
Abstract
The favorability of ring closure reactions as per Baldwin rules has gained immense importance recently. This is evident from the current literature such as research articles, reviews, and books that have been published in this area. This review covers the recent applications of 5-endo-dig cyclization in organic synthesis focusing in the last two decades. A variety of 5-membered heterocycles as well as carbocycles could be synthesized via 5-endo-dig cyclization reactions. The important applications of 5-endo-dig cyclization in organic synthesis covering different aspects have been summarized in this review.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
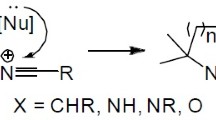
The article published in “Chemical Communication” more than 40 years ago summarized the Baldwin rules [1], that eventually became the most cited article of the journal. This article described a series of guidelines outlining the relative favorability of ring closure reactions. The favorability of ring closure reactions as per Baldwin’s guidelines gained valuable insight into the role of stereo-electronic factors. A cyclization reaction is designated by using three prefixes, the first of which gives the number of atoms forming a ring that can undertake any value ≥ 3. The second one prefix, exo- versus endo-, describes the placement of the bond that needs to be damaged in the cyclization relative to the forming ring. The prefix “exo” indicates that the bond being broken is outside of the new ring, while prefix “endo” indicates that the bond being broken is in the new ring (Fig. 1). The third prefix, tet, trig, and dig, refers back to the hybridization at the ring closure factor, tet (tetrahedral) for sp3, trig (trigonal) for sp2, and dig (digonal) for an sp-hybridized atom [2, 3].

The format of bond broken during ring closure
Radical cyclizations are generally used in the preparation of hetero- and carbocyclic compounds. In dig cyclizations the geometric restrictions are of minor importance, because the reacting orbitals may attack the in-plane π-system of acetylene functionality. This approach with less geometric strain and along with relatively low energy requirement for bending of the alkyne moiety provides many digonal cyclization ways [4, 5].
The alkynes are versatile functional groups that can undergo an array of reactions such as reductions, oxidations, additions, polymerizations, hydrations, metal-catalyzed carbon–carbon bond forming reactions. Carbocyclization of alkynes and alkenes is a highly useful reaction for the preparation of array of heterocyclic and carbocyclic compounds. Carbocyclization of alkynes is of special interest because it permits one to obtain hetero- and carbocycles with higher degree of unsaturation [6].
The scope of 5-endo-dig cyclization is constantly and speedily developing. Among the diversity of compounds, the 5-endo-dig cyclization is utilized for the synthesis of different classes of compounds such as indoles, cuomarins, cyclopentenes, benzofurans, furans. This review deals with fundamental applications of 5-endo-dig cyclization in organic synthesis.
Aims and scope of review
Given the marvelous impact of 5-endo-dig cyclization in the last few years, many publications in the last years covered different aspects. These tremendous applications of 5-endo-dig cyclization encouraged us to summarize the important applications in the field of synthetic organic chemistry.
Indole derivatives
Rode et al. [7] reported a metal-free approach for the synthesis of 2-aroyl-N-tosyl-1H-indole-3 carboxamides 2 with 91–94% yields, through 5-endo-dig carboxamidation reaction of β-(2-aminophenyl)-α,β-ynones 1 with a small excess of the tosyl-isocyanate and catalytic amount of Et3N in THF as solvent (Scheme 1).

Synthesis of 2-aroyl-N-tosyl-1H-indole-3 carboxamides 2
Rodriguez et al. reported the potassium or cesium-base-mediated synthesis of 2-substituted indoles 5 with 72–76% yields by the reaction of 4 with KOtBu or CsOtBu in N-methylpyrrolidinone (NMP) [8]. The precursors 4 were prepared by Sonogashira coupling of the corresponding (2-iodo or 2-bromoanilines) 3 with terminal alkynes in good yields (Scheme 2) [9].

Synthesis of 2-substituted indoles 5
Andreev et al. [10] reported an un-catalyzed thermally induced 5-endo-dig cyclization of amino propargylic alcohols 6 by heating at 160–170 °C for 1–3 h to give 4,5,6,7-tetrahydroindoles 7 with 73–90% yields. Further Dess–Martin periodinane oxidation gives 5,6-dihydro-1H-indol-2(4H)ones 8 (Scheme 3).

Synthesis of 5,6-dihydro-1H-indol-2(4H)ones 8
Naoe et al. reported the gold-catalyzed tandem cyclization of aniline derivatives having conjugated diyne functionality 9 into the indole merged with a seven-membered ring 10 with 86% yield and small amount of six-membered ring 11 (Scheme 4) [11].

Synthesis of indole fused with seven- and six-membered rings
Jalal et al. [12] reported the 1,5-enyne cycloisomerization of (3-methylene)indoline-tethered alkyne derivatives 12 catalyzed by Fe(OTf)3-catalyzed to give highly substituted 3-(1-indenyl)indole derivatives 13 with 80–89% yields (Scheme 5).

Synthesis of 3-(1-indenyl)indole derivatives 13
In a mechanistic explanation, iron coordinates with triple bond to form A which is followed by 5-endo-dig cyclization via intramolecular nucleophilic attack of the exo-cyclic double bond forming an intermediate B. Subsequently, isomerization of B leads to C, which on protonolysis affords the desired product 13 (Scheme 5).
Sharma et al. described the metal-/oxidant-free microwave-assisted synthesis of 3-sulfenylindoles 16 by cascade electrophilic sulfenylation/5-endo-dig cyclization reaction of 2-alkynylaniline 14 with sulfonyl hydrazide 15 in the presence of iodine and p-TsOH at 110 °C in 1,4-dioxane as solvent (Scheme 6) [13].

