
Abstract
Cerebral cavernous malformations (CCM) consist of clusters of irregular dilated capillaries and represent the second most common type of vascular malformation affecting the central nervous system. CCM might be asymptomatic or cause cerebral hemorrhage, seizures, recurrent headaches and focal neurologic deficits. Causative mutations underlining CCM have been reported in three genes: KRIT1/CCM1, MGC4607/CCM2 and PDCD10/CCM3. Therapeutic avenues are limited to surgery. Here we present clinical, neuroradiological and molecular findings in a cohort of familial and sporadic CCM patients. Thirty subjects underwent full clinical and radiological assessment. Molecular analysis was performed by direct sequencing and MLPA analysis. Twenty-eight of 30 subjects (93%) experienced one or more typical CCM disturbances with cerebral/spinal hemorrhage being the most common (43%) presenting symptom. A molecular diagnosis was achieved in 87% of cases, with three novel mutations identified. KRIT1/CCM1 patients displayed higher risk of de novo CCMs appearance and bleedings. Magnetic Resonance Imaging (MRI) showed that infratentorial region was more frequently affected in mutated subjects while brainstem was often spared in patients with negative genetic testing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral cavernous malformations (CCMs, OMIM 116,860) consist of delimited mulberry-shaped vascular anomalies characterized by clusters of enlarged, leaky capillaries mainly located in the brain. CCMs occur in 1% of the general population and represent the 10–20% of vascular malformations of the central nervous system (Zabramski et al. 1994; Moriarity et al. 1999).
The gold standard for detection of CCMs is magnetic resonance imaging (MRI) where they appear as focal lesions with a peripheral T2 hypointensity due to hemosiderin deposition which can be disclosed with higher sensitivity on T2* Gradient Echo (GRE) and Susceptibility Weighted Imaging (SWI) sequences. CCMs cannot be detected by Digital Subtraction Angiography (DSA) as they are angiographically silent. Given their fragility, they predispose to recurrent hemorrhage with an estimated rate of 4–11% in Caucasian patients (Al-Shahi Salman et al. 2012; Flemming et al. 2012). Symptomatic disease consists of hemorrhagic stroke, headaches, focal neurological deficits and seizures usually occurring between the third and fifth decade of life. However, CCM lesions can be also detected incidentally by MRI in up to 50% of asymptomatic subjects (Moore et al. 2014).
CCMs can occur sporadically or as a familial disorder (FCCM) (Labauge et al. 2007). The presence of multiple CCMs is a common evidence in FCCM and the number of lesions correlates with patients’ age. Few reports have described extra-neural presentations in FCCM mainly affecting the eye (retina), liver, kidney, vertebral bodies, and skin (Sirvente et al. 2009; Toll et al. 2009; Lan et al. 2010; Grippaudo et al. 2013). FCCM prevalence is variable ranging from 6 to 50% of patients (Rigamonti et al. 1988; Washington et al. 2010; Moore et al. 2014).
About 70% of FCCM patients harbor molecular defects in KRIT1/CCM1 (KREV-1/RAP1A Interaction Trapped 1, 7q21.2-q22, MIM #6,024,214) (Laberge-le Couteulx et al. 1999) while mutations in MGC4607/CCM2 (Malcavernin, 7p15-p13, MIM #603,284) (Liquori et al. 2003) and PDCD10/CCM3 (Programmed Cell Death 10, 3q25.2-q27, MIM #603,285) (Bergametti et al. 2005) account for 20% and 10% of genetically determined cases, respectively (Labauge et al. 2007). Germline mutations are dominantly inherited, but the analysis of affected tissues often discloses a second somatic mutation required to prime the pathogenetic cascade according a two-hit mechanism (Pagenstecher et al. 2009).
Clinical penetrance is incomplete for KRIT1/CCM1 (88%) and PDCD10/CCM3 (63%) defects (de Vos et al. 2017). Full penetrance for MGC4607/CCM2 was also recently questioned (Scimone et al. 2018). Conventional testing of CCM genes is negative in less than 10% of FCCM and 43% of sporadic subjects, suggesting that molecular defects escaping routinely testing (i.e., deep intronic mutations, complex genomic rearrangements) or somatic mutations in known genes might contribute to CCM genetics (Choquet et al. 2015). Alternatively, additional genes might be involved in CCM pathogenesis, as suggested by a recent report investigating CCM patients tested negative for both germline and somatic mutations (Scimone et al. 2020).
