
Abstract
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease that causes paralysis whose etiology and pathogenesis have not been fully elucidated. Presently it is incurable and rapidly progressive with a survival of 2–5 years from onset, and no treatments could cure it. Therefore, it is urgent to identify which therapeutic target(s) are more promising to develop treatments that could effectively treat ALS. So far, more than 90 novel treatments for ALS patients have been registered on ClinicalTrials.gov, of which 23 are in clinical trials, 12 have been terminated and the rest suspended. This review will systematically summarize the possible targets of these novel treatments under development or failing based on published literature and information released by sponsors, so as to provide basis and support for subsequent drug research and development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease, which the global incidence of 2–3/100,000 population over the age of 15 year (Tiryaki and Horak 2014; Kukharsky et al. 2021; Ustyantseva et al. 2020; Al-Chalabi and Hardiman 2013). Extensive loss of lower motor neurons in the anterior horn of spinal cord and brainstem is the main neuropathological feature of ALS (Grad et al. 2017). However, the specific pathogenesis is still unclear. ALS typically occurs in adults and presents as a relentlessly advanced loss of motor function and ultimately death, due to respiratory failure (Grad et al. 2017). Presently it is incurable and rapidly progressive with a mean survival of 2-5y from onset (Tiryaki and Horak 2014; Kukharsky et al. 2021; Ustyantseva et al. 2020; Grad et al. 2017). Thus, for most patients, ALS is a disease with short survival and poor prognosis.
Currently, there is no effective therapy available for ALS patients, only seven well-recognized disease-modify drugs. Three dosage of riluzole,, including tablet (Rilutek), suspension (Tiglutik) and oral film (Exservan), as well as edaravone (Radicava) and Nuedexta, have been approved for the treatment of ALS and its associated symptoms (Jaiswal 2019; Miller et al. 2012; Rothstein 2017). Riluzole, a glutamate antagonist, prolongs the patient survival of ALS patients by about 3 months. Edaravone, is a free radical/reactive oxygen species (ROS) scavenger, and improves the patient symptoms slightly. Nuedexta only improved swallowing function in patients in some degree. Relyvrio (oral fixed dose formulation of sodium phenylbutyrate and ursodoxicoltaurine, AMX0035), was approved for marketing in Canadian and the United States which playing a role by improving the health of endoplasmic reticulum and delaying the death of nerve cells. After a three-year follow-up of 137 patients with ALS, patients on active drug which compared with historical placebo groups from other trials, and this post-hoc analysis showed that the median survival time in the relyvrio group was 6.5 months longer than in these placebo groups (25.0 months vs. 18.5 months). Currently, a phase III clinical trial (Phoenix), is well underway, and this will establish whether relyvrio has a disease modifying effect (Paganoni et al. 2021). Subsequently, Qalsody (tofersen) has been approved by U.S. Food and Drug Administration (FDA), which can reduce SOD1 protein synthesis by promoting degradation of SOD1 messenger RNA. According to its phase III results, there was 38% and 26% difference in CSF total SOD1 proteins between the tofersen and placebo groups in patients with faster and slower progression, respectively. Plasma Nfl, a potential marker of neurodegeneration, differed 67% and 48% between the tofersen group and the placebo group in patients with faster and slower progression (Vacchiano et al. 2021). The efficacy of tofersen also needs phase III clinical trial (ATLAS) to further prove, and the drug is only suitable for ALS patients with SOD1 mutations (only 2% of 68,000 patients worldwide).
It may be due to the fact that the pathological mechanism underlying neurodegeneration in ALS are multifactorial and may function through interconnected molecular and genetic pathways. To be specific, neurodegeneration in ALS may be caused by a complex interaction of glutamate excitoxicity, generation of free radicals, cytoplasmic protein aggregates, superoxide dismutase1 (SOD1) enzymes, combined with mitochondrial dysfunction, and disruption of axonal transport processes through accumulation of neurofilament intracellular aggregates (Feldman et al. 2022). However, it is not clear which pathway is the key to the disease, and the drugs that on the clinical trial may target non-critical pathways and therefore have not shown efficacy. Therefore, there is still an urgent need to determine the key pathogenic mechanism and develop new therapeutic methods for the treatment of ALS. According to ClinicalTrials.gov and literature retrieval website (including PUBMED, Embase and Web of Science), we have selected 23 ongoing novel treatments (excluding old drugs for new approaches) with ALS as an indication, including innovative small molecule drugs, biological drugs, genetic therapies, cell therapies and non-drug approaches (Table 1). At present, 4 trials of these (17%) in the Phase I, 11 trials of these (48%) in the Phase II and 8 trials of these (35%) in the Phase III (Fig. 1A). Most of clinical trials for ALS are still in the Phase I and II, with some progressing to Phases III, possibly for four reasons below. First of all, the pathogenesis of ALS disease is multifarious, cellular and animal models that can characterize ALS disease cannot be constructed, and the efficacy results in the current models are not sufficient to predict the efficacy in heterogeneous human patients. Secondly, due to the unclear pathogenesis of ALS, effective drug targets for ALS cannot be determined at present. Researchers can only speculate about the mechanism and try different targets, some of which are low quality, and thus have a high failure rate. Thirdly, clinical trial design still has some drawbacks. There are many reasons for the onset of ALS, including genetic, environmental or age-related dysfunction. Different factors lead to different pathogenic mechanisms. In the early stage of clinical trials, it is impossible to distinguish different patient subgroups according to genetic background or other pathogenic factors, resulting in poor efficacy outcomes of drugs. Finally, clinical trials should reasonably use biomarkers as secondary endpoints of efficacy, including neurofilament heavy chain or neurofilament light chain (Nfl) et al.,. The results of biomarkers may provide a reference when primary efficacy indicators (such as survival time, etc.) are not met the requirements in clinical trials. However, whether biomarkers can independently be used as indicators to predict the efficacy of drugs for ALS remains controversial.

Distribution of novel treatments under development in different phases (A) and different pathways (B). Different colors represent different clinical trial phases (phase I, I, III) of drugs. The area of each color graph represents the proportion of drugs at different phases to the total number of ALS treatments under development (A); Different colors represent different targets of treatments. The area of each color graph represents the proportion of drugs acting on different targets to the total number of ALS treatments under development (B)
To clarify the probably pathogenesis of ALS disease and find possible targets for different patient subgroups is a reliable way to break the bottleneck of ALS treatment. This review will focus on summarizing novel treatments in clinical trials or terminations for ALS based on the possible signaling pathways of drugs action (Figs. 1B and 2). Meanwhile, under the corresponding potential targets, preclinical drugs with good effects will also be introduced, so as to analyze the future development prospects of the potential targets and provide ideas for the selection of them for subsequent drug development.

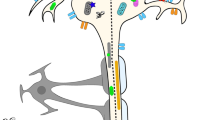
Mode of action of the drugs presented in this review. Red T-shaped arrow: inhibit; blue, green, and orange arrows: active
Genetic therapies (antisense oligonucleotide (ASO) therapies)
There are two types of ALS patients: familial amyotrophic lateral sclerosis (fALS) and sporadic ALS (sALS). The overall pooled mutation frequencies of ALS-related genes were 47.7% in fALS and 5.2% in sALS. In European populations, the most common mutations were the chromosome 9 open reading frame 72 (C9orf72) repeat expansions (fALS 33.7%, sALS 5.1%), followed by SOD1 (fALS 14.8%, sALS 1.2%), TARDNA-binding protein of 43 kDa (TDP43) (fALS 4.2%, sALS 0.8%) and fused-in-sarcoma (FUS) mutations (fALS 2.8%, sALS 0.3%) (Hardiman and Berg 2020). Therefore, gene targeting therapy, which reduces the mRNA level of the above proteins, may be a feasible approach. In 1993, the first gene associated with familiar ALS (fALS), SOD1 was discovered by a team of researchers at the University of Massachusetts Medical School. SOD1 is an antioxidant enzyme, which can directly participate in the inactivation of toxic superoxide radicals by binding copper and zinc ions, so as to protect cells from the damage of superoxide radicals (Kaur et al. 2016; Hayashi et al. 2016). Mutated SOD1 gene can obtain gain and loss of function mutations, and the functional toxic gain is the main part (Kaur et al. 2016; Miller et al. 2013). Tofersen is an intrathecally administered ASO, that worked primarily by inhibiting the gain of toxicity from the mutant SOD1.