Synthesis of 3-sulfenylindoles 16
Palimkar et al. [14] described one-pot preparation of 2-substituted indoles 19 in 71–90% yields by palladium-acetate-catalyzed domino Sonogashira coupling/5-endo-dig cyclization of 2-iodo-4-substituted-N-tosylbenzenamine 17 and terminal alkynes 18 with ultrasonic irradiation at room temperature (Scheme 7).

Synthesis of 2-substituted indoles 19
Palimkar et al. described the synthesis of KDR kinase inhibitor generating the indole moiety via domino Sonogashira coupling/5-endo-dig cyclization reaction of o-iodoanilide derivative 20 with terminal alkyne 21 in the presence of Pd(OAc)2 and Bu4NOAc as the base to give indole-chloroquinoline 22 in 80% yield [15]. Hydrolysis of indole-chloroquinoline 22 by acetic acid/water mixture (1:1) as reported in the previous literature [16] gives KDR kinase inhibitor 23 in 93% yield (Scheme 8).

Synthesis of KDR kinase inhibitor 23
Cai et al. described the gold-catalyzed synthesis of fused tetracyclic indoles 25 with 78–89% yield. The diaryl alkyne 24 undergoes 5-endo-dig cyclization to form the α-imino carbenoid and followed by O–H/N–H insertion to give the desired product (Scheme 9) [17].

Synthesis of fused tetracyclic indoles 25
Benzofuran derivatives
The synthesis of highly substituted benzo[b]furan 27 in 74% yield from ynamide 26 via gold(I)-catalyzed 5-endo-dig cyclization and subsequent alkylic oxonium intermediate rearrangement (Scheme 10) was described by Jaimes et al. [18].

Synthesis of benzo[b]furan 27
In a mechanistic explanation, the ynamide 26 was activated by gold species to give E. The alkyl-oxonium intermediate F formed through the nucleophilic attack of the oxygen atom and followed by the generation of stabilized alkyl carbocation and vinylgold(I) derivative G. The carbocation transfers to the most nucleophilic position on benzofuran ring to give more substituted benzo[b]furan 27 and gold species was regenerated (Scheme 10).
Saha et al. [19] reported a one-pot method for the preparation of 2-substituted benzo[b]furans 30 by the reaction of 2-iodophenol 28 with arylacetylenes 29 in the presence of Na2PdCl4 and sodium dodecyl sulfate (SDS) in water at 100 °C.
The Sonogashira coupling occurs through the oxidative addition of 2-iodophenol to Pd0 to afford H. Subsequently, transmetalation with phenylacetylene 29 gives I and then reductive elimination leads to J. Then J undergoes 5-endo-dig cyclization to furnish product 30 (Scheme 11).

Synthesis of 2-substituted benzo[b]furans 30
Mantovani et al. described the mechanochemical cyclization reaction of 2-(arylethynyl)anisoles 31 to give 3-iodobenzofurans 32 in 78–83% yields by solvent-less milling of 31 with equimolar amount of iodine at 15 Hz for 1 h (Scheme 12) [20]. The molecular iodine alkyne interaction leads to intermediate L, which on simultaneous demethylation gives 3-iodobenzofurans 32. In solvent-less mechanochemical conditions, charge stabilizing effects are fewer, while, in solution conditions, charge-separated species as well as iodide would experience considerable interaction by solution molecules.

Synthesis of 3-iodobenzofurans 32
Kim et al. [21] synthesized the 2-substituted 5-hydroxybenzofurans 34 by Pt-catalyzed cascade dienone-phenol rearrangement/5-endo-dig cyclization reaction of quinols 33 having alkyne moiety in dimethoxyethane/methanol (20:1) at 40 °C. The alkyne moiety in M shifts to adjacent carbon atom to give alkynone N. Subsequent 5-endo-dig cyclization furnishes 2-substituted 5-hydroxy benzofuran 34 (Scheme 13).

Synthesis of 2-aryl-5-hydroxybenzofurans 34
Palimkar et al. [22] synthesized benzofuran derivatives 37 via Sonogashira cross-coupling/5-endo-dig cyclization reaction of 35 with phenylacetylene 36 under ultrasonic irradiation using Bu4NOAc and Pd(OAc)2 in CH3CN at room temperature (Scheme 14).

Synthesis of benzofuran derivatives 37
Coumarin derivatives
Majumdar et al. synthesized 3-iodopyrrolocoumarin 40 by Sonogashira coupling [23] of 5-bromo-6-aminocoumarin 38 with different alkynes to afford 39, followed by iodine-induced 5-endo-dig cyclization reaction of 39 in acetonitrile at room temperature (Scheme 15) [24].

Synthesis of 3-iodopyrrolocoumarin derivatives 40
Cyclopentene derivatives
Staben et al. developed a gold(I)-catalyzed 5-endo-dig carbocyclization reaction of dicarbonyl compound 41 onto internal alkyne to give cyclopentene derivative 42 under open-flask conditions in dichloromethane at room temperature (Scheme 16) [25].

Synthesis of cyclopentene derivative 42
Suzuki et al. [26] reported the 5-endo-dig carbocyclization of β-ketoesters 43 onto internal alkyne to afford cyclopentene derivatives 44 by using (S,S)-Box-Ph/Zn(II)/HFIP/Yb-(OTf)3, a four-component system in dichloromethane at ambient temperature (Scheme 17).

Synthesis of cyclopentene derivative 44
Dolaine and Gleason reported the dicobalt octacarbonyl [Co2(CO)8]-mediated synthesis of 5-vinylcyclopentene 46 in good yield from 1,6-enynes 45 under argon atmosphere (Scheme 18).