Here we describe the clinical and neuroradiological features of 30 CCM patients in view of their genetic backgrounds.
Methods
This study was approved by Ethical Committee Area 2 Milan (ID:721). The experimental protocols were approved by Ethical Committee Area 2 Milan (ID:721) and performed in accordance with the institutional review board of the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico. The patients provided written informed consent for all aspects of the study.
We evaluated a series of 30 patients with multiple CCMs (25 symptomatic, 5 incidental findings) referred to our Institution over a four-year period (2014–2017).
All subjects underwent a full clinical evaluation, detailed clinical history was collected targeted to specific CCM related symptoms (headache, focal neurological deficits, seizures, cerebral and spinal hemorrhage) at onset and during the follow-up period, extra-neurological CCM location (eye, skin) and positive familial history.
MRI examinations were retrospectively collected and reviewed. Only MRI examinations with at least T1 weighted images, Turbo Spin Echo (TSE) T2 weighted images, FLAIR images and one of the two, GRE T2 weighted images or SWI images, were included in this review. Expert reading was performed by two experienced radiologists in consensus and CCMs classified according to Zabramski et al. (1994). Our analysis included: number of CCMs according to the anatomical site (brainstem, cerebellum, basal ganglia and thalami, cerebral hemispheres), acute hemorrhage and their location. In consecutive MRI scans, we recorded the modification in size and MRI signal of CCMs, the MRI signal and location of de novo CCMs (if present), the appearance and location of new acute/subacute hemorrhage.
CCMs were numbered according to numerosity classes for each anatomical site: 0 (absence of CCMs in that region), 1 (1–10 CCMs), 2 (11–20 CCMs), 3 (21–30 CCMs), 4 (31–40 CCMs) and 5 (> 41 CCMs). In addition, a target CCM was chosen for each anatomical site according to criteria derived from the RECIST guidelines (Eisenhauer et al. 2009). For the MRI review one target lesion was chosen for each anatomical site according to the following criteria, derived from the RECIST guideline criteria 1: when more than one measurable CCMs were present at baseline in one of the four regions assessed, one CCM lesion representative of that region was identified as target lesion and was recorded and measured at baseline and in the follow up MR studies. Considering the specific risk of hemorrhage of CCMs, target lesions were selected also on the basis of the presence of MRI signs of acute/subacute hemorrhage and not only on the basis of their size, as for RECIST criteria: the CCM with MRI signs of acute/subacute hemorrhage was identified as target lesion and, if none of the CCMs of one region presented MRI signs of acute/subacute hemorrhage, the target lesion was depicted as the lesion with the longest diameter. Additionally, when the largest CCM did not allow a reproducible measurement then the next largest lesion with better reproducible measurement was selected.
Written informed consent for genetic testing was provided by all the patients. Genomic DNA was extracted from peripheral blood lymphocytes of probands and available relatives. KRIT1 (NM_194456.1), CCM2 (NM_031443.3) and PDCD10 (NM_007217.3) coding exons and their intronic boundaries were PCR-amplified and sequenced using primers and conditions available on request. Exon or multiexon deletions/duplications were investigated by using multiple ligation-dependent probe amplification (MLPA). MLPA reactions were performed as previously described using probe mixes SALSA MLPA Kits P130 (A4-0119) & P131 (B1-0918) (MRC Holland, Netherlands) and electrophoresed on an ABI PRISM 3130 Genetic analyzer, in accordance with the manufacturer’s instructions. Data analysis was performed by using Coffalyser Software (MRC-Holland BV).
Total RNA, obtained from peripheral blood of the probands (Tempus Blood RNA Isolation Kit, Thermo Fisher) and healthy controls, was retrotranscribed (HiFi Transcriptor cDNA Synthesis kit, Roche) into cDNA. RT-PCR amplicons were obtained and sequenced by using specific primers.