WVE-004 is a stereopure oligonucleotide designed to selectively target transcriptional variants containing the C9orf72 gene-associated hexanucleotide repeat expansion (G4C2), thereby leaving the C9orf72 protein intact (Phase I/II, NCT04931862) (Liu et al. 2022). However, phase I results of BIIB078, an ASO in development for the treatment of C9orF72-associated ALS, showed no clinical benefit and the company decided to terminate the clinical trial. More insight is needed why them had poor efficacy, may be due to the late treatment, which targeted only one chain, rather than the reverse stand of C9orf72. There were indications that C9orF72-associated ALS and frontotemporal dementia (FTD) was in part also a developmental disease (e.g. very early other abnormalities can be found like lower body mass index (BMI), or magnetic resonance imaging (MRI) changes without having ALS or FTD) (Table 1) (Nolan et al. 2016; Nolan et al. 2016; Pang and Hu 2021). So further trials are needed to verify if including some early-stage patients in the trial will make a difference. FUS is an RNA-binding protein with 526 amino acids encoded by 15 exons. It belongs to the FUS/EWSR1/TAF15 (FET) family and contains a few different functional domains, including RNA recognition motifs and highly conserved C-terminal nuclear localization signals. FUS gene mutations were considered to be a pathogenic factor in a few cases of ALS in 2009 (Nolan et al. 2016). ION363 is an ASO targeting FUS RNA that reduces FUS protein production. Antisense-mediated reduction of mutant FUS proteins prevents motor neuron loss in a mouse model of FUS-associated ALS (Korobeynikov et al. 2022). By targeting the underlying cause of FUS-associated ALS, ION363 has the potential to reduce or prevent disease progression in patients, and a Phase III trial is currently underway (NCT04768972). In addition to ASO, which directly reduces the mRNA level of the above protein, has achieved some effect, ASO, which reduces the toxicity of the toxic protein, is also being further verified. TDP-43 is a nucleic acid binding protein that exists mainly in the nucleus but can travel between the cytoplasm and the nucleus. However, TDP-43 mistakenly self-aggregates in the cytoplasm of motor neurons in ALS patients, causing toxicity. We also found that overexpression of the yeast ortholog human Ataxin-2 (ATXN2) enhanced TDP-43 toxicity, while null mutation of ATXN2 ortholog suppressed TDP-43 toxicity (St Martin et al. 2020). Drugs that target it are being developed. Moreover, dysfunction of TDP-43 leads to missplice of the precursor mRNA of the stathmin-2 (STMN2) gene, which leads to loss of STMN2 protein (required for axon regeneration), as well as further loss of motor ability and paralysis specific to ALS. QRL-201, an ASO drug, containing the human STMN2 gene sequence and injected it into a mouse model with TDP-43 dysfunction, which can successfully correct the splicing of STMN2 precursor mRNA, restore normal STMN2 protein levels in the nervous system of mice, and promote neuronal axon regeneration, thereby treating ALS (Baughn et al. 2023). For ALS, the first ASO drug was already on the market, and QRL-201 was in phase I clinical trial (NCT05633459), which is also hopeful based on its mechanism of action.
Drugs targeting protein aggregates
Protein misfolding, aggregation, and deposition of specific proteins are biochemical and histopathological hallmarks of ALS (McAlary et al. 2019). The pathological specific protein inclusions observed in ALS patients are immunoreactive for SOD1, TDP-43, and FUS etc. (Kim et al. 2020). Therefore, TDP-43, SOD1, and FUS etc., are disease-associated proteins. By reducing the amounts of aggregated proteins and waning the degree of reactions they cause, there may also be an impact on disease. Heat shock proteins (HSPs) can prevent and clear inclusions such as SOD1 and TDP-43 etc., suggesting that the ability of ALS motor neurons to induce the heat shock response (HSR) is impair (Seminary et al. 2018). Likewise, the induction rate of HSPs was reduced in the human post-mortem samples and mouse models compared to the unaffected control group. Arimoclomol acts by amplifying the production of HSPs and has completed its pivotal clinical study (NCT03491462), but was discontinued due to its failure to meet the primary endpoint (The completed data have not yet been published). SLS-005 Trehalose is a low molecular weight disaccharide with a variety of uncertain drug action mechanisms, including modulation of autophagy, gut brain microbiome, and targeting TDP43 mislocalization and accumulation et al., which is also undergoing clinical trials (Phase II/III, NCT04297683) (Wang et al. 2018).
Accumulation and aggregation of mutated proteins accumulation and aggregation can cause endoplasmic reticulum stress (ERS), unfolded protein response (UPR) and diversiform intracellular stress responses, a mechanism that a self-defense mechanism that reduces misfolded protein load (Kukharsky et al. 2021; Cai et al. 2016). Furthermore, UPR initiates apoptotic death of affected cells in response to sustained stress (Sano and Reed 2013; Bogorad et al. 2018; Bugiani et al. 2010). GDC-0134, a dual leucine zipper kinase (DLK) inhibitor that regulates cell response to the ERS through c-Jun N-terminal kinases (JNK) and protein kinase R-like endoplasmic reticulum kinase (PERK) (Kukharsky et al. 2021). In addition, DLK inhibitors can regulate axonal degeneration and GDC-0134 is being investigated for safety and efficacy in Phase I clinical trial (NCT02655614). TCH346 (Supplementary Table 1) improves protein aggregation, energy metabolism, oxidative stress and anti- apoptotic in ALS patients by binding to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Lazarev et al. 2013; Pierce et al. 2008; Desseille et al. 2017). Unfortunately, the results of its phase II trial showed no significant differences between placebo and active treatment groups in mean rate of decline in the primary outcome ALSFRS-R at 24 weeks and secondary outcomes (survival, lung function, and manual muscle testing). TCH346 has no beneficial effect on the disease progression of ALS (NCT00230074), but based on previous data, the beneficial effect was not observed until patients were treated with riluzole for 9 months, so TCH346 may also be treatment for too short to see no therapeutic effect (Miller et al. 2007).
Drugs targeting oxidative stress
Oxidative stress refers to the damage of the intracellular antioxidant defense system due to the excessive production of oxidants, resulting in the excessive accumulation of oxygen, hydroxyl free radical, and so on, thereby causing damage to cellular macromolecules such as DNA, lipids and proteins, and ultimately lead to necrotic and apoptotic cell death (Singh et al. 2019). Thus, oxidative stress is another potential therapeutic target for ALS (Table 1). The brain is particularly vulnerable to oxidation due to the abundance of polyunsaturated fatty acids, which can give rise to several oxidized metabolites. Myeloperoxidase (MPO) promotes their production and accumulation, leading to oxidative stress (Xiong et al. 2022). Verdiperstat is an inhibitor of MPO and was declared a failure in its Phase II/III (NCT04297683) development. According to the results of its Phase II/III clinical trial, there were no statistically significant differences between the verdiperstat and placebo groups in primary outcome (Change from Baseline in ALSFRS-R Total Score at week 24) and survival, as well as key secondary outcome (including respiratory function, muscle strength, and survival). As with TCH346, it may also be because the follow-up time was too short to see the drug effect. Therefore, when designing ALS clinical trials, it is necessary to clarify the follow-up time of treatment for patients, otherwise it is impossible to judge the efficacy of drugs. And given the longstanding interest and lack of success for molecules that putatively modulate oxidative stress a more critical appraisal of this biology as a therapeutic target would be appropriate. A common cause of neuronal oxidative stress is also disordered metabolism of metal ions, mainly copper, iron, and zinc. CNM-Au8 is a clean-surfaced, multi-faceted gold nanocrystals aqueous suspension with extraordinary catalytic capacity to improve the efficiency of key metabolic reactions while reducing reactive oxygen species levels (Table 1). In addition, CNM-Au8 can also promote the transcription of Heat Shock Factor 1 (HSF1) gene (Vucic et al. 2021). In addition to the protection of neurons in ALS models by upregulation of HSF1 activity, increased NAD + and ATP have also been shown to play the same role (Vucic et al. 2021; Southon et al. 2020). However, the Phase II trial of CNM-Au8 (NCT04098406) failed, mainly because it did not meet the biomarker primary (Motor Unit Index) and secondary FVC endpoint at week 36. The good news is that it may be effective in ALS limb type patients and some degree of protective effect was observed on lower motor neurons (week 12, p = 0.0385), requiring further verification. However, the primary endpoint of ALS clinical trials is survival or ALSFRS-R Total Score, but the primary endpoint of this trial was biomarker, which may be lack of power. Glutathione peroxidase 4 (GPX4), an anti-oxidant enzyme and central repressor of ferroptosis, occurred in post-mortem spinal cords of both sporadic and familial ALS patients. GPX4 depletion is also an early and common feature of the spinal cord and brain of SOD1 G93A, TDP-43, and C9orf72 ALS mice. Overexpression of human GPX4 in SOD1G93A mice significantly delayed disease onset, improved motor function and prolonged life, which has been shown to be associated with reduced lipid peroxidation and motor neuron preservation. Therefore, GPX4 agonist may be a promising approach for ALS treatment (Hilton et al. 2017; Wang et al. 2021; Liu and Wang 2017; Vallarola et al. 2018; Liu and Wang 2017).