Synthesis of 5-vinylcyclopentene 46
Mechanistically, the cobalt complex O formed from substrate 45 at room temperature. One CO ligand is lost on heating and alkene coordinates in place of that ligand to form complex P. An allylic C–H oxidative addition generates η3-allylcobalt hydride Q and on reductive elimination a C–C bond between C1 and C5 was generated (complex R). It is followed by reductive elimination to generate a C–H bond to get complex S, then after decomplexation gives cyclopentene 46 [27].
Iwasawa et al. described the annulations of cyclopentene onto α,β-unsaturated ketones. 1,4-Propargylation onto α,β-unsaturated ketone 47 was carried out by reacting t-butyldimethylsilyl triflate and dimethyl sulfide with 47 and in situ generated organoindium reagent (previously reported literature) [28] at room temperature in THF to afford 48 with 76% yield. Followed by 5-endo-dig cyclization reaction of 48 catalyzed by W(CO)5(thf) to afford the cyclopentene derivative 49 in 72% yield (Scheme 19) [29].

Synthesis of cyclopentene derivative 49
Fujino et al. [30] synthesized 1,2-disubstituted cyclopentenes 52 via 5-endo-dig cyclization of homopropargyl-substituted dicarbonyl compounds 50 with organic halides 51 catalyzed by Pd(dba)3 in the presence of XPhos and NaHMDS in DMF at 60 °C.
In mechanistic explanation oxidative addition of ‘Pd’ to aryl halide gives arylpalladium intermediate T, which activates the alkyne functionality of complex U via π-coordination. The complex U undergoes 5-endo-dig cyclization to give vinylpalladium complex V. Followed by reductive elimination to afford phenyl-substituted cyclopentene 52 and palladium complex was regenerated (Scheme 20).

Synthesis of 1,2-disubstituted cyclopentene 52
Dehydropyrrolidine derivatives
French and Diver reported the effective synthetic route to dehydropyrrolidine 54 under gold(I) catalysis. The unsubstituted propargylic acetate 53 undergoes direct 5-endo-dig cyclization under gold(I) catalysis to give 54 (Scheme 21). Mechanistically gold(I) coordinate with alkyne moiety (complex W) and in the absence of substitution on the acetyl group leads to direct 5-endo-dig cyclization to give X which subsequently gives dehydropyrrolidine 54 [31].

Synthesis of dehydropyrrolidine derivative 54
Furan derivatives
Majumdar et al. [32] reported the synthesis of pyrimidine-annulated spiro-dihydrofurans 56 with 69–87% yields by silver-catalyzed 5-endo-dig cyclization of various O-propargylated uracil derivatives 55 in AcOH as solvent at 80 °C.
In a mechanistic explanation initially a π-complex Y is suggested to be formed which rearranges to carbocation Z. The N-atom of pyrimidine ring undergoes 5-endo-dig cyclization by the formation of Mannich species A1. Then water attacks the imino bond to afford the spiro-dihydrofuran 56 (Scheme 22).

Synthesis of pyrimidine-annulated spiro-dihydrofurans 56
Sniady et al. described the synthesis of 2,5-disubstituted 3-halofurans 58 and 59 in 84–89% yields through the electrophilic 5-endo-dig cyclization of 1,4-aryl-butynone 57 with N-bromosuccinimide (NBS) or N-iodosuccinimide (NIS) in acetone as solvent (Scheme 23) [33].

Preparation of 2,5-disubstituted-3-halofurans 58 and 59
Sniady et al. [34] described zinc chloride-catalyzed cycloisomerization of 1,4-disubstituted-butynones 60 in the presence of a catalytic amount of zinc chloride in dichloromethane at room temperature to afford 2,5-disubstituted furans 61 in good yields (Scheme 24).

Preparation of 2,5-disubstituted furans 61
El-Taeb et al. reported the iodine-mediated 5-endo-dig cyclization of 3-alkyne-1,2-diols 62 prepared by previously reported method [35]. The reaction of 62 with 3 equivalents of I2 and NaHCO3 in dichloromethane as solvent at ambient temperature provides β-iodofurans 63 with high yields (Scheme 25) [36].

Synthesis of β-iodofurans 63
Shi et al. described the synthesis of 2,5-dihydrofurans 66 with 69–78% yield through the Pd(II)-catalyzed [4 + 1] cycloaddition of aryl diazoacetates 64 and aryl propargyl alcohols 65 in dichloromethane at room temperature (Scheme 26) [37].

Synthesis of 2,5-dihydrofurans 66
Heteroacene derivatives
Oechsle et al. [38] synthesized sulfur-containing heteroacenes via palladium-catalyzed C–S cross-coupling/5-endo-dig cyclization of bis-alkynes. The Sonogashira coupling of 67 with 4-diphenylaminophenylacetylene 68 furnished monoalkyne 69 and bis-alkyne 70 in 70 and 76% yield, respectively. Consequent reaction of 70 with potassium thioacetate (KSAc) in the presence of DPPF and Pd(OAc)2 gives heteroacene 71 in 73% yield. Monoalkyne 69 was further used to get the zinc reagent by magnetization using iPrMgCl and transmetalation using zinc chloride. Negishi coupling of this reagent with one equivalent of 69 or 0.5 equivalents of 67 furnishes the biphenyl 72 or terphenyl 73 in 90 and 58% yield, respectively. Then the reaction of 72 and 73 with KSAc in the presence of DiPrPF and [Pd(P(oTol)3)2] at 120 °C afforded heteroacene 74 and 75 in 56 and 45% yield, respectively, (Scheme 27).

Synthesis of heteroacenes 71, 74 and 75
Indene derivatives
Khan and Wirth [39] described the preparation of 3-Iodo-1H-indene derivative 77 in good yield by iodine-mediated 5-endo-dig carbocyclization of 2-substituted ethynylmalonates 76 in the presence of I2 and NaH as base in THF. Mechanistically, the base deprotonated the malonate 76 to furnish B1. Then deprotonated malonate 76 undergoes 5-endo-dig cyclization to give 3-Iodo-1H-indene derivatives 77 via intermediate C1 (Scheme 28).