Results
Thirty subjects from 20 independent families (male to female ratio 1.7) carrying multiple (> 3) CCMs underwent clinical, genetic and radiological evaluation at Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan. Independent cases were classified as familial (FCCM, n = 10) or sporadic cases (n = 10). Mean age of clinical onset was 39.6 (range 4–63). All the patients underwent cerebral MRI (100%). Whole spinal MRI was performed in 16 cases (53%) showing spinal CCMs in 10 patients. Cutaneous angiomas were observed in 8 cases (27%). Vertebral and liver angiomas were observed in 3 and 2 patients, respectively.
Clinical evaluation
One or more typical CCM clinical manifestations including seizures, recurrent headache, intracerebral hemorrhage (ICH), and focal neurological deficits (FND) occurred in 28/30 subjects (93%). In 13 cases (43%) the presenting manifestation was a symptomatic cerebral or medullary hemorrhage manifesting with FND (n = 11); in the remaining subjects, hemorrhage caused seizures or headache. No literature data are available about the incidence of hemorrhage causing headache and no cases were previously reported in CCM cohort of patients (de Vos et al. 2017).
57% of patients (n = 17) did not experience hemorrhagic events during the whole observational time. In this group, the first clinical manifestation was seizures (n = 5), FND (n = 4), headache (n = 3). Five asymptomatic patients were diagnosed incidentally or in the context of a radiological screening for positive familial history: only two of them remained asymptomatic during the whole observational time.
In symptomatic patients, the most frequent manifestations were ICH (50%) and headache (43%). One patient suffered from migraine, four patients presented headache after ICH (one suffered trigeminal neuralgia), eight patients suffered from chronic tensive headache). Other frequent symptoms included: sensory loss (30%), limb weakness (23%), visual disorders (23%), dizziness (23%), ataxia or postural instability (20%), speech disturbance (17%), facial palsy (17%) and other symptoms such as nausea and vomiting (17%). Rare manifestations included insomnia, cognitive impairment, tinnitus and dysphagia.
At follow-up only three subjects showed hemorrhagic events: two patients presenting epilepsy and FND at onset experienced spinal and cerebral hemorrhage. One patient presented left parieto-occipital hemorrhage requiring surgery 9 years after the first ICH. Additional events recorded during the follow-up were: onset of recurrent headache (n = 6) and seizures (n = 2) or FND (n = 1).
Molecular findings
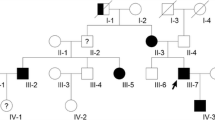
A molecular diagnosis was established in 26 patients (87%). Molecular screening in familial cases disclosed causative mutations in KRIT1 (n = 6, 60%) and CCM2 (n = 4, 40%) segregating with the phenotype in available family members. Germline mutations were found in 6 sporadic patients (60%, 4 KRIT1 mutations, 1 splice-site mutation in CCM2 and 1 PDCD10 large deletion). Four isolated subjects did not display any defect in CCM genes (Fig. 1).

Distribution of variants by genetic results in our cohort of CCM patients (20 independent cases)
Fifteen different mutations were identified in this study (Supplementary Table 1). As expected, most of them likely result in the loss of the affected allele: large deletions (n = 5), frameshift (n = 3) or non-sense (n = 1) mutations. Five mutations affect conserved splicing sites.
In this report we detected three molecular defects which were not previously published and are absent in public-available databases (LOVD, gnomAD) (Supplementary Fig. 1A). Segregation studies were performed in available family members (Supplementary Fig. 2).
Two cis-linked single-base substitutions (Tandem Base Substitution, TBS) were observed in KRIT1 in two affected siblings. This variant of unknown significance (VUS), as classified according ACGM guidelines, results in the amino acid change p.Ala13Asn which is predicted as pathogenetic by multiple tools (Polyphen, SIFT, CADD, Mutation Taster). Interestingly, somatic and germline TBS mutations have been previously observed in several genes involved in cellular proliferation, including KRIT1 (Chen et al. 2013).
In CCM2, the c.915 + 2 T > C substitution resulted in the loss of a conserved donor site and the partial retention of Intron 8–9 in CCM2 transcript (Supplementary Fig. 1B). The c.745 + 1G > T mutation was found in 3 patients from 2 families. RT-PCR analysis disclosed the reduction of the full-length CCM2 isoform in favor of two alternative transcripts (Supplementary Fig. 1C).