Mitochondria are the main sites of intracellular oxygen consumption. Under normal circumstances, about 1–4% of the oxygen molecules in mitochondria are reduced by one electron to form superoxide, which makes mitochondria become an important source of ROS (Sinha et al. 2013). Mitochondrial-specific antioxidant mechanisms do exist, including the aforementioned SOD1, which can reduce ROS production (Hayashi et al. 2016). However, oxidative damage that accumulates over time is a major cause of mitochondrial dysfunction which triggers oxidative stress and glutamate excitotoxicity in ALS. RT001 is the first drug in the new drug class deuterated polyunsaturated fatty acids (D-PUFAs) (Yerton et al. 2022; Pallardó et al. 2021). D-PUFAs are a novel approach to prevent mitochondrial lipid peroxidative damage leading to cell death (Phase II, NCT04762589) (Pang and Hu 2021). TRO19622 is a novel mitochondria-targeted neuroprotective compound belonging to a novel family of cholesterol oximes. Proteins outside the mitochondrial membrane are targeted to prevent the opening of the permeability transition pore mediated by factors such as oxidative stress (Table 1) (Bordet et al. 2010). TRO19622 has been shown to exert neuroprotective effect in various studies of in vitro and in vivo (Bordet et al. 2010). However, patients who received TRO19622 did not show a significant increase in survival compared to those who received placebo and riluzole (The completed data have not yet been published). Another drug in preclinical development that can improve mitochondrial dysfunction, 7, 8-dihydroxyflavone prodrug, which promotes mitochondrial biogenesis by activating tyrosine kinase receptor B (TrkB), and prolongs the survival rate of worms with stable expression of SOD1 G93A, which needs to be further validated clinically (Li et al. 2021).
Drugs targeting neuroinflammation
Neuroinflammation in patients with ALS is characterized by astrocyte and microglial activation, lymphocyte infiltration, and excessive production of inflammatory cytokines (Liu and Wang 2017). At the same time, there is accumulating evidence has implicated that the normal immune cells may play a protective role on the survival of motor. Consequently, some compounds affecting neuroinflammation have been the candidates for drugs in ALS (Table 1). On the whole, the drugs that target neuroinflammation (25%, 6) are the most, followed by gene therapies (17%, 3) and protein aggregate (13%, 2). NP001 is a small molecule that modulates inflammatory cells, transforming macrophages and monocytes from an inflammatory phenotype to a basal noninflammatory phenotype (Zhang et al. 2022). NP001 was found to be safe and well tolerated, showing positive trends in slowing disease progression, especially in patients 40–65 years of age, according to the results of phase I and II trials completed in ALS patients (NCT01091142, NCT01281631 and NCT02794857) (Liu and Wang 2017). Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) is a critical regulator of cell death and inflammation (Ito et al. 2016). And, numerous studies have shown that the expression of RIPK1 were increased in the spinal cords of SOD1G93A transgenic mice and human ALS pathological samples (Ito et al. 2016). RIPK1 inhibitor DNL788 and SAR443820 were going on their phase I and phase II clinical trial, respectively, but another RIPK1 inhibitor DNL747 was suspended in phase I due to safety concerns (Supplementary Table 1). Therefore, the future of RIPK inhibitors in ALS field still needs to be explored.
IPL344 is a novel drug targeting the the phosphatidylinositol-3-kinase (PI3K)/ v-akt murine thymoma viral oncogene (Akt) signaling pathway to reduce the expression of pro-inflammatory molecules and is undergoing Phase II clinical trials (NCT03652805). On February 2, 2020, the FDA and the European Medicines Agency (EMA) granted IPL344 orphan drug designation for the treatment of ALS. MN-166 is a non-selective phosphodiesterase 4 inhibitor that affects the survival and activation of resident immune cells by regulating their production of pro-inflammatory cytokines (Liu and Wang 2017). In vitro evidence suggests that MN-166 is neuroprotective by suppressing neuronal cell death induced by microglial activation (Oskarsson et al. 2021). But MN-166 was not well tolerated as it required multiple dose reductions for adverse events (Babu et al. 2021). In a similar vein, Masitinib, a tyrosine kinase inhibitor known for its potential to provide neuroprotection by effectively inhibiting the activity of microglia, macrophages, and mast cells in both the central and peripheral nervous systems. This inhibition serves to mitigate neuroinflammation. A significant survival benefit of 25 months (p = 0.037) and 47% reduced risk of death (p = 0.025) was observed for patients receiving 4.5 mg/kg/day masitinib (n = 45) versus placebo (n = 62) in an enriched cohort with ⩾2 on each baseline ALSFRS-R individual component score (i.e. prior to any complete loss or severe impairment of functionality) (Mora et al. 2021).
Complement provides the first line of defense against microorganisms and is a crucial branch of the innate immune system. There are multiple pathways of complement activation, but all ultimately converge on the central C3 and C5 invertase, promoting inflammation and apoptosis (Carpanini et al. 2019). Human studies proved the activation of the classical complement pathway and upregulation of C3 in the spinal cord, motor cortex and CSF of ALS patients. Elevated levels of C5a and sC5b-9 were found in the serum of ALS patients suggesting that terminal complement components play the most important role (Gavriilaki et al. 2020). Ravulizuma and zilucoplan are C5 invertase inhibitors, and pegcetacoplan (Phase II, NCT04579666) is C3 invertase inhibitor, which improves the efficacy of ALS patients by inhibiting complement activation (Howard Jr et al. 2020). Also, C1q participates in the classical pathway of complement activation. After the antigen is combined with the antibody, C1q can recognize and bind to the complement binding site on the antibody. ANX005 is a C1q invertase inhibitor that inhibits amplification of immune responses (Phase II, NCT04569435). However, Astrazeneca announced the termination of the development of the C5 convertase inhibitor ravulizumab (Chen 2020) because the the mid-stage analysis of its Phase III trial (NCT04248465) did not meet the clinical endpoint (Table 1). Subsequently, zilucoplan was also discontinued, which is not optimistic for the treatment of ALS with complement inhibitors (Table 1). Thus, given multiple molecular failures in relatively large, well-executed phase II or III trials, complement may not be a viable target for ALS.
Stem cells
Stem cells are emerging a prospective treatment means for neuronal replacement and regeneration in multifarious neurodegenerative diseases, including ALS (Bonafede and Mariotti 2017). Neuronata-R, mesenchymal stem cells (MSCs), collected from patient bone marrow, has been marketed in South Korea with ALS as an indication. And according to ClinicalTrials.gov, at least 37 stem cells clinical trials have been registered for ALS. However, the Phase III trial of NurOwn® (bone marrow-derived autologous MSCs) did not meet its primary endpoint (ALSFRS-R post-treatment slope improved by 1.25/ month at 28 weeks, Table 1). But surprisingly, patients with milder disease experienced better treatment outcomes, with 34.6% of patients in the NurOwn® administration group experiencing slower disease progression compared to 15.6% in the placebo group (p = 0.29) (Berry et al. 2019). This suggested that it may be the heterogeneity of the disease and short follow-up time that led to NurOwn®’s effect not being seen. Overall, there may be some hope for stem cell treatment of ALS after all. While autologous MSCs have the advantage of retaining genetic information from the donor, patient-derived MSCs derived from fALS patients may carry over the pathological effects of mutated genes. But advances in CRISPR/Cas-9, a genome-editing tool, can repair the gene mutations of the patient-derived MSCs (Wang et al. 2017). Therefore, more researches are needed to confirm the efficacy of optimized stem cell products in the treatment of ALS patients.
Usually, stem cells are administered intrathecally (Fig. 3A), which solved the problem of difficult in swallowing for ALS patients. However, intrathecal administration of drugs requires a hospital visit, which also increases the inconvenience of patients. At present, riluzole has three dosage forms and has been listed successively: tablet, suspension and oral film. Therefore, in addition to the mechanism of action, the development of innovative drugs for ALS can also start from the pharmaceutical dosage form.

The number of novel treatments under development for ALS in different routes of administration (A) and at different phases in the past 14 years (B). Different colors represent different treatments delivery routes. The area of each color plot represents the proportion of drugs in different routes of delivery ((excluding drugs with unknown routes of delivery)) (B); The horizontal axis is the year, the vertical axis is the number of treatments, and different colors represent different clinical trial phases (phase I, II and III). The height of each bar chart represents the number of ALS treatments developed each year, and the height of each color in each bar chart represents the number of ALS treatments at different phases in that year (A)
Drugs targeting excitotoxicity
Excitatory toxicity of neuronal circuits can eventually lead to motor neuron death, and excitatory alterations have been found to be a common feature in almost all cases of ALS, so excitatory toxicity is considered to be one of the possible factors leading to the disease (Kukharsky et al. 2021; Kawahara anf Kwak 2005). Consequently, the compounds that affect neuronal excitability and/or neuronal synaptic transmission are potential candidates for drugs capable of preventing motor neuron death in ALS (Kukharsky et al. 2021; Kawahara and Kwak 2005).
