, Dallas A, Emtenäs H, Mårtensson J (2008) Two competing mechanisms for the copper-free Sonogashira cross-coupling reaction. Organometallics 27:2490–2498. https://doi.org/10.1021/om800251s
Zhou M, Miao MM, Du G, Li XN, Shang SZ, Zhao W, Liu ZH, Yang GY, Che CT, Hu QF, Gao XM (2014) Aspergillines A-E, highly oxygenated hexacyclic indole–tetrahydrofuran–tetramic acid derivatives from Aspergillus versicolor. Org Lett 16:5016–5019. https://doi.org/10.1021/ol502307u
Nakhla MC, Wood JL (2017) Total Synthesis of (±)-Aspergilline A. J Am Chem Soc 139:18504–18507. https://doi.org/10.1021/jacs.7b12570
Uka V, Moore GG, Arroyo-Manzanares N, Nebija D, De Saeger S, Diana Di Mavungu J (2017) Unravelling the diversity of the cyclopiazonic acid family of mycotoxins in Aspergillus flavus by UHPLC Triple-TOF HRMS. Toxins 9:35. https://doi.org/10.3390/toxins9010035
Nakhla MC, Weeks KN, Villalobos MN, Wood JL (2018) Total synthesis of cyclopiamide A and speradine E. Tetrahedron 74:5085–5088. https://doi.org/10.1016/j.tet.2018.02.039
James KJ, Crowley J, Duphard J, Lehane M, Furey A (2007) Phycotoxins: chemistry and biochemistry. In: Botana LM (ed), Blackwell Publishing, Ames, Iowa, USA, pp 141–158
Metcalf JS, Codd GA (2012) Ecology of cyanobacteria II: their diversity in space and time. In: Whitton BA (ed), Springer, pp 651–665
James KJ, Dauphard J, Crowley J, Furey A (2008) Seafood and freshwater toxins. Pharmacology, physiology and detection. In: Botana LM (ed), CRC Press, Boca Raton, pp 809–822
Thomas P, Stephens M, Wilkie G, Amar M, Lunt GG, Whiting P, Gallagher T, Pereira E, Alkondon M, Albuquerque EX, Wonnacott S (1993) (+)-Anatoxin-a is a potent agonist at neuronal nicotinic acetylcholine receptors. J Neurochem 60:2308–2311. https://doi.org/10.1111/j.1471-4159.1993.tb03519.x
Addante-Moya LG, Agulló C, Quiñones-Reyes G, Mercader JV, Abad-Fuentes A, Abad-Somovilla A (2018) A unified approach to the synthesis of both enantiomers of anatoxin-a and homoanatoxin-a cyanotoxins. Tetrahedron 74:5022–5031. https://doi.org/10.1016/j.tet.2018.04.057
Bohlmann F, Zdero C, King RM, Robinson H (1981) The first acetylenic monoterpene and other constituents from senecio clevelandii. Phytochemistry 20:2425–2427. https://doi.org/10.1016/S0031-9422(00)82681-3
Rossi R, Bellina F, Biagetti M (1999) A concise and efficient novel synthesis of cleviolide. Synth Commun 29:3415–3420. https://doi.org/10.1080/00397919908085969
Cheval NP, Hoffmann B, Dikova A, Sirindil F, Bertus P, Blanc A, Weibel JM, Pale P (2018) Vinyl nosylates as partner in copper and silver co-catalyzed Sonogashira cross-coupling reactions. Tetrahedron 74:7111–7119. https://doi.org/10.1016/j.tet.2018.10.017
Sarotti AM (2018) Structural revision of two unusual rhamnofolane diterpenes, curcusones I and J, by means of DFT calculations of NMR shifts and coupling constants. Org Biomol Chem 16:944–950. https://doi.org/10.1039/c7ob02916k
Wang HB, Wang XY, Liu LP, Qin GW, Kang TG (2015) Tigliane diterpenoids from the euphorbiaceae and thymelaeaceae families. Chem Rev 115:2975–3011. https://doi.org/10.1021/cr200397n
Li Y, Wei M, Dai M (2017) Gold catalysis-facilitated rapid synthesis of the daphnane/tigliane tricyclic core. Tetrahedron 73:4172–4177. https://doi.org/10.1016/j.tet.2016.11.005
Mitani H, Matsuo T, Kodama T, Nishikawa K, Tachi Y, Morimoto Y (2016) Total synthesis of nitropyrrolins A, B, and D. Tetrahedron 72:7179–7184. https://doi.org/10.1016/j.tet.2016.09.058
Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR (2012) Marine natural products. Nat Prod Rep 29:144–222. https://doi.org/10.1039/C2NP00090C