Synthesis of 3-Iodo-1H-indene derivatives 77
Ma et al. [40] reported the preparation of indene derivatives 80 with 75–87% yield via gold(I)-catalyzed domino C–H functionalization/5-endo-dig cyclization reaction of o-alkynylaryl α-diazoesters 78 and arene 79 in dichloromethane at room temperature (Scheme 29).

Synthesis of indene derivatives 80
Indolizine derivatives
Smith et al. [41] described the Pt(II)-catalyzed cyclization of pyridine propargylic alcohols 81 using Cs2CO3 as base to afford indolizinone 82 with 85–95% yield (Scheme 30).

Synthesis of indolizinone 82
Pandya et al. described the silver-promoted preparation of indolizines 85 through tandem oxidative C–H functionalization/5-endo-dig cyclization. The reaction of 2-pyridylacetate 83 with phenylacetylene 84 in the presence of 2 equivalents of Ag2CO3 and KOAc in DMF at room temperature gives indolizines 85 with 84–89% yield.
Mechanistically, one equivalent of Ag2CO3 activates ethyl 2-pyridylacetate 83 via chelation to furnish D1, while second equivalent of Ag2CO3 reacts with phenylacetylene 84 by chelation to give E1. The coupling of two intermediates (D1 and E1) furnishes intermediate F1, which undergoes 5-endo-dig cyclization to give the required product 85 (Scheme 31) [42].

Silver-mediated synthesis of indolizines 85
Kim et al. reported the synthesis of highly substituted indolizines 88 through iodine-promoted 5-endo-dig cyclization of propargylic acetate 87. The propargylic acetate 87 was first synthesized from the reaction of 2-pyridinecarboxaldehyde 86 and 1-alkynyllithium via earlier reported method [43]. Exposure of acetate 87 to iodine in dichloromethane at room temperature undergoes 5-endo-dig cyclization to afford indolizines 88 (Scheme 32) [44].

Iodine mediated synthesis of indolizines 88
Isoxazole derivatives
Sugita et al. [45] reported a gold-catalyzed reaction of O-allyl hydroxamates 89 having an alkyne functionality for the synthesis of 3-hydroxyisoxazoles 90 and isoxazole-3-ones 91. The cyclization of O-allyl hydroxamates 89 in the presence of PicAuCl2 (Pic = 2-picolinate) in (CH2Cl)2 gives 3-hydroxyisoxazoles 90 with 78–86% yield and small amount of isoxazole-3-ones 91.
Mechanistically, the addition of oxygen atom of hydroxamate to Au(III)-activated alkyne moiety through 5-endo-dig pathway gives oxonium intermediate H1. The consequent [3,3] sigmatropic rearrangement of allyl group to C-4 carbon proceeded through conformation I1 to afford intermediate J1, which undergoes an aromatization reaction to furnish the 3-hydroxyisoxazole 90. The rearrangement through conformation K1 gives the intermediate L1, which undergoes an aromatization reaction to afford isoxazole-3-ones 91 (Scheme 33).

Gold-catalyzed synthesis of 3-hydroxyisoxazoles 90 and isoxazole-3-ones 91
Isoxazolines derivatives
Wang and Tsui [46] described the Cu(OTf)2-promoted preparation of trifluoromethylated 4-isoxazolines 93 with 50–65% yield by domino 5-endo-dig cyclization/trifluoromethylation of propargylic N-hydroxylamines 92, using Cu(OTf)2, TMSCF3, KF and AgOAc in DMF at room temperature.
In a mechanistic explanation, the alkyne functionality of 92 was activated using Cu(OTf)2 as a Lewis acid. The 5-endo-dig cyclization was carried out by nucleophilic attack of the oxygen atom onto the triple bond furnishing the 4-cuprated isoxazoline N1. The 4-cuprated isoxazoline N1 reacted with CF3-(generated in situ from TMSCF3 and KF), in the presence of AgOAc to give the extremely reactive trifluoromethylated Cu(III) species O1. Finally reductive elimination forms the C-CF3 bond to give required compound 93 (Scheme 34).

Synthesis of trifluoromethylated 4-isoxazolines 93
N-Fused heterocyclic derivatives
Xiao et al. synthesized N-fused heterocycles 96 through Csp–S coupling/5-endo-dig cyclization. The reaction of 2-mercaptobenzoimidazole 94 and arylethyne 95 in the presence of CuCl, N,Nʹ-dicyclohexylimidazolium chloride (ICy·HCl) and Et3N in toluene at 80 °C afforded N-fused heterocycles 96 with 42–74% yield (Scheme 35).

Synthesis of N-fused heterocycles 96
Mechanistically, precursor P1 formed easily and subsequent oxidative coupling with 94 gives the intermediate Q1, which is then converted into the alkynyl structure R1 through transmetalation. Consequently, the N-fused heterocyclic compound S1 is formed via 5-endo-dig cyclization, followed by Cu+–H+ exchange to furnish the N-fused heterocycle 96 (Scheme 35) [47].
Organogold complexes
Chen et al. [48] reported the ionic organogold complexes 98 with 88–90% yield synthesized through triazole-alkyne 5-endo-dig cyclization by the reaction of substituted triazole 97 with one equivalent of Ph3PAu-SbF6 in dichloromethane at room temperature. Mechanistically, 5-endo-dig cyclization of propargyl triazole 97 gives 5-5-bicyclic intermediate T1, which further rearranges to give organogold complexes 98 through either 1,5-H shift or proton transfer (Scheme 36).

Synthesis of ionic organogold complexes 98
Pyrrole derivatives
Ueda et al. [49] reported the gold(I)-catalyzed preparation of substituted pyrroles 101. The precursor 99 reacted with substituted acetylene 100 in the presence of AgOTf, gold(I) catalyst and KHSO4 in xylene at 140 °C gives substituted pyrrole 101 with 52–85% yield. In mechanistic explanation, gold acetylide is added to oxonium ion U1 to furnish the alkyne adduct V1. The electrophilicity of alkyne moiety of V1 was activated by π-coordination of the gold catalyst to give W1, which undergoes 5-endo-dig cyclization to afford X1. At the end, protonolysis of the carbon–gold bond in X1 and subsequent aromatization furnishes pyrrole 101 (Scheme 37).