No clear association emerged by matching clinical features with genetic background (Fig. 2).

Relative frequencies of patients’ demographical and clinical features according to patients’ genotype
Neuroradiological findings
MRI examinations were available for systematic revision in 28 patients (single MRI scan: 10 patients, 2 MRI scans: 9 patients, 3 MRI scans: 9 patients).
All subjects harbored multiple CCMs, ranging from hundreds (CCM2 mutated) to only 5 CCMs (negative genetic testing). In FCCM patients, CCMs were observed in the supratentorial compartment and at least one CCM in the cerebral hemispheres was detected, irrespective of the molecular defect (Supplementary Fig. 3). The infratentorial compartment was more frequently affected in patients with proven genetic disease (more than 80% in CCM1 and CCM2 vs 50%). Moreover, we observed that brainstem was usually spared in patients without proven mutation and frequently involved in CCM1 (62.5%) and CCM2 patients (86%). The cerebellar hemispheres and vermis were more frequently involved in CCM1 patients (73% of patients) compared to other subgroups.
Seven of 16 (44%) CCM1 patients and 3 of 7 (43%) CCM2 patients had at least 20 CCMs in the cerebral hemispheres, even when the infratentorial compartment was relatively spared.
To better describe the lesion evolution over time, we decided to select a target lesion, usually the largest one and/or the one with signs of bleeding, in each cerebral area evaluated (hemispheres, basal ganglia, brainstem and cerebellum). All target lesions were then classified according to Zabramski classification (Zabramski et al. 1994). Fifteen CCMs were seen as hyperintense lesions on T1 and T2 sequences (type I), of which only 2 CCMs had signs of extralesional bleeding (type Ia). CCMs with signs of acute bleeding were located in the brainstem (5) and in the cerebral hemispheres (10).
Seven CCMs were seen as mixed hyper- and hypointense signals (type II), 27 were seen as homogeneous hypointense signal (type III), and 34 were seen as hypointense signals visible only on GRE sequences (type IV) (Supplementary Fig. 4).
Acute intralesional bleeding was observed in 8 of 16 (50%) CCM1-mutated patients, in 2 of 7 (29%) CCM2-mutated patients, in the single CCM3-mutated case and in 2 of 4 (50%) patients without molecular diagnosis. CCMs that acutely bled were located exclusively in the brainstem or in the cerebral hemispheres (Supplementary Table 2). Signs of acute extralesional bleeding were observed in 2 CCM1 patients.
Follow-up MRI scan was available in a small number of subjects and the mean follow-up period was 4 years. Changes in serial MRI, including new hemorrhagic changes and appearance of de novo CCMs, were observed in 9 patients of 18 with at least one follow-up MRI available (Supplementary Table 2). New acute bleeding was observed in 22% CCM patients (15% CCM1-, 25% CCM2- and in the single CCM3-mutated case) in the brainstem, basal ganglia or in the cerebral hemispheres. New acute bleeding was detected outside the target CCMs in 5 patients.
De novo CCMs were depicted in 50% patients (53% CCM1-, 25% CCM2. and in the CCM3-mutated patient) and more frequently observed in the cerebellum and in the cerebral hemispheres (Supplementary Fig. 4).
Changes in signal intensity of target CCMs were observed in 10% of target CCMs (Supplementary Table 3). Signs of new acute bleeding were more frequently seen in non-target CCMs (n = 3) compared to target CCMs (n = 1).
CCMs size ranged between 1 and 27 mm in the first MRI scan available and between 1 and 30 mm in the follow up scans. A modification in size was observed in 23% of lesions. We did not observe any correlation with signs of previous bleeding or clinical evolution.
Discussion
Cerebral cavernous malformations constitute a relevant cerebrovascular disorder characterized by acute and chronic neurological symptoms. Even in asymptomatic subjects, CCMs might evolve towards clinical conditions associated with severe morbidity and mortality. A rapid diagnosis is fundamental to establish an effective clinical and radiological surveillance (Mouchtouris et al. 2015). The neuroradiological diagnosis is strengthened by the detection of germline mutations in familial forms, allowing genetic counselling and extending precautionary monitoring to proband’s relatives. Besides, germline mutation can be also detected in isolated patients, suggesting a common mechanism underlining sporadic and familial forms (McDonald et al. 2014).