Hepatocyte growth factor (HGF), a novel neurotrophic factor, acts on astrocytes in SOD1-G93A transgenic mice to maintain recombinant excitatory amino acid transporter 2 (EAAT2) levels in addition to direct neurotrophic activity (Funakoshi et al. 2007). EAAT2 is a glial-specific glutamate transporter that may be responsible for reducing glutamatergic neurotoxicity in motor neurons (Funakoshi et al. 2007). VM202 (Sufit et al. 2017) reduce excitotoxicity in ALS patients by producing HGF in humans, and currently in Phase II clinical trials (NCT03705390). ILB is another drug that promotes HGF protein production, and has completed the phase II clinical trial. The result showed that ILB improved clinical conditions and decreased neuronal damage in ALS patients (NCT03705390) (Logan et al. 2022; Lazzarino et al. 2021).
In addition to the medications mentioned above, another way to treat ALS is to use non-invasive brain stimulation (NIBS), which regulates brain activity through different forms of energy, such as electrical currents, magnetic pulses, or focused ultrasound through the scalp and skull. Corticospinal excitability can be inhibited or enhanced using NIBS techniques, namely repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS), as well as invasive brain and spinal cord stimulation (Ranieri et al. 2021). At present, this technique is still in the preliminary trial stage and needs further verification with a large sample size.
Sigma non-opioid intracellular receptor 1 (Sigma-1 receptor) is a protein expressed in motor neurons, mainly in close contact with cholinergic postsynaptic sites or in the endoplasmic reticulum (ER) on the mitochondria-associated ER membrane (MAM) (Herrando-Grabulosa et al. 2021).
The sigma-1 receptor regulates calcium homeostasis, excitatory toxicity, ER stress and other basic mechanisms of survival in neurons. Therefore, it can be used as a target for the treatment of ALS (Herrando-Grabulosa et al. 2021). Pridopidine, a Sigma-1 receptor agonist originally developed for Huntington’s disease, had a negative Phase II/III trial in ALS (NCT04297683). Pridopidine did not meet the primary endpoint of change from baseline to week 24 in the ALSFRS-R, rapidly declining participants on pridopidine with definite or probable ALS who were early in the disease (less than 18 months from symptom onset) had substantially less decline in the ALSFRS-R total score (-7.51 points) compared to placebo (-12.71 points, unadjusted p-value = 0.04) in post-hoc analyses. What’s more, pridopidine reduced NfL levels in rapidly declining patients with disease duration less than 18 months (average reduction of 40%) at 24 weeks compared to placebo in post-hoc analyses. This suggested that the effect may not be observed due to the heterogeneity of the disease, and the short follow-up time may also be one of the factors. Later, patients can be enrolled in the early stages and treated for enough time to confirm its efficacy.
Drugs targeting muscle strength
ALS leads to muscle functional capacity decline, and then generate great impact in quality of life. Some research teams have focused on improving muscle mass and function in ALS patients, and hope to develop treatments for the corresponding symptom (Ferreira et al. 2016). Therefore, this is also a possible research target.
Reldesemtiv and Tirasemtiv are fast skeletal muscle troponin activators (FSTA), and sensitize the sarcomere to calcium and increase muscle force. The Phase II clinical trial of Reldesemtiv (NCT03160898) has completed. Although the primary efficacy (change in percent predicted slow vital capacity (SVC) at 12 weeks) analysis did not demonstrate statistical significance, there were trends in favor of Reldesemtiv for all three endpoints (change in percent predicted SVC at 12 weeks, ALSFRS-R and muscle strength megascore) (Shefner et al. 2021; Keifer Jr et al. 2014; Vijayalakshmi et al. 2015). However, results from Phase III trials of tirasemtiv showed that it failed to meet the primary endpoint (slow vital capacity at 24 weeks) and secondary endpoints (improvement in muscle function and strength at 48 weeks), which is not a good signal for the development of such targeted drugs. Whereas the primary endpoint of ALS clinical trials was usually survival or the ALSFRS-R total score, the primary endpoint in this trial was slow vital capacity at 24 weeks which was a more relevant measure of the drug’s mechanism of action but may not fully characterize the drug’s efficacy. Later, the selection of end points can be optimized to confirm its efficacy.
Drugs targeting metabolism
Epidemiological evidence suggests that ALS patients suffer from catabolism and begin to lose weight > 10 years before the onset of motor symptoms. In addition, the increased risk of ALS is associated with lower body mass index and abnormal levels of circulatory hormones, so increasing body mass index with high-caloric nutrition may be a possible way to benefit patients with ALS. High-caloric nutrition conducted a randomized controlled study with a large sample size after completing its phase I safety assessment (NCT04172792). The study enrolled 221 patients who were randomly assigned (1:1) to receive high-caloric nutrition (405 kcal/ day, 100% fat) or placebo in addition to riluzole (100 mg/ day). The primary endpoint was survival, defined as time of death or study cutoff time. The findings suggest evidence that high-caloric nutrition did not prolong life in the overall ALS population. However, postmortem analysis showed significant survival benefits in the fast- progressing patients. Furthermore, in the subgroup of patients with high NfL serum levels, patients in the high-caloric nutrition group had a significantly prolonged survival, corroborating a potential effect of high-caloric nutrition on fast- progressing patients during the 18 months of intervention (Kawahara and Kwak 2005; Funakoshi et al. 2007).
Supportive and palliative care
Paying attention to the many symptoms that can occur during the course of the disease is critical to improving the quality of life for people with ALS. Therefore, symptomatic treatment is an integral aspect of ALS treatment. Such as: thickened saliva, emotional lability and pain (i.e., musculoskeletal pain and cramps, fasciculations and spasticity, skin pressure pain caused by immobility) (Kiernan et al. 2011). Salivation, or excessive production of saliva, is one of the most disturbing symptoms of ALS patients, and is commonly seen in patients with globular onset and in advanced stages of the disease (Kiernan et al. 2011). Anticholinergic drugs such as atropine, hyrisine, amitriptyline can be used to treat the disease. For emotional lability, treatments such as Amitriptyline, Benzodiazepines, Dextromethorphan hydrobromide/quinidine sulfate can be choice (Kiernan et al. 2011). Pain is reported in 15–85% of ALS patients, and medications include gabapentin, pregabalin, and tricyclic antidepressants. Muscle spasms are the main cause of pain in about a quarter of ALS patients, especially those with spinal onset, and drugs commonly used to treat muscle spasms include quinine sulfate, levetiracetam, and mexetine (Kiernan et al. 2011). In addition, ALS patients may experience fecal incontinence as the disease progresses, delayed colon transport time, and gastric empting (Mazzini et al. 2021). At the same time, some researchers found that SOD1G93A mice also had intestinal leakage, an increase in the number of abnormal intestinal Pan’s cells, and changes in microbial communities (Mazzini et al. 2021). Gut flora can communicate with the central nervous system via the gut-brain axis, a two-way pathway between the central and enteric nervous systems that links the brain’s higher abilities to peripheral intestinal function (Mazzini et al. 2021). Therefore, whether the gut flora can be used as a therapeutic target needs to be further verified.
Others
The peptide drug GM604 was developed as a candidate ALS therapy and is hypothesized to bolster neuron survival through the multi-target regulation of developmental pathways, but mechanisms of action are not fully understood (Table 1) (Swindell et al. 2018). Recently, a Phase II clinical trial was reported, which compared the 2-week results of 8 patients with ALS who received GM604 and 4 patients who received placebo, and GM604 had a favorable safety profile but no efficacy (NCT01854294) (Kindy et al. 2017).
Moreover, it has been demonstrated that an anticoagulation-deficient form of activated protein C (APC), 3K3A-APC, rescued two defects in C9ORF72 and sporadic ALS induced motor neuron models (iMNs): the abnormal accumulation of glutamate receptors and impaired autophagosome formation (Shi et al. 2019). Concomitantly, APC therapy reduced C9ORF72 dipeptide-repeat protein (DPR) levels, restored nuclear localization of TDP-43, and saved the survival of both C9ORF72 and sporadic ALS iMNs(Shi et al. 2019). The phase II clinical trial of 3K3A-APC Protein is undergoing for ALS treatment (NCT05039268).