Synthesis of substituted pyrroles 101
Bharathiraja et al. [50] described the preparation of pentasubstituted pyrroles 104 through a cascade reaction involving aza-Michael addition, iodocyclization, and oxidative aromatization. The reaction of 1,3-enynes 102 and amines 103 in the presence of molecular iodine and K2CO3 in dichloromethane at room temperature gives pentasubstituted pyrroles 104 with 72–82% yield.
Mechanistically, the intermolecular aza-Michael addition of the amine 103 to the electron-deficient conjugated 1,3-enyne 102 in the presence of iodine furnishes the intermediate Z1, which in the presence of base gives the iodonium intermediate C2. The intermediate C2 undergoes intramolecular cyclization to afford the dihydropyrrole derivative D2. The iodine-promoted oxidative aromatization of D2 gives the required product 104 (Scheme 38).

Synthesis of pentasubstituted pyrroles 104
Queiroz et al. [51] described the ‘Pd’/CuI catalyzed preparation of substituted pyrroles 107 in 33–70% yield by reaction of N-Boc-β-iododehydroamino acid methyl ester 105 and different terminal alkynes 106 using Pd(II) catalyst, Cs2CO3 and CuI in dry DMF at 70 °C. It is one-pot, two-step reaction, in first step Sonogashira coupling gives intermediate E2 and tandem 5-endo-dig cyclization gives target compound 107 (Scheme 39).

Synthesis of pyrrole derivatives 107
Yan et al. [52] reported the preparation of 2,3-dihydro-1H-pyrrolizines 109 with 84–96% yield having electron-withdrawing groups at 5-position via gold-catalyzed 5-endo-dig cyclization reaction of azidoenynes 108 in toluene at 80 °C (Scheme 40).

Synthesis of 2,3-dihydro-1H-pyrrolizines 109
Gorin et al. [53] reported the preparation of substituted pyrroles 111 with 76–88% yield via gold(I)-catalyzed reaction of homopropargyl azides 110 in dichloromethane at 35 °C (Scheme 41). Herein gold(I) catalyst plays dual role, to activate the alkyne for nucleophilic addition and to shift the electron density into electron-deficient π-system.

Synthesis of 2,5-disubstituted pyrroles 111
Selenophene derivatives
Pistoia et al. [54] reported the iodine-mediated synthesis of 3-iodo-selenophenes 113 through nucleophilic cyclization of selenoenynes 112 in the presence of iodine and nucleophile in dichloromethane. Initial iodine activates carbon–carbon triple bond of selenoenynes 112 and subsequent nucleophilic attack of the selenium atom onto activated iodonium intermediate F2 affords the salt G2. The elimination of the alkyl group through SN2 displacement by iodide anion, present in the reaction mixture, gives the dihydroselenophene H2. The aromatization of selenophene provides allylic cation I2, which trapped by a nucleophile to give selenophene 113 (Scheme 42).

Synthesis of 3-iodo-selenophenes 113
Spiro-compounds
Adler et al. described cerium(IV) ammonium nitrate (CAN) promoted 5-endo-dig cyclization of α-amino allenylphosphonates 114 using CAN at room temperature to give spirodienones 115 with 80–84% yield. Mechanistically, the para-methoxybenzyl ring oxidized to cyclohexadienone carbocation J2 and subsequent 5-endo-dig cyclization to give iminium ion K2. The nucleophilic addition of water to K2 and subsequent prototropy transforms the phosphonate moiety into a leaving group to give the lactam 115 (Scheme 43) [55].

Synthesis of spirodienones 115
Wu et al. [56] described the gold-catalyzed intramolecular dearomatization reaction of naphthols 116 using Ph3PAuCl and AgOMs in dichloromethane at room temperature to afford spirocarbocycles 117 via 5-endo-dig cyclization with excellent yield. In mechanistic explanation, the in situ-generated cationic gold(I) complex activated the C–C triple bond in 116, resulting in 5-endo-dig cyclization with the help of parallel deprotonation by the counter anion MsO−, yielding the spirocyclic gold intermediate O2. Protodemetallation of O2 via in situ-generated MsOH gives the desired spirocarbocyclic product 117 (Scheme 44).

Synthesis of spirocarbocycles 117
Reddy et al. [57] reported the gold-catalyzed 5-endo-dig/spirocyclization of 2-[(2-aminophenyl)ethynyl]phenylamine 118 with isatins 119 by using NaAuCl4·2H2O in EtOH at 25 °C to afford corresponding spiroindolone derivatives 120 with 70–88% yield. Mechanistically, the gold(III) species coordinates to the alkyne part of 118 and subsequent nucleophilic attack of the tethered amino group, followed by protodemetalation to give N-(2-aminophenyl)indole R2. Thus imine S2 obtained by the condensation of activated isatin and R2. Subsequently, gold activated the imine leading to the nucleophilic attack of the indole onto imine S2 to give 120 (Scheme 45).