Previous studies have described the clinical, neuroradiological and genetic findings in large cohorts of CCM patients (Zabramski et al. 1994; Moriarity et al. 1999; Sirvente et al. 2009; Nardella et al. 2018), but these aspects were rarely addressed in the same study (Denier et al. 2006; de Vos et al. 2017).
Overall mutation detection rate was 87% and 100% for FCCM patients, which is in line with literature data (Labauge et al. 1998; Spiegler et al. 2014) but largely higher compared to a recently published Italian cohort which considered subjects presenting single lesions as sporadic CCM (Nardella et al. 2018). This is likely due to the accurate selection of patients and the use of more stringent criteria of inclusions.
The most common clinical manifestations of the disease are seizure (40–70%), followed by FND (25–50%), headache (10–30%) and hemorrhage (25–32%) (Stapleton and Barker 2018). In our cohort only 2 of 30 subjects remained asymptomatic during the whole observational period (7%). Among symptomatic patients the most frequent manifestations were ICH (50%) and headache (43%). In our cohort we did not find any specific clinical features to be predictive of a specific genetic defect.
The limited number of patients doesn’t allow a definite correlation between neuroradiological and molecular findings. Although variable, the number of CCMs was lower in patients with negative genetic testing and higher in patients affected with FCCM compared to sporadic form. The supratentorial region was affected in all patients irrespective of the molecular defect and supratentorial CCMs were found to outnumber the infratentorial CCMs, as previously reported (Spiegler et al. 2014). The infratentorial region was more frequently affected in patients with a molecular diagnosis respect to subjects without proven mutation. Additional studies in larger groups of patients are needed to ascertain this regional selectivity.
Spinal cavernous malformations are less frequently investigated and have been reported in a small proportion of subjects. In this study only 16 patients underwent at least one spinal MRI (53%): in ten of them (62%) spinal involvement was evident and three patients showed multiple spinal cavernous malformations (18%). This proportion is higher than previously reported (Labauge et al. 1998; Spiegler et al. 2014, Cigoli et al. 2014).
The incomplete knowledge of factors predisposing to hemorrhage still make clinical management challenging. To date two main factors have been linked to increased hemorrhagic risk, previous bleeding and brainstem location (Horne et al. 2016).
Prospective studies reported annual bleeding rates between 0.7 and 6.5% in the general population. Higher hemorrhagic rate was observed in symptomatic FCCM patients than in asymptomatic familial cases (9.1% of patients with a rate of hemorrhage equal to 4.3% for patient per year) (Zabramski et al. 1994; Labauge et al. 2001). No difference in the bleeding rate was observed between asymptomatic and sporadic CCM patients (Robinson et al. 1991). Malformations located in the brainstem or basal ganglia have been reported to have a substantial increase in the risk of rebleeding (Fritschi et al. 1994). Little is known about radiological predictors of bleeding risk, which may be important in the context of clinical management and research setting. In this study hemorrhage occurred in a single type II (target) CCM as well as in one type III and one type IV CCMs: this observation favors the hypothesis that the types of CCMs classified by Zabramsky should be grouped as a single entity as proposed by some authors rather than divided into vascular malformations (types I and II) and related vascular malformations (types III and IV), as previously proposed (Labauge et al. 2001; Lehnhardt et al. 2005; Rigamonti et al. 1991).
Interestingly, in our cohort only one target CCM bled in follow up MRI while new acute bleeding was detected outside the target CCMs in 5 patients. This may suggest that the risk of hemorrhage is not linked to size or previous hemorrhagic changes depicted in the CCMs, in opposition to a published study where bleeding occurred only in type II CCMs and no hemorrhagic de novo CCMs (type I) was observed (Labauge et al. 2001). Additionally, MRI data showed a higher rate of bleeding and a higher rate of de novo CCMs in CCM1 patients suggesting a more dynamic progression of the disease.
Several issues limit the range of our conclusions: (i) the relatively small number of patients investigated, (ii) the different timespan between first and last MRI follow up; (iii) the retrospective design of our study. Our efforts to overcome these limitations included: the recording of every change suggestive of subacute/chronic hemorrhage inside a CCM lesion, as an indirect sign of previous hemorrhagic event, and the presence of at least either of GRE or SWI sequences in the MR scans, to achieve a sensitive detection of CCM.