Conclusion
According to the registration from clinicaltrials.gov, the number of ALS drugs under development fluctuated from 2008 to 2019, with a significant increase between 2019 and 2021 (Fig. 3B). Also, there were many drug development failures (Supplementary Table 1), which may be due to the target itself is not effective, or it may be due to the design of clinical trials (e.g. heterogeneity of the disease, lack of power, questionable endpoint, short follow-up time, etc.), so standardized clinical trial design will help researchers better judge the advantages and disadvantages of the target. In addition, the fundamental pathophysiological mechanisms underlying ALS are not well understood, but the neuropathological hallmark of disease is the aggregation and accumulation of ubiquitylated proteinaceous inclusions in motor neurons. In 97% of ALS subtypes, TDP43 is the primary component of these inclusion bodies. In specific ALS subtypes, other types of protein aggregation, such as neurofilamentous hyaline conglomerate inclusions, sequestosome 1 positive, and the accumulation of misfolded SOD1 can be observed. However, drugs designed to remove ubiquitylated proteinaceous inclusions in motor neurons have not worked well in ALS patients. Therefore, it is necessary to identify the key pathological mechanism of the disease, so as to explicit pathways and targets that drugs can target, and achieve breakthroughs. Currently, tofersen was the first FDA-approved ASO for ALS, and possibly the second was for FUS mutations, which will stimulate research into gene-targeted therapies. Meanwhile, relyvrio’s approval spurred the development of ALS treatments and encourage sponsors to try to break the bottleneck by combining different target drugs.
Data Availability
All data generated or analyzed during this study were included in Table 1 and Supplementary Table 1.
Abbreviations
- ALS:
-
amyotrophic lateral sclerosis
- ROS:
-
radical/reactive oxygen species
- TDP-43:
-
TARDNA-binding protein of 43 kDa
- SOD1:
-
superoxide dismutase1
- FUS:
-
fused-in-sarcoma
- ERS:
-
endoplasmic reticulum stress
- UPR:
-
unfolded protein response
- fALS:
-
familiar ALS
- ASO:
-
antisense oligonucleotide
- ALSFRS-R:
-
Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised
- Nfl:
-
neurofilament light chain
- OLE:
-
non-blind extended study
- ATXN2:
-
Ataxin-2
- FET:
-
FUS/EWSR1/TAF15
- Exportin 1:
-
XPO1
- eIF2B:
-
Eukaryotic translation initiation factor 2B
- HSPs:
-
heat shock proteins
- HSR:
-
heat shock response
- DLK:
-
dual leucine zipper kinase
- JNK:
-
c-Jun N-terminal kinases
- PERK:
-
protein kinase R-like endoplasmic reticulum kinase
- MPO:
-
myeloperoxidase
- GPX4:
-
Glutathione peroxidase 4
- TrkB:
-
tyrosine kinase receptor B
- CuII (atsm):
-
bis(thiosemicarbazone) copper (II)compound
- AuNPs:
-
gold nanoparticles
- D-PUFAs:
-
deuterated polyunsaturated fatty acids
- RIPK1:
-
receptor-interacting serine/threonine-protein kinase 1
- FDA:
-
the U.S. Food and Drug Administration
- EMA:
-
the European Medicines Agency
- MSCs:
-
Mesenchymal stem cells
- NTFs:
-
neurotrophic factors
- HGF:
-
Hepatocyte growth factor
- NIBS:
-
non-invasive brain stimulation
- rTMS:
-
repetitive transcranial magnetic stimulation
- tDCS:
-
transcranial direct current stimulation
- EAAT2:
-
excitatory amino acid transporter2
- FSTAs:
-
fast skeletal muscle troponin activators
- SVC:
-
slow vital capacity
- VEGF:
-
vascular endothelial growth factor
- NADH:
-
nicotinamide adenine dinucleotide hydride
- HSF1:
-
Heat Shock Factor 1
- NDA:
-
new drug applications
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- TCP:
-
Taylor-Couette-Poiseuille
- CSNs:
-
charge-stabilized nanostructures
- PI3K:
-
phosphatidylinositol-3-kinase
- Akt:
-
v-akt murine thymoma viral oncogene homologue
- Foxp3+:
-
forkhead box P3
- APC:
-
activated protein C
- iMNs:
-
induced motor neuron models
- DPR:
-
dipeptide-repeat protein
- ER:
-
endoplasmic reticulum
- Sigma-1 receptor:
-
Sigma non-opioid intracellular receptor 1
- MAM:
-
mitochondria-associated ER membrane
References
Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a Conspiracy of genes, environment and time. Nat Rev Neurol 9(11):617–628. https://doi.org/10.1038/nrneurol.2013.203
Babu S, Hightower BG, Chan J, Zürcher NR, Kivisäkk P, Tseng CJ, Sanders DL, Robichaud A, Banno H, Evora A, Ashokkumar A, Pothier L, Paganoni S, Chew S, Dojillo J, Matsuda K, Gudesblatt M, Berry JD, Cudkowicz ME, Hooker JM, Atassi N (2021) Ibudilast (MN-166) in amyotrophic lateral sclerosis- an open label, safety and pharmacodynamic trial. Neuroimage Clin 30:102672. https://doi.org/10.1016/j.nicl.2021.102672
Baughn MW, Melamed Z, López-Erauskin J, Beccari MS, Ling K, Zuberi A, Presa M, Gonzalo-Gil E, Maimon R, Vazquez-Sanchez S, Chaturvedi S, Bravo-Hernández M, Taupin V, Moore S, Artates JW, Acks E, Ndayambaje IS, Agra de Almeida Quadros AR, Jafar-Nejad P, Rigo F, Bennett CF, Lutz C, Lagier-Tourenne C, Cleveland DW (2023) Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science 379(6637):1140–1149. https://doi.org/10.1126/science.abq5622
Berry JD, Cudkowicz ME, Windebank AJ, Staff NP, Owegi M, Nicholson K, McKenna-Yasek D, Levy YS, Abramov N, Kaspi H, Mehra M, Aricha R, Gothelf Y, Brown RH (2019) NurOwn, phase 2, randomized, clinical trial in patients with ALS: Safety, clinical, and biomarker results. Neurolog 93(24):e2294–e2305. https://doi.org/10.1212/WNL.0000000000008620
Bogorad AM, Lin KY, Marintchev A (2018) eIF2B mechanisms of action and regulation: a thermodynamic view. Biochemistry 57(9):1426–1435. https://doi.org/10.1021/acs.biochem.7b00957
Bonafede R, Mariotti R (2017) ALS Pathogenesis and Therapeutic approaches: the role of mesenchymal stem cells and extracellular vesicles. Front Cell Neurosci 11:80. https://doi.org/10.3389/fncel.2017.00080
Bordet T, Berna P, Abitbol JL, Pruss RM (2010) Olesoxime (TRO19622): a novel mitochondrial-targeted neuroprotective compound. Pharmaceuticals (Basel) 3(2):345–368. https://doi.org/10.3390/ph3020345
Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS (2010) Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol 69(10):987–996. https://doi.org/10.1097/NEN.0b013e3181f2eafa
Cai Y, Arikkath J, Yang L, Guo ML, Periyasamy P, Buch S (2016) Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 12(2):225–244. https://doi.org/10.1080/15548627.2015.1121360
Carpanini SM, Torvell M, Morgan BP (2019) Therapeutic inhibition of the Complement System in Diseases of the Central Nervous System. Front Immunol 10:362. https://doi.org/10.3389/fimmu.2019.00362
Chen JJ (2020) Overview of current and emerging therapies for amytrophic lateral sclerosis. Am J Manag Care 26(9 Suppl). https://doi.org/10.37765/ajmc.2020.88483. S191-S197
Desseille C, Deforges S, Biondi O, Houdebine L, D’amico D, Lamazière A, Caradeuc C, Bertho G, Bruneteau G, Weill L, Bastin J, Djouadi F, Salachas F, Lopes P, Chanoine C, Massaad C, Charbonnier F (2017) Specific physical Exercise improves energetic metabolism in the skeletal muscle of amyotrophic-lateral- sclerosis mice. Front Mol Neurosci 10:332. https://doi.org/10.3389/fnmol.2017.00332
Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, Sobue G (2022) Amyotrophic lateral sclerosis. Lancet. 15;400(10360):1363–1380. https://doi.org/10.1016/S0140-6736(22)01272-7
Ferreira GD, Costa AC, Plentz RD, Coronel CC, Sbruzzi G (2016) Respiratory training improved ventilatory function and respiratory muscle strength in patients with multiple sclerosis and lateral amyotrophic sclerosis: systematic review and meta-analysis. Physiotherapy 102(3):221–228. https://doi.org/10.1016/j.physio.2016.01.002
Funakoshi H, Ohya W, Kadoyama K, Nakamura T (2007) ALS and neurotrophic factors–HGF as a novel neurotrophic and neuroregenerative factor. Brain Nerve 59(10):1195–1202 Japanese. PMID: 17969361