Ding XB, Furkert DP, Brimble MA (2017) General synthesis of the nitropyrrolin family of natural products via Regioselective CO2-mediated alkyne hydration. Org Lett 19:5418–5421. https://doi.org/10.1021/acs.orglett.7b02687
Raju R, Piggott AM, Diaz LXB, Khalil Z, Capon RJ (2010) Heronapyrroles A− C: farnesylated 2-nitropyrroles from an Australian marine-derived Streptomyces sp. Org Lett 12:5158–5161. https://doi.org/10.1021/ol102162d
Schmidt J, Khalil Z, Capon RJ, Stark CB (2014) Heronapyrrole D: A case of co-inspiration of natural product biosynthesis, total synthesis and biodiscovery. Beilstein J Org Chem 10:1228–1232. https://doi.org/10.3762/bjoc.10.121
Matsuo T, Hashimoto S, Nishikawa K, Kodama T, Kikuchi S, Tachi Y, Morimoto Y (2015) Total synthesis and complete stereochemical assignment of heronapyrroles A and B. Tetrahedron Lett 56:5345–5348. https://doi.org/10.1016/j.tetlet.2015.07.0940040-4039
Ding XB, Brimble MA, Furkert DP (2018) Reactivity of 2-nitropyrrole systems: development of improved synthetic approaches to nitropyrrole natural products. J Org Chem 83:12460–12470. https://doi.org/10.1021/acs.joc.8b01692
Zhang Y, Danishefsky SJ (2010) Total Synthesis of (()-aplykurodinone-1: traceless stereochemical guidance. J Am Chem Soc 132:9567–9569. https://doi.org/10.1021/ja1035495
Peixoto PA, Jean A, Maddaluno J, De Paolis M (2013) Formal enantioselective synthesis of aplykurodinone-1. Angew Chem Int Ed 125:7109–7111. https://doi.org/10.1002/anie.201301465
Tao C, Zhang J, Chen X, Wang H, Li Y, Cheng B, Zhai H (2017) Formal synthesis of (±)-aplykurodinone-1 through a HeteroPauson−Khand Cycloaddition approach. Org Lett 19:1056–1059. https://doi.org/10.1021/acs.orglett.7b00068
Liu G, Mei G, Chen R, Yuan H, Yang Z, Li CC (2014) Total synthesis of aplykurodinone-1. Org Lett 16:4380–4383. https://doi.org/10.1021/ol502220r
Wu YZ, Zhang HW, Sun ZH, Dai JG, Hu YC, Li R, Lin PC, Xia GY, Wang LY, Qiu BL, Zhang JF (2018) Bysspectin A, an unusual octaketide dimer and the precursor derivatives from the endophytic fungus Byssochlamys spectabilis IMM0002 and their biological activities. Eur J Med Chem 145:717–725. https://doi.org/10.1016/j.ejmech.2018.01.030
Yang F, Liu X, Shi X, Wang Y, Hu G, Lin S, Jiao X, Xie P (2019) Total synthesis of bysspectin A. Tetrahedron 75:3101–3107. https://doi.org/10.1016/j.tet.2019.04.047
Zhao J, Bane S, Snyder JP, Hu H, Mukherjee K, Slebodnick C, Kingston DG (2011) Design and synthesis of simplified taxol analogs based on the T-Taxol bioactive conformation. Bioorg Med Chem 19:7664–7678. https://doi.org/10.1016/j.bmc.2011.10.010
Schäckermann JN, Lindel T (2018) Macrocyclic core of Salarin C: synthesis and oxidation. Org Lett 20:6948–6951. https://doi.org/10.1021/acs.orglett.8b03094
Bishara A, Rudi A, Aknin M, Neumann D, Ben-Califa N, Kashman Y (2008) Salarin C, a new cytotoxic sponge-derived nitrogenous macrolide. Tetrahedron Lett 49:4355–4358. https://doi.org/10.1016/j.tetlet.2008.05.038
Schäckermann JN, Lindel T (2017) Synthesis and photooxidation of the trisubstituted oxazole fragment of the marine natural product Salarin C. Org Lett 19:2306–2309. https://doi.org/10.1021/acs.orglett.7b00845
Silveira N, Sandjo LP, Biavatti MN (2018) Spilanthol-containing products: a patent review (1996–2016). Trends Food Sci Technol 74:107–111. https://doi.org/10.1016/j.tifs.2018.02.012