To date the only curative option for CCM is surgical removal: alternative therapeutic avenues are needed. In vitro data and clinical findings support the use of propranolol as a potential treatment for CCM (Apra et al. 2019) and prompted the start of two randomized prospective clinical trials in the United States (NCT03523650) and Italy (NCT03589014, Lanfranconi et al. 2020).
Novel clinical, neuroradiological and molecular findings collected in CCM patients might be helpful to improve the design of future interventional clinical trials.
Data availability
Not applicable.
Code availability
Not applicable.
References
Al-Shahi Salman R, Hall JM, Horne MA, Moultrie F, Josephson CB, Bhattacharya JJ, Counsell CE, Murray GD, Papanastassiou V, Ritchie V, Roberts RC, Sellar RJ, Warlow CP, Scottish Audit of Intracranial Vascular Malformations (SAIVMs) collaborators (2012) Untreated clinical course of cerebral cavernous malformations: a prospective, population-based cohort study. Lancet Neurol 11:217–224. https://doi.org/10.1016/S1474-4422(12)70004-2
Apra C, Dumot C, Bourdillon P, Pelissou-Guyotat I (2019) Could propranolol be beneficial in adult cerebral cavernous malformations? Neurosurg Rev 42:403–408. https://doi.org/10.1007/s10143-018-01074-0
Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E, Société Française de Neurochirurgie (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76:42–51. https://doi.org/10.1086/426952
Chen JM, Férec C, Cooper DN (2013) Patterns and mutational signatures of tandem base substitutions causing human inherited disease. Hum Mutat 34:1119–1130. https://doi.org/10.1002/humu.22341
Choquet H, Pawlikowska L, Lawton MT, Kim H (2015) Genetics of cerebral cavernous malformations: current status and future prospects. J Neurosurg Sci 59:211–220
Cigoli MS, Avemaria F, De Benedetti S, Gesu GP, Accorsi LG, Parmigiani S, Corona MF, Capra V, Mosca A, Giovannini S, Notturno F, Ciccocioppo F, Volpi L, Estienne M, De Michele G, Antenora A, Bilo L, Tavoni A, Zamponi N, Alfei E, Penco S (2014) PDCD10 gene mutations in multiple cerebral cavernous malformations. PLoS ONE 9:e110438. https://doi.org/10.1371/journal.pone.0110438
de Vos IJ, Vreeburg M, Koek GH, van Steensel MA (2017) Review of familial cerebral cavernous malformations and report of seven additional families. Am J Med Genet A 173:338–351. https://doi.org/10.1002/ajmg.a.38028
Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, Maciazek J, Vicaut E, Brunereau L, Tournier-Lasserve E (2006) Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol 60:550–556. https://doi.org/10.1002/ana.20947
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Flemming KD, Link MJ, Christianson TJ, Brown RD (2012) Prospective hemorrhage risk of intracerebral cavernous malformations. Neurology 78:632–636. https://doi.org/10.1212/WNL.0b013e318248de9b
Fritschi JA, Reulen HJ, Spetzler RF, Zabramski JM (1994) Cavernous malformations of the brain stem. A review of 139 cases. Acta Neurochir (wien) 130:35–46. https://doi.org/10.1007/BF01405501
Grippaudo FR, Piane M, Amoroso M, Longo B, Penco S, Chessa L, Giubettini M, Santanelli F (2013) Cutaneous venous malformations related to KRIT1 mutation: case report and literature review. J Mol Neurosci 5:442–445. https://doi.org/10.1007/s12031-013-0053-1
Horne MA, Flemming KD, Su IC, Stapf C, Jeon JP, Li D, Maxwell SS, White P, Christianson TJ, Agid R, Cho WS, Oh CW, Wu Z, Zhang JT, Kim JE, Ter Brugge K, Willinsky R, Brown RD, Murray GD, Al-Shahi SR (2016) Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. Lancet Neurol 15:166–173. https://doi.org/10.1016/S1474-4422(15)00303-8
Labauge P, Laberge S, Brunereau L, Levy C, Tournier-Lasserve E (1998) Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Lancet 352:1892–1897. https://doi.org/10.1016/s0140-6736(98)03011-6