Gavriilaki M, Kimiskidis VK, Gavriilaki E (2020) Precision Medicine in Neurology: the inspirational paradigm of complement therapeutics. Pharmaceuticals (Basel) 13(11):341. https://doi.org/10.3390/ph13110341
Gothelf Y, Abramov N, Harel A, Offen D (2014) Safety of repeated transplantations of neurotrophic factors-secreting human mesenchymal stromal stem cells. Clin Transl Med 3:21. https://doi.org/10.1186/2001-1326-3-21
Grad LI, Rouleau GA, Ravits J, Cashman NR (2017) Clinical spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb Perspect Med 7(8):a024117. https://doi.org/10.1101/cshperspect.a024117
Hardiman O, van den Berg LH (2020) The beginning of genomic therapies for ALS. N Engl J Med 383(2):180–181. https://doi.org/10.1056/NEJMe2012930
Hayashi Y, Homma K, Ichijo H (2016) SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul 60:95–104. https://doi.org/10.1016/j.jbior.2015.10.006
Herrando-Grabulosa M, Gaja-Capdevila N, Vela JM, Navarro X (2021) Sigma 1 receptor as a therapeutic target for Amyotrophic Lateral Sclerosis. Br J Pharmacol 178(6):1336–1352. https://doi.org/10.1111/bph.15224
Hilton JB, Mercer SW, Lim NK, Faux NG, Buncic G, Beckman JS, Roberts BR, Donnelly PS, White AR, Crouch PJ (2017) CuII(atsm) improves the neurological phenotype and survival of SOD1G93A mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci Rep 13:7:42292. https://doi.org/10.1038/srep42292
Howard JF Jr, Nowak RJ, Wolfe GI, Freimer ML, Vu TH, Hinton JL, Benatar M, Duda PW, MacDougall JE, Farzaneh-Far R, Kaminski HJ, Zilucoplan MGS, Group, Barohn R, Dimachkie M, Pasnoor M, Farmakidis C, Liu T, Colgan S, Benatar MG, Bertorini T, Pillai R, Henegar R, Bromberg M, Gibson S, Janecki T, Freimer M, Elsheikh B, Matisak P, Genge A, Guidon A, David W, Habib AA, Mathew V, Mozaffar T, Hinton JL, Hewitt W, Barnett D, Sullivan P, Ho D, Howard JF Jr, Traub RE, Chopra M, Kaminski HJ, Aly R, Bayat E, Abu-Rub M, Khan S, Lange D, Holzberg S, Khatri B, Lindman E, Olapo T, Sershon LM, Lisak RP, Bernitsas E, Jia K, Malik R, Lewis-Collins TD, Nicolle M, Nowak RJ, Sharma A, Roy B, Nye J, Pulley M, Berger A, Shabbir Y, Sachdev A, Patterson K, Siddiqi Z, Sivak M, Bratton J, Small G, Kohli A, Fetter M, Vu T, Lam L, Harvey B, Wolfe GI, Silvestri N, Patrick K, Zakalik K, Duda PW, MacDougall J, Farzaneh-Far R, Pontius A, Hoarty M (2020) Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized Myasthenia gravis: results of a phase 2 Randomized, Double-Blind, Placebo-Controlled, Multicenter Clinical Trial. JAMA Neurol 77(5):582–592. https://doi.org/10.1001/jamaneurol.2019.5125
Ionescu A, Gradus T, Altman T, Maimon R, Saraf Avraham N, Geva M, Hayden M, Perlson E (2019) Targeting the Sigma-1 receptor via Pridopidine Ameliorates Central Features of ALS Pathology in a SOD1G93A model. Cell Death Dis 10(3):210. https://doi.org/10.1038/s41419-019-1451-2
Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan JB, Pasparakis M, Kelliher MA, Ravits J, Yuan J (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353(6299):603–608. https://doi.org/10.1126/science.aaf6803
Jaiswal MK (2019) Riluzole and edaravone: a tale of two Amyotrophic Lateral Sclerosis Drugs. Med Res Rev 39(2):733–748. https://doi.org/10.1002/med.21528
Jiang J, Wang Y, Deng M (2022) New developments and opportunities in Drugs being trialed for Amyotrophic Lateral Sclerosis from 2020 to 2022. Front Pharmacol 13:1054006. https://doi.org/10.3389/fphar.2022.1054006
Juntas-Morales R, Pageot N, Bendarraz A, Alphandéry S, Sedel F, Seigle S, Camu W (2020) High-dose pharmaceutical grade biotin (MD1003) in Amyotrophic Lateral Sclerosis: a pilot study. EClinicalMedicine 19:100254. https://doi.org/10.1016/j.eclinm.2019.100254
Katz JS, Rothstein JD, Cudkowicz ME, Genge A, Oskarsson B, Hains AB, Chen C, Galanter J, Burgess BL, Cho W, Kerchner GA, Yeh FL, Ghosh AS, Cheeti S, Brooks L, Honigberg L, Couch JA, Rothenberg ME, Brunstein F, Sharma KR, van den Berg L, Berry JD, Glass JD (2022) A phase 1 study of GDC-0134, a dual leucine zipper kinase inhibitor, in ALS. Ann Clin Transl Neuro 9(1):50–66. https://doi.org/10.1002/acn3.51491
Kaur SJ, McKeown SR, Rashid S (2016) Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene577(2):109 – 18. https://doi.org/10.1016/j.gene.2015.11.049
Kawahara Y, Kwak S (2005) Excitotoxicity and ALS: what is unique about the AMPA receptors expressed on spinal motor neurons? Amyotroph lateral Scler Other Motor Neuron Disord. 6(3):131–144. https://doi.org/10.1080/14660820510037872
Keifer OP Jr, O’Connor DM, Boulis NM (2014) Gene and protein therapies utilizing VEGF for ALS. Pharmacol Ther 141(3):261–271. https://doi.org/10.1016/j.pharmthera.2013.10.009
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis. Lancet 12;377(9769):942 – 55. https://doi.org/10.1016/S0140-6736(10)61156-7
Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD (2020) ALS Genetics: gains, losses, and implications for future therapies. Neuro 108(5):822–842. https://doi.org/10.1016/j.neuron.2020.08.022
Kindy M, Lupinacci P, Chau R, Shum T, Ko D (2017) A phase 2A randomized, double-blind, placebo-controlled pilot trial of GM604 in patients with Amyotrophic Lateral Sclerosis (ALS Protocol GALS-001) and a single compassionate patient treatment (protocol GALS-C). https://doi.org/10.12688/f1000research.10519.1. F1000Res 6:230
Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA (2022) Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in Amyotrophic Lateral Sclerosis. Nat Med 28(1):104–116. https://doi.org/10.1038/s41591-021-01615-z
Kukharsky MS, Skvortsova VI, Bachurin SO, Buchman VL (2021) In a search for efficient treatment for Amyotrophic Lateral Sclerosis: old Drugs for new approaches. Med Res re 41(5):2804–2822. https://doi.org/10.1002/med.21725
Lazarev VF, Sverchinskyi DV, Ippolitova MV, Stepanova AV, Guzhova IV, Margulis BA (2013) Factors affecting aggregate formation in cell models of Huntington’s Disease and Amyotrophic Lateral Sclerosis. Acta Naturae 5(2):81–89
Lazzarino G, Mangione R, Belli A, Di Pietro V, Nagy Z, Barnes NM, Bruce L, Ropero BM, Persson LI, Manca B, Saab MW, Amorini AM, Tavazzi B, Lazzarino G, Logan A (2021) ILB® attenuates clinical symptoms and Serum Biomarkers of Oxidative/Nitrosative Stress and Mitochondrial Dysfunction in patients with Amyotrophic Lateral Sclerosis. J Pers Med 11(8):794. https://doi.org/10.3390/jpm11080794
Li X, Chen C, Zhan X, Li B, Zhang Z, Li S, Xie Y, Song X, Shen Y, Liu J, Liu P, Liu GP, Yang X (2021) R13 preserves motor performance in SOD1G93A mice by improving mitochondrial function. Theranostics 11(15):7294–7307. https://doi.org/10.7150/thno.56070
Liu J, Wang F (2017) Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front Immunol. 2017;8:1005. https://doi.org/10.3389/fimmu.2017.01005
Liu Y, Andreucci A, Iwamoto N, Yin Y, Yang H, Liu F, Bulychev A, Hu XS, Lin X, Lamore S, Patil S, Mohapatra S, Purcell-Estabrook E, Taborn K, Dale E, Vargeese C (2022) Preclinical evaluation of WVE-004, aninvestigational stereopure oligonucleotide forthe treatment of C9orf72-associated ALS or FTD. Mol Ther Nucleic Acids 28:558–570. https://doi.org/10.1016/j.omtn.2022.04.007
Logan A, Nagy Z, Barnes NM, Belli A, Di Pietro V, Tavazzi B, Lazzarino G, Lazzarino G, Bruce L, Persson LI (2022) A phase II open label clinical study of the safety, tolerability and efficacy of ILB® for Amyotrophic Lateral Sclerosis. PLoS ONE 17(5):e0267183. https://doi.org/10.1371/journal.pone.0267183
Mazzini L, De Marchi F, Niccolai E, Mandrioli J, Amedei A (2021) Gastrointestinal status and microbiota shaping in Amyotrophic Lateral Sclerosis: a New Frontier for Targeting? In: Araki T (ed) Amyotrophic Lateral Sclerosis [Internet]. Exon Publications, Brisbane (AU). Chap. 8. PMID: 34473437.