Synthesis of spiroindolone derivatives 120
Conclusion
This review illustrates the recent applications of 5-endo-dig cyclizations in organic synthesis for last two decades and clearly depicted by the class of formed heterocyclic and carbocyclic compounds such as indoles, cuomarins, cyclopentenes, benzofurans, furans, pyrroles. These heterocycles and carbocycles are often observed in many pharmaceutical substances, functional materials and biologically active compounds. Also this review expresses the synthesis of organogold complexes as well as spiro-compounds.
References
Baldwin JE (1976) Rules for ring closure. J Chem Soc Chem Comm. https://doi.org/10.1039/C39760000734
Gilmore K, Alabugin IV (2011) Cyclizations of alkynes: revisiting Baldwin’s rules for ring closure. Chem Rev 111:6513–6556. https://doi.org/10.1021/cr200164y
Gilmore K, Mohamed RK, Alabugin IV (2016) The Baldwin rules: revised and extended. WIREs Comput Mol Sci 6:487–514. https://doi.org/10.1002/wcms.1261
Alabugin IV, Timokhin VI, Abrams JN, Manoharan M, Abrams R, Ghiviriga I (2008) In search of efficient 5-endo-dig cyclization of a carbon-centered radical: 40 Years from a prediction to another success for the Baldwin rules. J Am Chem Soc 130:10984–10995. https://doi.org/10.1021/ja801478n
Alabugin IV, Manoharan M (2005) 5-Endo-dig radical cyclizations: “The poor cousins” of the radical cyclizations family. J Am Chem Soc 127:9534–9545. https://doi.org/10.1021/ja050976h
Ojima I, Tzamarioudaki M, Li Z, Donovan RJ (1996) Transition metal-catalyzed carbocyclizations in organic synthesis. Chem Rev 96:635–662. https://doi.org/10.1021/cr950065y
Rode ND, Aschi M, Chiarini M, Vecchio LD, Marinelli F, Arcadi A (2018) Reaction of β-(2-aminophenyl)-α,β-ynones with tosyl isocyanate: experimental and computational investigations. Adv Synth Catal 360:3672–3676. https://doi.org/10.1002/adsc.201800733
Rodriguez AL, Koradin C, Dohle W, Knochel P (2000) Versatile indole synthesis by a 5-endo-dig cyclization mediated by potassium or cesium bases. Angew Chem Int Ed 39:2488–2490. https://doi.org/10.1002/1521-3773(20000717)39:14%3c2488:AID-ANIE2488%3e3.0.CO;2-E
Takahashi S, Kuroyama Y, Sonogashira K, Hagihara N (1980) A convenient synthesis of ethynylarenes and di-ethynylarenes. Synthesis 1980:627–630. https://doi.org/10.1055/s-1980-29145
Andreev IA, Ratmanova NK, Novoselov AM, Belov DS, Seregina IF, Kurkin AV (2016) Oxidative dearomatization of 4,5,6,7-tetrahydro-1H-indoles obtained by metal- and solvent-free thermal 5-endo-dig cyclization: the route to erythrina and lycorine alkaloids. Chem Eur J 22:7262–7267. https://doi.org/10.1002/chem.201600273
Naoe S, Saito T, Uchiyama M, Oishi S, Fujii N, Ohno H (2015) Direct construction of fused indoles by gold-catalyzed cascade cyclization of conjugated diynes. Org Lett 17:1774–1777. https://doi.org/10.1021/acs.orglett.5b00550
Jalal S, Paul K, Jana U (2016) Iron-catalyzed 1,5-enyne cycloisomerization via 5-endo-dig cyclization for the synthesis of 3-(inden-1-yl)indole derivatives. Org Lett 18:6512–6515. https://doi.org/10.1021/acs.orglett.6b03544
Sharma S, Pathare RS, Sukanya Maurya AK, Goswami B, Agnihotri VK, Sawant DM, Pardasani RT (2017) Microwave assisted metal-/oxidant-free cascade electrophilic sulfenylation/5-endo-dig cyclization of 2-alkynylanilines to generate diversified 3-sulfenylindoles. Tetrahedron Lett 58:3823–3826. https://doi.org/10.1016/j.tetlet.2017.08.046
Palimkar SS, Kumar PH, Lahoti RJ, Srinivasan KV (2006) Ligand-, copper-, and amine-free one-pot synthesis of 2-substituted indoles via Sonogashira coupling 5-endo-dig cyclization. Tetrahedron 62:5109–5115. https://doi.org/10.1016/j.tet.2006.03.035
Palimkar SS, More VS, Kumar PH, Srinivasan KV (2007) Synthesis of an indole containing KDR kinase inhibitor by tandem Sonogashira coupling-5-endo-dig-cyclization as a key step. Tetrahedron 63:12786–12790. https://doi.org/10.1016/j.tet.2007.09.075
Kuethe JT, Wong A, Qu C, Smitrovich J, Davies IW, Hughes DL (2005) Synthesis of 5-substituted-1H-indol-2-yl-1H-quinolin-2-ones: a novel class of KDR kinase inhibitors. J Org Chem 70:2555–2567. https://doi.org/10.1021/jo0480545
Cai J, Wu B, Rong G, Zhang C, Qiu L, Xu X (2018) Gold-catalyzed bicyclization of diaryl alkynes: synthesis of polycyclic fused indole and spirooxindole derivatives. Org Lett 20:2733–2736. https://doi.org/10.1021/acs.orglett.8b00939
Jaimes MCB, Weingand V, Rominger F, Hashmi ASK (2013) From ynamides to highly substituted benzo[b]furans: gold(I)-catalyzed 5-endo-dig-cyclization/rearrangement of alkylic oxonium intermediates. Chem Eur J 19:12504–12511. https://doi.org/10.1002/chem.201301595
Saha D, Dey R, Ranu BC (2010) A simple and efficient one-pot synthesis of substituted benzo[b]furans by Sonogashira coupling-5-endo-dig cyclization catalyzed by palladium nanoparticles in water under ligand- and copper-free aerobic conditions. Eur J Org Chem 2010:6067–6071. https://doi.org/10.1002/ejoc.201000980