Labauge P, Brunereau L, Laberge S, Houtteville JP (2001) Prospective follow-up of 33 asymptomatic patients with familial cerebral cavernous malformations. Neurology 57:1825–1828. https://doi.org/10.1212/wnl.57.10.1825
Labauge P, Denier C, Bergametti F, Tournier-Lasserve E (2007) Genetics of cavernous angiomas. Lancet Neurol 6:237–244. https://doi.org/10.1016/S1474-4422(07)70053-4
Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E (1999) Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 23:189–193. https://doi.org/10.1038/13815
Lan MY, Liu YF, Huang CC, Peng CH, Liu JS, Chang YY (2010) Cavernous malformations of the central nervous system combined with cutaneous vascular lesions due to KRIT1 mutation: a case report. Clin Neurol Neurosurg 112:729–732. https://doi.org/10.1016/j.clineuro.2010.05.010
Lanfranconi S, Scola E, Bertani GA, Zarino B, Pallini R, d’Alessandris G, Mazzon E, Marino S, Carriero MR, Scelzo E, Faragò G, Castori M, Fusco C, Petracca A, d’Agruma L, Tassi L, d’Orio P, Lampugnani MG, Nicolis EB, Vasamì A, Novelli D, Torri V, Meessen JMTA, Salman RA, Dejana E, Latini R (2020) Propranolol for familial cerebral cavernous malformation (Treat_CCM): study protocol for a randomized controlled pilot trial. Trials 21:401. https://doi.org/10.1186/s13063-020-4202-x
Lehnhardt FG, von Smekal U, Rückriem B, Stenzel W, Neveling M, Heiss WD, Jacobs AH (2005) Value of gradient-echo magnetic resonance imaging in the diagnosis of familial cerebral cavernous malformation. Arch Neurol 62:653–658. https://doi.org/10.1001/archneur.62.4.653
Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L, Marchuk DA (2003) Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 73:1459–1464. https://doi.org/10.1086/380314
McDonald DA, Shi C, Shenkar R, Gallione CJ, Akers AL, Li S, De Castro N, Berg MJ, Corcoran DL, Awad IA, Marchuk DA (2014) Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet 23:4357–4370. https://doi.org/10.1093/hmg/ddu153
Moore SA, Brown RD, Christianson TJ, Flemming KD (2014) Long-term natural history of incidentally discovered cavernous malformations in a single-center cohort. J Neurosurg 120:1188–1192. https://doi.org/10.3171/2014.1.JNS131619
Moriarity JL, Wetzel M, Clatterbuck RE, Javedan S, Sheppard JM, Hoenig-Rigamonti K, Crone NE, Breiter SN, Lee RR, Rigamonti D (1999) The natural history of cavernous malformations: a prospective study of 68 patients. Neurosurgery 44:1166–1173
Mouchtouris N, Chalouhi N, Chitale A, Starke RM, Tjoumakaris SI, Rosenwasser RH, Jabbour PM (2015) Management of cerebral cavernous malformations: from diagnosis to treatment. Sci World J 2015:808314. https://doi.org/10.1155/2015/808314
Nardella G, Visci G, Guarnieri V, Castellana S, Biagini T, Bisceglia L, Palumbo O, Trivisano M, Vaira C, Scerrati M, Debrasi D, D’Angelo V, Carella M, Merla G, Mazza T, Castori M, D’Agruma L, Fusco C (2018) A single-center study on 140 patients with cerebral cavernous malformations: 28 new pathogenic variants and functional characterization of a PDCD10 large deletion. Human Mutat 39:1885–1900. https://doi.org/10.1002/humu.23629
Pagenstecher A, Stahl S, Sure U, Felbor U (2009) A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet 18:911–918. https://doi.org/10.1093/hmg/ddn420
Rigamonti D, Spetzler RF, Drayer BP, Bojanowski WM, Hodak J, Rigamonti KH, Plenge K, Powers M, Rekate H (1988) Appearance of venous malformations on magnetic resonance imaging. J Neurosurg 69:535–539. https://doi.org/10.3171/jns.1988.69.4.0535
Rigamonti D, Johnson PC, Spetzler RF, Hadley MN, Drayer BP (1991) Cavernous malformations and capillary telangiectasia: a spectrum within a single pathological entity. Neurosurgery 28:60–64