McAlary L, Plotkin SS, Yerbury JJ, Cashman NR (2019) Prion-like propagation of protein misfolding and aggregation in Amyotrophic Lateral Sclerosis. Front Mol Neurosci 12:262. https://doi.org/10.3389/fnmol.2019.00262
Medical Advisory Secretariat (2021) Endovascular radiofrequency ablation for varicose veins: an evidence-based analysis. Ont Health Technol Assess Ser 11(1):1–93
Meininger V, Genge A, van den Berg LH, Robberecht W, Ludolph A, Chio A, Kim SH, Leigh PN, Kiernan MC, Shefner JM, Desnuelle C, Morrison KE, Petri S, Boswell D, Temple J, Mohindra R, Davies M, Bullman J, Rees P, Lavrov A (2017) Safety and efficacy of ozanezumab in patients with Amyotrophic Lateral Sclerosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol 16(3):208–216. https://doi.org/10.1016/S1474-4422(16)30399-4
Miller R, Bradley W, Cudkowicz M, Hubble J, Meininger V, Mitsumoto H, Moore D, Pohlmann H, Sauer D, Silani V, Strong M, Swash M, Vernotica (2007) Phase II/III randomized trial of TCH346 in patients with ALS. Neurology 69(8):776–784. https://doi.org/10.1212/01.wnl.0000269676.07319.09
Miller RG, Mitchell JD, Moore DH (2012) Riluzole for Amyotrophic Lateral Sclerosis (ALS)/motor neuron Disease (MND). Cochrane Database Syst Rev2012 3CD001447. https://doi.org/10.1002/14651858.CD001447.pub3
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial Amyotrophic Lateral Sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12(5):435–442. https://doi.org/10.1016/S1474-4422(13)70061-9
Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, Chiò A, Van Damme P, Ludolph AC, Glass JD, Andrews JA, Babu S, Benatar M, McDermott CJ, Cochrane T, Chary S, Chew S, Zhu H, Wu F, Nestorov I, Graham D, Sun P, McNeill M, Fanning L, Ferguson TA, Fradette S (2022) Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Me 387(12):1099–1110. https://doi.org/10.1056/NEJMoa2204705
Miller RG, Zhang R, Bracci PM, Azhir A, Barohn R, Bedlack R, Benatar M, Berry JD, Cudkowicz M, Kasarskis EJ, Mitsumoto H, Manousakis G, Walk D, Oskarsson B, Shefner J, McGrath MS (2022a) Phase 2B randomized controlled trial of NP001 in Amyotrophic Lateral sclerosis: pre-specified and post hoc analyses. Muscle Nerve 66(1):39–49. https://doi.org/10.1002/mus.27511
Mora JS, Bradley WG, Chaverri D, Hernández-Barral M, Mascias J, Gamez J, Gargiulo-Monachelli GM, Moussy A, Mansfield CD, Hermine O, Ludolph AC (2021) Long-term survival analysis of masitinib in Amyotrophic Lateral Sclerosis. Ther Adv Neurol Disord 19:14:17562864211030365. https://doi.org/10.1177/17562864211030365
Nolan M, Talbot K, Ansorge O (2016) Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun 4(1):99. https://doi.org/10.1186/s40478-016-0358-8
Oskarsson B, Maragakis N, Bedlack RS, Goyal N, Meyer JA, Genge A, Bodkin C, Maiser S, Staff N, Zinman L, Olney N, Turnbull J, Brooks BR, Klonowski E, Makhay M, Yasui S, Matsuda K (2021) MN-166 (ibudilast) in Amyotrophic Lateral Sclerosis in a phase IIb/III study: COMBAT-ALS study design. Neurodegener Dis Manag 11(6):431–443. https://doi.org/10.2217/nmt-2021-0042
Paganoni S, Hendrix S, Dickson SP, Knowlton N, Macklin EA, Berry JD, Elliott MA, Maiser S, Karam C, Caress JB, Owegi MA, Quick A, Wymer J, Goutman SA, Heitzman D, Heiman-Patterson TD, Jackson CE, Quinn C, Rothstein JD, Kasarskis EJ, Katz J, Jenkins L, Ladha S, Miller TM, Scelsa SN, Vu TH, Fournier CN, Glass JD, Johnson KM, Swenson A, Goyal NA, Pattee GL, Andres PL, Babu S, Chase M, Dagostino D, Hall M, Kittle G, Eydinov M, McGovern M, Ostrow J, Pothier L, Randall R, Shefner JM, Sherman AV, St Pierre ME, Tustison E, Vigneswaran P, Walker J, Yu H, Chan J, Wittes J, Yu ZF, Cohen J, Klee J, Leslie K, Tanzi RE, Gilbert W, Yeramian PD, Schoenfeld D, Cudkowicz ME (2021) Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in Amyotrophic Lateral Sclerosis. Muscle Nerve 63(1):31–39. https://doi.org/10.1002/mus.27091
Pallardó FV, Pagano G, Rodríguez LR, Gonzalez-Cabo P, Lyakhovich A, Trifuoggi M (2021) Friedreich Ataxia: current state-of-the-art, and future prospects for mitochondrial-focused therapies. Transl Res 229:135–141. https://doi.org/10.1016/j.trsl.2020.08.009
Pang W, Hu F (2021) Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J Neurochem 157(3):334–350. https://doi.org/10.1111/jnc.15255
Petrou P, Kassis I, Yaghmour NE, Ginzberg A, Karussis D (2021) A phase II clinical trial with repeated intrathecal injections of autologous mesenchymal stem cells in patients with Amyotrophic Lateral Sclerosis. Front Biosci (Landmark Ed) 26(10):693–706. https://doi.org/10.52586/4980
Pierce A, Mirzaei H, Muller F, De Waal E, Taylor AB, Leonard S, Van Remmen H, Regnier F, Richardson A, Chaudhuri A (2008) GAPDH is conformationally and functionally altered in association with oxidative stress in mouse models of Amyotrophic Lateral Sclerosis. J Mol Biol 382(5):1195–1210. https://doi.org/10.1016/j.jmb.2008.07.088
Pognan F, Buono C, Couttet P, Galarneau JR, Timsit Y, Wolf A (2022) Liver enzyme delayed clearance in rat treated by CSF1 receptor specific antagonist Sotuletinib. Curr Res Toxicol 3:100091. https://doi.org/10.1016/j.crtox.2022.100091
Ranieri F, Mariotto S, Dubbioso R, Di Lazzaro V (2021) Brain stimulation as a therapeutic Tool in Amyotrophic Lateral Sclerosis: current Status and Interaction with mechanisms of altered cortical excitability. Front Neurol 11:605335. https://doi.org/10.3389/fneur.2020.605335
Rhinn H, Tatton N, McCaughey S, Kurnellas M, Rosenthal A (2022) Progranulin as a therapeutic target in neurodegenerative Diseases. Trends Pharmacol Sci 43(8):641–652. https://doi.org/10.1016/j.tips.2021.11.015
Rothstein JD (2017) Edaravone: a new drug approved for ALS. Cell 171(4):725. https://doi.org/10.1016/j.cell.2017.10.011
Sano R, Reed JC (2013) ER stress-induced cell death mechanisms. Biochim Biophys Act 1833(12):3460–3470. https://doi.org/10.1016/j.bbamcr.2013.06.028
Seminary ER, Sison SL, Ebert AD (2018) Modeling protein aggregation and the heat shock response in ALS iPSC-Derived motor neurons. Front Neurosci 12:86. https://doi.org/10.3389/fnins.2018.00086
Shefner JM, Andrews JA, Genge A, Jackson C, Lechtzin N, Miller TM, Cockroft BM, Meng L, Wei J, Wolff AA, Malik FI, Bodkin C, Brooks BR, Caress J, Dionne A, Fee D, Goutman SA, Goyal NA, Hardiman O, Hayat G, Heiman-Patterson T, Heitzman D, Henderson RD, Johnston W, Karam C, Kiernan MC, Kolb SJ, Korngut L, Ladha S, Matte G, Mora JS, Needham M, Oskarsson B, Pattee GL, Pioro EP, Pulley M, Quan D, Rezania K, Schellenberg KL, Schultz D, Shoesmith C, Simmons Z, Statland J, Sultan S, Swenson A, Berg LHVD, Vu T, Vucic S, Weiss M, Whyte-Rayson A, Wymer J, Zinman L, Rudnicki SA (2021) A phase 2, Double-Blind, randomized, dose-ranging trial of Reldesemtiv in patients with ALS. Amyotroph Lateral Scler Frontotemporal Degene 22(3–4):287–299. https://doi.org/10.1080/21678421.2020.1822410