Mantovani AC, Hernandez JG, Bolm C (2018) Synthesis of 3-iodobenzofurans by electrophilic cyclization under solventless conditions in a ball mill. Eur J Org Chem 2018:2458–2461. https://doi.org/10.1002/ejoc.201800027
Kim I, Kim K, Choi J (2009) A direct approach to 5-hydroxybenzofurans via a platinum-catalyzed domino rearrangement/5-endo-dig cyclization reaction of quinols. J Org Chem 74:8492–8495. https://doi.org/10.1021/jo901937u
Palimkar SS, More VS, Srinivasan KV (2008) Ultrasound promoted copper-, ligand- and amine-free synthesis of benzo[b]furans/nitro benzo[b]furans via Sonogashira coupling-5-endo-dig-cyclization. Ultrason Sonochem 15:853–862. https://doi.org/10.1016/j.ultsonch.2007.10.006
Majumdar KC, Chattopadhyay B, Samanta S (2009) A short route to the synthesis of pyrrolocoumarin and pyrroloquinolone derivatives by sonogashira cross-coupling and gold-catalyzed cycloisomerization of acetylenic amines. Synthesis 2:311–317. https://doi.org/10.1055/s-0028-1083295
Majumdar KC, De N, Sinha B, Roy B (2012) Synthesis of 3-iodopyrrolocoumarins via iodine-induced 5-endo-dig electrophilic cyclization. Monatsh Chem 143:1067–1073. https://doi.org/10.1007/s00706-011-0694-0
Staben ST, Kennedy-Smith JJ, Toste FD (2004) Gold(I)-catalyzed 5-endo-dig carbocyclization of acetylenic dicarbonyl compounds. Angew Chem 116:5464–5466. https://doi.org/10.1002/ange.200460844
Suzuki S, Tokunaga E, Reddy DS, Matsumoto T, Shiro M, Shibata N (2012) Enantioselective 5-endo-dig carbocyclization of β-ketoesters with internal alkynes employing a four-component catalyst system. Angew Chem Int Ed 51:4131–4135. https://doi.org/10.1002/anie.201201060
Dolaine R, Gleason JL (2000) Diastereoselective formation of 5-vinylcyclopentenes from 1,6-enynes: cobalt-mediated C–H allylic activation and formal 5-endo-dig cyclization. Org Lett 2:1753–1756. https://doi.org/10.1021/ol005928a
Lee PH, Lee K, Kim S (2001) A novel nucleophilic substitution of in situ generated 3-tert-butyldimethyl-silyloxyalk-2-enylsulfonium salts with allylindium reagents. Org Lett 3:3205–3207. https://doi.org/10.1021/ol016542i
Iwasawa N, Miura T, Kiyota K, Kusama H, Lee K, Lee PH (2002) An efficient method for cyclopentene annulation onto α,β-unsaturated ketones: W(CO)5(L)-catalyzed 5-endo-dig cyclization of 6-siloxy-5-en-1-ynes. Org Lett 4:4463–4466. https://doi.org/10.1021/ol026993i
Fujino D, Yorimitsu H, Osuka A (2012) Synthesis of 1,2-disubstituted cyclopentenes by palladium-catalyzed reaction of homopropargyl-substituted dicarbonyl compounds with organic halides via 5-endo-dig cyclization. Org Lett 14:2914–2917. https://doi.org/10.1021/ol301257m
French JM, Diver ST (2014) Gold(I)-promoted heterocyclization of internal alkynes: a comparative study of direct metallate 5-endo-dig cyclization versus a stepwise cyclization. J Org Chem 79:5569–5585. https://doi.org/10.1021/jo500748e
Majumdar KC, Ganai S, Nandi RK (2011) Regioselective synthesis of pyrimidine-annulated spiro-dihydrofurans by silver-catalyzed 5-endo-dig cyclization. New J Chem 35:1355–1359. https://doi.org/10.1039/c1nj20121b
Sniady A, Wheeler KA, Dembinski R (2005) 5-Endo-dig electrophilic cyclization of 1,4-disubstituted but-3-yn-1-ones: regiocontrolled synthesis of 2,5-disubstituted 3-bromo- and 3-iodofurans. Org Lett 7:1769–1772. https://doi.org/10.1021/ol050372i
Sniady A, Durham A, Morreale MS, Wheeler KA, Dembinski R (2007) Room temperature zinc chloride-catalyzed cycloisomerization of alk-3-yn-1-ones: synthesis of substituted furans. Org Lett 9:1175–1178. https://doi.org/10.1021/ol062539t
Wakabayashi Y, Fukuda Y, Shiragami H, Utimoto K, Nozaki H (1985) Preparation of furans from alkynols utilizing palladium catalyzed intramolecular addition of alcohol to acetylene as a key reaction. Tetrahedron 41:3655–3661. https://doi.org/10.1016/S0040-4020(01)91384-5
El-Taeb GMM, Evans AB, Jones S, Knight DW (2001) Practical alternatives for the synthesis of β-iodofurans by 5-endo-dig cyclisations of 3-alkyne-1,2-diols. Tetrahedron Lett 42:5945–5948. https://doi.org/10.1016/S0040-4039(01)01112-1
Shi T, Guo X, Teng S, Hu W (2015) Pd(II)-catalyzed formal [4 + 1] cycloadditions of diazoacetates and aryl propargyl alcohols to form 2,5-dihydrofurans. Chem Commun 51:15204–15207. https://doi.org/10.1039/C5CC05000F
Oechsle P, Florke U, Egold H, Paradies J (2016) Heteroacene synthesis through C–S cross-coupling/5-endo-dig cyclization. Chem Eur J 22:18559–18563. https://doi.org/10.1002/chem.201603737
Khan ZA, Wirth T (2009) Synthesis of indene derivatives via electrophilic cyclization. Org Lett 11:229–231. https://doi.org/10.1021/ol8024956