Robinson JR, Awad IA, Little JR (1991) Natural history of the cavernous angioma. J Neurosurg 75:709–714. https://doi.org/10.3171/jns.1991.75.5.0709
Scimone C, Donato L, Katsarou Z, Bostantjopoulou S, D’Angelo R, Sidoti A (2018) Two novel KRIT1 and CCM2 mutations in patients affected by cerebral cavernous malformations: new information on CCM2 penetrance. Front Neurol 29:953. https://doi.org/10.3389/fneur.2018.00953
Scimone C, Donato L, Alibrandi S, Esposito T, Alafaci C, D’Angelo R, Sidoti A (2020) Transcriptome analysis provides new molecular signatures in sporadic Cerebral Cavernous Malformation endothelial cells. BBA Mol Basis Dis 12:165956. https://doi.org/10.1016/j.bbadis.2020.165956
Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E, Labauge P (2009) Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol 23:1066–1072. https://doi.org/10.1111/j.1468-3083.2009.03263.x
Spiegler S, Najm J, Liu J, Gkalympoudis S, Schröder W, Borck G, Brockmann K, Elbracht M, Fauth C, Ferbert A, Freudenberg L, Grasshoff U, Hellenbroich Y, Henn W, Hoffjan S, Hüning I, Korenke GC, Kroisel PM, Kunstmann E, Mair M, Munk-Schulenburg S, Nikoubashman O, Pauli S, Rudnik-Schöneborn S, Sudholt I, Sure U, Tinschert S, Wiednig M, Zoll B, Ginsberg MH, Felbor U (2014) High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one-third of probands are minors. Mol Genet Genomic Med 2:176–185. https://doi.org/10.1002/mgg3.60
Stapleton CJ, Barker FG (2018) Cranial cavernous malformations: natural history and treatment. Stroke 49:1029–1035. https://doi.org/10.1161/STROKEAHA.117.017074
Toll A, Parera E, Giménez-Arnau AM, Pou A, Lloreta J, Limaye N, Vikkula M, Pujol RM (2009) Cutaneous venous malformations in familial cerebral cavernomatosis caused by KRIT1 gene mutations. Dermatology 218:307–313. https://doi.org/10.1159/000199461
Washington CW, McCoy KE, Zipfel GJ (2010) Update on the natural history of cavernous malformations and factors predicting aggressive clinical presentation. Neurosurg Focus 29:E7. https://doi.org/10.3171/2010.5.FOCUS10149
Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, Drayer BP, Brown B, Rigamonti D, Brown G (1994) The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg 80:422–432. https://doi.org/10.3171/jns.1994.80.3.0422
Acknowledgements
The support of Treat-CCM: Propranolol in Cerebral Cavernous Malformation (Treat_CCM, ClinicalTrials.gov Identifier: NCT03589014) and ANACC (Associazione Nazionale Angioma Cavernoso Cerebrale ONLUS) is gratefully acknowledged.
Funding
This work was partially supported by grants from the Ministero della Salute (Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Ricerca Corrente 2020) to GPC and NB.
Author information
Authors and Affiliations
Contributions
LP, GC, ES: neuroradiological data collection and analysis. DR, GM: molecular studies. GAB, GV, EM, SL: clinical assessment of patients. FT, NB, GPC: manuscript revision. SL, ES, DR, were responsible for drafting the manuscript and preparing figures. ES, SL: study concept and design and full responsibility of data.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This study was approved by Ethical Committee Area 2 Milan (ID:721). The experimental protocols were approved by Ethical Committee Area 2 Milan (ID:721) and performed in accordance with the institutional review board of the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico.
Consent to participate
The patients provided written informed consent for all aspects of the study.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lanfranconi, S., Piergallini, L., Ronchi, D. et al. Clinical, neuroradiological and genetic findings in a cohort of patients with multiple Cerebral Cavernous Malformations. Metab Brain Dis 36, 1871–1878 (2021). https://doi.org/10.1007/s11011-021-00809-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-021-00809-1