Shi Y, Hung ST, Rocha G, Lin S, Linares GR, Staats KA, Seah C, Wang Y, Chickering M, Lai J, Sugawara T, Sagare AP, Zlokovic BV, Ichida JK (2019) Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 5(15):e127736. https://doi.org/10.1172/jci.insight.127736
Singh A, Kukreti R, Saso L, Kukreti S (2019) Oxidative stress: a key modulator in neurodegenerative Diseases. Molecules 24(8):1583. https://doi.org/10.3390/molecules24081583
Sinha K, Das J, Pal PB, Sil PC (2013) Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 87(7):1157–1180. https://doi.org/10.1007/s00204-013-1034-4
Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, Barnham KJ, Crouch PJ, Ayton S, Donnelly PS, Bush AI (2020) CuII (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative Disease. Br J Pharmacol 177(3):656–667. https://doi.org/10.1111/bph.14881
St Martin JL, Wang L, Kaprielian Z (2020) Toxicity in ALS: TDP-43 modifiers and C9orf72. Neurosci Lett 716:134621. https://doi.org/10.1016/j.neulet.2019.134621
Sufit RL, Ajroud-Driss S, Casey P, Kessler JA (2017) Open label study to assess the safety of VM202 in subjects with Amyotrophic Lateral Sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 18(3–4):269–278. https://doi.org/10.1080/21678421.2016.1259334
Swindell WR, Bojanowski K, Kindy MS, Chau RMW, Ko D (2018) GM604 regulates developmental neurogenesis pathways and the expression of genes associated with Amyotrophic Lateral Sclerosis. Transl Neurodegene 7:30. https://doi.org/10.1186/s40035-018-0135-7
Tiryaki E, Horak HA (2014) ALS and other motor neuron diseases. Continuum (Minneap Minn) 20(5 Peripheral Nervous System Disorders):1185 – 207. https://doi.org/10.1212/01.CON.0000455886.14298.a4
Ustyantseva EI, Medvedev SP, Zakian SM (2020) Studying ALS: current approaches, effect on potential treatment strategy. Adv Exp Med Biol 1241:195–217. https://doi.org/10.1007/978-3-030-41283-8_11
Vacchiano V, Mastrangelo A, Zenesini C, Masullo M, Quadalti C, Avoni P, Polischi B, Cherici A, Capellari S, Salvi F, Liguori R, Parchi P (2021) Plasma and CSF neurofilament light chain in Amyotrophic Lateral Sclerosis: a cross-sectional and longitudinal study. Front Aging Neurosc 13:753242. https://doi.org/10.3389/fnagi.2021.753242
Vallarola A, Sironi F, Tortarolo M, Gatto N, De Gioia R, Pasetto L, De Paola M, Mariani A, Ghosh S, Watson R, Kalmes A, Bonetto V, Bendotti C (2018) RNS60 exerts therapeutic effects in the SOD1 ALS mouse model through protective glia and peripheral nerve rescue. J Neuroinflammation 15(1):65. https://doi.org/10.1186/s12974-018-1101-0
Vijayalakshmi K, Ostwal P, Sumitha R, Shruthi S, Varghese AM, Mishra P, Manohari SG, Sagar BC, Sathyaprabha TN, Nalini A, Raju TR, Alladi PA (2015) Role of VEGF and VEGFR2 receptor in reversal of ALS-CSF Induced Degeneration of NSC-34 Motor Neuron Cell line. Mol Neurobiol 51(3):995–1007. https://doi.org/10.1007/s12035-014-8757-y
Vucic S, Kiernan MC, Menon P, Huynh W, Rynders A, Ho KS, Glanzman R, Hotchkin MT (2021) Study protocol of RESCUE-ALS: a phase 2, randomised, double-blind, placebo-controlled study in early symptomatic Amyotrophic Lateral Sclerosis patients to assess bioenergetic catalysis with CNM-Au8 as a mechanism to slow Disease progression. BMJ Open 11(1):e041479. https://doi.org/10.1136/bmjopen-2020-041479
Wang SM, Wu HE, Yasui Y, Geva M, Hayden M, Maurice T, Cozzolino M, Su TP (2013) Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophag 19(1):126–151. https://doi.org/10.1080/15548627.2022.2063003
Wang L, Yi F, Fu L, Yang J, Wang S, Wang Z, Suzuki K, Sun L, Xu X, Yu Y, Qiao J, Belmonte JCI, Yang Z, Yuan Y, Qu J, Liu GH (2017) CRISPR/Cas9-mediated targeted gene correction in Amyotrophic Lateral Sclerosis patient iPSCs. Protein Cell 8(5):365–378. https://doi.org/10.1007/s13238-017-0397-3
Wang Y, Liu FT, Wang YX, Guan RY, Chen C, Li DK, Bu LL, Song J, Yang YJ, Dong Y, Chen Y, Wang J (2018) Autophagic modulation by Trehalose reduces Accumulation of TDP-43 in a cell model of Amyotrophic Lateral Sclerosis via TFEB activation. Neurotox Res 34(1):109–120. https://doi.org/10.1007/s12640-018-9865-7
Wang T, Tomas D, Perera ND, Cuic B, Luikinga S, Viden A, Barton SK, McLean CA, Samson AL, Southon A, Bush AI, Murphy JM, Turner BJ (2021) Ferroptosis mediates selective motor neuron death in Amyotrophic Lateral Sclerosis. Cell Death Differ 29(6):1187–1198. https://doi.org/10.1038/s41418-021-00910-z
Xiong L, McCoy M, Komuro H, West XZ, Yakubenko V, Gao D, Dudiki T, Milo A, Chen J, Podrez EA, Trapp B, Byzova TV (2022) Inflammation-dependent oxidative stress metabolites as a hallmark of Amyotrophic Lateral Sclerosis. Free Radic Biol Med 178:125–133. https://doi.org/10.1016/j.freeradbiomed.2021.11.031
Yerton M, Winter A, Kostov A, Lieberman C, Gelevski D, Weber H, Doyle M, Kane G, Parikh N, Burke KM, Rohrer M, Stirrat T, Bruno M, Hochman A, Luppino S, Scalia J, Skoniecki D, D’Agostino D, Sinani E, Yu H, Sherman AV, Babu S, Berry JD, Midei MG, Milner PG, Cudkowicz ME, Paganoni S (2022) An expanded access protocol of RT001 in amyotrophic lateral sclerosis-initial experience with a lipid peroxidation inhibitor. Muscle Nerve 66(4):421–425. https://doi.org/10.1002/mus.27672
Zhang R, Bracci PM, Azhir A, Forrest BD, McGrath MS (2022) Macrophage-targeted Sodium Chlorite (NP001) slows progression of Amyotrophic Lateral Sclerosis (ALS) through regulation of Microbial translocation. Biomedicines 10(11):2907. https://doi.org/10.3390/biomedicines10112907
Acknowledgements
Not applicable.
Funding
This project was supported by the grants from National Natural Sciences Foundation of China (No. 82073835, and 81872855), CAMS Innovation Fund for Medical Sciences (No. 2021-I2M-1-054), and Disciplines construction project (201920200802).
Author information
Authors and Affiliations
Contributions
Zhuo Sun performed the literature search, created the table and figures. Ying Peng and Bo Zhang contributed to the writing and editing of the manuscript. All authors read and approved this manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Table 1.
The overview of novel treatments for ALS that are terminated. The table summarizes the specific information of treatments terminated for lack of efficacy or safety
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sun, Z., Zhang, B. & Peng, Y. Development of novel treatments for amyotrophic lateral sclerosis. Metab Brain Dis 39, 467–482 (2024). https://doi.org/10.1007/s11011-023-01334-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-023-01334-z