Alonso LG, Yamane LT, de Freitas-Blanco VS, Novaes LF, Franz-Montan M, de Paula E, Rodrigues MV, Rodrigues RA, Pastre JC (2018) A new approach for the total synthesis of spilanthol and analogue with improved anesthetic activity. Tetrahedron 74:5192–5199. https://doi.org/10.1016/j.tet.2018.06.034
Dai C, Fu GC (2001) The first general method for palladium-catalyzed Negishi cross-coupling of aryl and vinyl chlorides: use of commercially available Pd(P(t-Bu3)2 as a catalyst. J Am Chem Soc 123:2719–2724. https://doi.org/10.1021/ja003954y
Liu Z, Dong N, Xu M, Sun Z, Tu T (2013) Mild Negishi cross-coupling reactions catalyzed by acenaphthoimidazolylidene palladium complexes at low catalyst loadings. J Org Chem 78:7436–7444. https://doi.org/10.1021/jo400803s
McCann LC, Hunter HN, Clyburne JA, Organ MG (2012) Higher-order zincates as transmetalators in alkyl-alkyl Negishi cross-coupling. Angew Chem Int Ed 51:7024–7027. https://doi.org/10.1002/anie.201203547
Haas D, Hammann JM, Greiner R, Knochel P (2016) Recent developments in Negishi cross-coupling reactions. ACS Catal 6:1540–1552. https://doi.org/10.1021/acscatal.5b02718
Pogorevc D, Tang Y, Hoffmann M, Zipf G, Bernauer HS, Popoff A, Steinmetz H, Wenzel SC (2019) Biosynthesis and heterologous production of argyrins. ACS Synth Biol 8:1121–1133. https://doi.org/10.1021/acssynbio.9b00023
Smolyar IV, Yudin AK, Nenajdenko VG (2019) Heteroaryl rings in peptide macrocycles. Chem Rev 119:10032–10240. https://doi.org/10.1021/acs.chemrev.8b00789
Stempel E, Kaml RFX, Budisa N, Kalesse M (2018) Painting argyrins blue: Negishi cross-coupling for synthesis of deep-blue tryptophan analogue β-(1-azulenyl)-˪ alanine and its incorporation into argyrin. Bioorg Med Chem 26:5259–5269. https://doi.org/10.1016/j.bmc.2018.03.037
Davin LB, Lewis NG (2003) An historical perspective on lignan biosynthesis: monolignol, allylphenol and hydroxycinnamic acid coupling and downstream metabolism. Phytochem Rev 2:257. https://doi.org/10.1023/B:PHYT.0000046175.83729.b5
Saleem M, Kim HJ, Ali MS, Lee YS (2005) An update on bioactive plant lignans. Nat Prod Rep 22:696–716. https://doi.org/10.1039/B514045P
Kc S, Basnet P, Thapa S, Shrestha B, Giri R (2018) Ni-catalyzed regioselective dicarbofunctionalization of unactivated olefins by tandem cyclization/cross-coupling and application to the concise synthesis of lignan natural products. J Org Chem 83:2920–2936. https://doi.org/10.1021/acs.joc.8b00184
Tanaka C, Tanaka J, Bolland RF, Marriott G, Higa T (2006) Seragamides A-F, new actin targeting depsipeptides from the sponge Suberites japonicus Thiele. Tetrahedron 62:3536–3542. https://doi.org/10.1016/j.tet.2006.01.099
Lang JH, Lindel T (2019) Synthesis of the polyketide section of seragamide A and related cyclodepsipeptides via Negishi cross coupling. Beilstein J Org Chem 15:577–583. https://doi.org/10.3762/bjoc.15.53
Taishi T, Takechi S, Mori S (1998) First total synthesis of (±)-Stachyflin. Tetrahedron Lett 39:4347–4350. https://doi.org/10.1016/S0040-4039(98)00769-2
Haut FL, Speck K, Wildermuth R, Möller K, Mayer P, Magauer T (2018) A Negishi cross-coupling reaction enables the total synthesis of (+)-stachyflin. Tetrahedron 74:3348–3357. https://doi.org/10.1016/j.tet.2018.02.048
Wildermuth R, Speck K, Haut FL, Mayer P, Karge B, Brönstrup M, Magauer T (2017) A modular synthesis of tetracyclic meroterpenoid antibiotics. Nat Commun 8:1–9. https://doi.org/10.1038/s41467-017-02061-7
Moore BS, Chen JL, Patterson GML, Moore RE, Brinen LS, Kato Y, Clardy J (1990) [7.7]Paracyclophanes from blue-green algae. J A Chem Soc 112:4061–4063. https://doi.org/10.1021/ja00166a066