Ma B, Wu Z, Huang B, Liu L, Zhang J (2016) Gold-catalysed facile access to indene scaffold via sequential C–H functionalization and 5-endo-dig carbocyclization. Chem Commun 52:9351–9354. https://doi.org/10.1039/C6CC04034A
Smith CR, Bunnelle EM, Rhodes AJ, Sarpong R (2007) Pt-Catalyzed cyclization/1,2-migration for the synthesis of indolizines, pyrrolones, and indolizinones. Org Lett 9:1169–1171. https://doi.org/10.1021/ol0701971
Pandya AN, Fletcher JT, Villa EM, Agrawal DK (2014) Silver-mediated synthesis of indolizines via oxidative C–H functionalization and 5-endo-dig cyclization. Tetrahedron Lett 55:6922–6924. https://doi.org/10.1016/j.tetlet.2014.10.112
Seregin IV, Gevorgyan V (2006) Gold-catalyzed 1,2-migration of silicon, tin, and germanium en route to C-2 substituted fused pyrrole-containing heterocycles. J Am Chem Soc 128:12050–12051. https://doi.org/10.1021/ja063278l
Kim I, Choi J, Won HK, Lee GH (2007) Expeditious synthesis of indolizine derivatives via iodine mediated 5-endo-dig cyclization. Tetrahedron Lett 48:6863–6867. https://doi.org/10.1016/j.tetlet.2007.07.180
Sugita S, Ueda M, Doi N, Takeda N, Miyata O (2016) Gold-catalyzed sequential cyclization/rearrangement reaction of O-allyl hydroxamates: atom economical synthesis of 3-hydroxyisoxazoles. Tetrahedron Lett 57:1786–1789. https://doi.org/10.1016/j.tetlet.2016.03.032
Wang Q, Tsui GC (2018) Copper-mediated domino cyclization/trifluoromethylation of propargylic N-hydroxylamines: synthesis of 4-trifluoromethyl-4-isoxazolines. J Org Chem 83:2971–2979. https://doi.org/10.1021/acs.joc.7b03191
Xiao D, Han L, Sun Q, Chen Q, Gong N, Lv Y, Suzenet F, Guillaumet G, Chenga T, Li R (2012) Copper-mediated synthesis of N-fused heterocycles via Csp–S coupling reaction and 5-endo-dig cyclization sequence. RSC Adv 2:5054–5057. https://doi.org/10.1039/c2ra20254a
Chen Y, Wang D, Petersen JL, Akhmedov NG, Shi X (2010) Synthesis and characterization of organogold complexes containing an acid stable Au–C bond through triazole-yne 5-endo-dig cyclization. Chem Commun 46:6147–6149. https://doi.org/10.1039/c0cc01338b
Ueda H, Yamaguchi M, Kameya H, Sugimoto K, Tokuyama H (2014) Autotandem catalysis: synthesis of pyrroles by gold-catalyzed cascade reaction. Org Lett 16:4948–4951. https://doi.org/10.1021/ol5024695
Bharathiraja G, Sakthivel S, Sengoden M, Punniyamurthy T (2013) A novel tandem sequence to pyrrole syntheses by 5-endo-dig cyclization of 1,3-enynes with amines. Org Lett 15:4996–4999. https://doi.org/10.1021/ol402305b
Queiroz M-JRP, Begouin A, Pereira G, Ferreira PMT (2008) New synthesis of methyl 5-aryl or heteroaryl pyrrole-2-carboxylates by a tandem Sonogashira coupling/5-endo-dig-cyclization from β-iododehydroamino acid methyl esters and terminal alkynes. Tetrahedron 64:10714–10720. https://doi.org/10.1016/j.tet.2008.08.105
Yan Z-Y, Xiao Y, Zhang L (2012) Gold-catalyzed one-step construction of 2,3-dihydro-1H-pyrrolizines with an electron-withdrawing group in the 5-position: a formal synthesis of 7-methoxymitosene. Angew Chem Int Ed 51:8624–8627. https://doi.org/10.1002/anie.201203678
Gorin DJ, Davis NR, Toste FD (2005) Gold(I)-catalyzed intramolecular acetylenic Schmidt reaction. J Am Chem Soc 127:11260–11261. https://doi.org/10.1021/ja053804t
Postoia RA, Roehrs JA, Back DF, Zeni G (2017) Iodine-mediated regioselective 5-endo-dig electrophilic cyclization reaction of selenoenynes: synthesis of selenophene derivatives. Org Chem Front 4:277–282. https://doi.org/10.1039/x0xx00000x
Adler P, Fadel A, Rabasso N (2015) Cerium(IV) ammonium nitrate mediated 5-endo-dig cyclization of α-amino allenylphosphonates to spirodienones. Chem Commun 51:3612–3615. https://doi.org/10.1039/c5cc00281h
Wu W-T, Xu R-Q, Zhang L, You S-L (2016) Construction of spirocarbocycles via gold-catalyzed intramolecular dearomatization of naphthols. Chem Sci 7:3427–3431. https://doi.org/10.1039/c5sc04130a
Reddy BVS, Swain M, Reddy SM, Yadav JS, Sridhar B (2014) Gold-catalyzed 5-endo-dig cyclization of 2-[(2-aminophenyl)-ethynyl]phenylamine with ketones for the synthesis of spiroindolone and indolo[3,2-c]quinolone scaffolds. Eur J Org Chem 2014:3313–3318. https://doi.org/10.1002/ejoc.201402006
Acknowledgements
The second and third authors are grateful to Professor Dr. Thomas Wirth, Cardiff University, UK, for proof reading and valuable suggestions to improve the manuscript. The corresponding author is grateful to Higher Education Commission, Pakistan (Project No. PM-IPFP/HRD/HEC/2011/2063) and Government College University, Faisalabad (GCUF-RSP Project No. 61-CHM-1) for providing the facilities to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sajid, M.A., Khan, Z.A., Shahzad, S.A. et al. 5-Endo-dig cyclizations in organic syntheses. Mol Divers 24, 295–317 (2020). https://doi.org/10.1007/s11030-019-09930-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-019-09930-x