Moore BS, Chen JL, Patterson GM, Moore RE (1992) Structures of cylindrocyphanes a-f. Tetrahedron 48:3001–3006. https://doi.org/10.1016/S0040-4020(01)92244-6
Nakamura H, Schultz EE, Balskus EP (2017) A new strategy for aromatic ring alkylation in cylindrocyclophane biosynthesis. Nat Chem Biol 13:916–921. https://doi.org/10.1038/nchembio.2421
Berthold B, Breit B (2018) Total synthesis of (−)-cylindrocyclophane F: A Yardstick for probing new catalytic C–C bond forming methodologies. Chem Eur 24:16770–16773. https://doi.org/10.1002/chem.201804585
Loska R, Volla CMR, Vogel P (2008) Iron-catalyzed Mizoroki-Heck cross-coupling reaction with styrenes. Adv Synth Catal 350:2859–2864. https://doi.org/10.1002/adsc.200800662
Islam MS, Nahra F, Tzouras NV, Barakat A, Cazin CSJ, Nolan SP, Al-Majid AM (2019) Mizoroki-Heck cross-coupling of acrylate derivatives with aryl halides catalyzed by palladate pre-catalysts. Eur J Inorg Chem 2019:4695–4699. https://doi.org/10.1002/ejic.201901075
Inuki S, Oishi S, Fujii N, Ohno H (2008) Total synthesis of (±)-lysergic acid, lysergol, and isolysergol by palladium-catalyzed domino cyclization of amino allenes bearing a bromoindolyl group. Org Lett 10:5239–5242. https://doi.org/10.1021/ol8022648
Milde B, Pawliczek M, Jones PG, Werz DB (2017) Enantioselective total synthesis of (+)-lysergol: a formal anti-carbopalladation/heck cascade as the key step. Org Lett 19:1914–1917. https://doi.org/10.1021/acs.orglett.7b00675
Angle SR, Boyce JP (1995) A stereoselective formal synthesis of (+)-(γ)-lycorane. Tetrahedron Lett 36:6185–6188. https://doi.org/10.1016/0040-4039(95)01245-D
Monaco A, Szulc BR, Rao ZX, Barniol-Xicota M, Sehailia M, Borges BM, Hilton ST (2017) A short total synthesis of (±)-lycorane by a sequential intramolecular acylal cyclisation (IAC) and intramolecular heck addition reaction. Chem Eur J 23:4750–4755. https://doi.org/10.1002/chem.201700143
Kumar MMK, Naik JD, Satyavathi K, Ramana H, Varma PR, Nagasree KP, Smitha D, Rao DV (2014) Denigrins A-C: new antitubercular 3,4-diarylpyrrole alkaloids from Dendrilla nigra. Nat Prod Res 28:888–894. https://doi.org/10.1080/14786419.2014.891112
Karak M, Oishi T, Torikai K (2018) Synthesis of anti-tubercular marine alkaloids denigrins A and B. Tetrahedron Lett 59:2800–2803. https://doi.org/10.1016/j.tetlet.2018.06.013
Ayers S, Graf TN, Adcock AF, Kroll DJ, Matthew S, Carcache de Blanco EJ, Shen Q, Swanson SM, Wani MC, Pearce CJ, Oberlies NH (2011) Resorcylic acid lactones with cytotoxic and NF-κB inhibitory activities and their structure–activity relationships. J Nat Prod 74:1126–1131. https://doi.org/10.1021/np200062x
Doda SR, Raghavendar A, Haridasyam SB, Putta CS, Kadari S (2019) Asymmetric total synthesis of filamentous fungi related resorcylic acid lactones 7-epi-zeaenol and zeaenol. Heterocycl Commun 25:78–84. https://doi.org/10.1515/hc-2019-0015
Leung J, Giraudat J (1998) Abscisic acid signal transduction. Annu Rev Plant Biol 49:199–222. https://doi.org/10.1146/annurev.arplant.49.1.199
Dumonteil G, Hiebel MA, Berteina-Raboin S (2018) Solvent-free mizoroki-heck reaction applied to the synthesis of abscisic acid and some derivatives. Catalysts 8:115. https://doi.org/10.3390/catal8030115
Gribanov PS, Golenko YD, Topchiy MA, Minaeva LI, Asachenko AF, Nechaev MS (2018) Stannylation of aryl halides, stille cross-coupling, and one-pot two-step stannylation/stille cross-coupling reactions under solvent-free conditions. Eur J Org Chem 2018:120–125. https://doi.org/10.1002/ejoc.201701463
Huang X, Yao F, Wei T, Cai M (2017) A highly efficient and recyclable Pd(PPh3)4 /PEG-400 system for Stille crosscoupling reactions of organostannanes with aryl bromides. J Chem Res 41:547–550. https://doi.org/10.3184/174751917X15045169836226
Ishigami K, Yamamoto M, Watanabe H (2015) Synthesis and revision of the relative configuration of glabramycin B. Tetrahedron Lett 56:6290–6293. https://doi.org/10.1016/j.tetlet.2015.09.149
Yamamoto M, Ishigami K, Watanabe H (2017) First total synthesis of glabramycin B and revision of its relative configuration. Tetrahedron 73:3271–3280. https://doi.org/10.1016/j.tet.2017.04.061
Ueno K, Furumoto T, Umeda S, Mizutani M, Takikawa H, Batchvarova R, Sugimoto Y (2014) First total synthesis of glabramycin B and revision of its relative configuration. Phytochemistry 108:122–128. https://doi.org/10.1016/j.tet.2017.04.061
Woo S, McErlean CS (2019) Total synthesis and stereochemical confirmation of heliolactone. Org Lett 21:4215–4218. https://doi.org/10.1021/acs.orglett.9b01402
Huang B, Xiao CJ, Huang ZY, Tian XY, Cheng X, Dong X, Jiang B (2014) Hispidanins A−D: four new asymmetric dimeric diterpenoids from the rhizomes of Isodon hispida. Org Lett 16:3552–3555. https://doi.org/10.1021/ol501417k
Oyama H, Sassa T, Ikeda M (1978) Structures of new plant growth inhibitors, trans-and cis-resorcylide. Agric Biol Chem 42:2407–2409. https://doi.org/10.1271/bbb1961.42.2407
Barrow CJ (1997) New macrocyclic lactones from a Penicillium species. J Nat Prod 60:1023–1025. https://doi.org/10.1021/np970200x
Deng H, Cao W, Liu R, Zhang Y, Liu B (2017) Asymmetric total synthesis of hispidanin A. Angew Chem Int Ed 56:5849–5852. https://doi.org/10.1002/ange.201700958
Luo Y, Yin X, Dai M (2019) Total synthesis of trans-resorcylide via macrocyclic stille carbonylation. J Antibiot 72:482–485. https://doi.org/10.1038/s41429-019-0145-4
Halle MB, Yudhistira T, Lee WH, Mulay SV, Churchill DG (2018) Diels−Alder and stille coupling approach for the short protectinggroup-free synthesis of mycophenolic acid, its phenylsulfenyl and phenylselenyl analogues, and reactive oxygen species (ROS) probing capacity in water. Org Lett 20:3557–3561. https://doi.org/10.1021/acs.orglett.8b01327
Venkanna A, Poornima B, Siva B, Babu BH, Babu KS (2018) Towards the total synthesis of schisandrene: stereoselective synthesis of the dibenzocyclooctadiene lignan core. Synlett 29:908–911. https://doi.org/10.1055/s-0036-1591539
Duquette DC, Jensen T, Stoltz BM (2018) Progress towards the total synthesis of hamigerans C and D: a direct approach to an elaborated 6–7-5 carbocyclic core. J Antibiot 71:263–267. https://doi.org/10.1038/ja.2017.96
Phillips AW, Anketell MJ, Balan T, Lam NY, Williams S, Paterson I (2018) Toward the total synthesis of patellazole B: synthesis of an advanced C1–C25 fragment corresponding to the macrocyclic skeleton. Org Biomol Chem 16:8286–8291. https://doi.org/10.1039/c8ob01621f
Kock M, Fresia M, Jones PG, Lindel T (2019) Synthesis of raputimonoindoles A-C and congeners. Eur J Org Chem 2019:4061–4065. https://doi.org/10.1002/ejoc.201900583
Singh DK, Kim I (2019) Convergent synthesis of diptoindonesin G. Tetrahedron Lett 60:300–301. https://doi.org/10.1016/j.tetlet.2018.12.036
Acknowledgements
The authors are thankful to Government College University, Faisalabad to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tabassum, S., Zahoor, A.F., Ahmad, S. et al. Cross-coupling reactions towards the synthesis of natural products. Mol Divers 26, 647–689 (2022). https://doi.org/10.1007/s11030-021-10195-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10195-6