
Abstract
Pompe disease (PD) is a rare autosomal recessive multi-systemic lysosomal storage disorder, caused by mutations in the acid alpha-glucosidase (GAA) gene located on 17q25.2-q25.3. It is one of about 50 rare genetic diseases categorized as lysosomal storage disorders. This disease is characterized by a range of different symptoms related to acid alpha-glucosidase deficiency. Mutation recognition in the GAA gene can be very significant for purposes such as therapeutic interference, early diagnosis and genotype-phenotype relationship. In the current study, peripheral blood samples were gathered from patients with PD and healthy members of three families. Enzymatic activity of GAA was checked. Then, mutation detection was performed by polymerase chain reaction followed by direct sequencing of all exons in samples with decreased enzyme activity. The identified mutations were investigated using bioinformatics tools to predict possible effects on the protein product and also to compare the mutated sequence with near species. Three novel mutations (c.1966-1968delGAG, c.2011-2012delAT and c.1475-1481dupACCCCAC) were identified in the GAA gene. Assessment of the effects of these mutations on protein structure and function showed the possibility of harmful effects and their significant alterations in the protein structure. The three novel GAA gene mutations detected in this study expand the information about the molecular genetics of PD and can be used to helpdiagnosis and genetic counseling of affected families.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glycogen storage disease type II (GSD II, OMIM # 232300), also called Pompe disease (PD), is an uncommon autosomal recessive deadly muscle disorder (Schoser et al. 2019), that is caused by acid alpha-glucosidase (GAA acid maltase, EC 3.2.1.20, Swiss-Prot P10253) deficiency due to pathogenic variations in the corresponding GAA gene (GAA, OMIM # 606800). Its product degrades α −1,4 and α −1,6 linkages in glycogen, maltose, and isomaltose, thus absence or low-level of GAA activity blocks glycogen breakage into the glucose which leads to aggregation of glycogen in lysosomes in all of the body tissues. It is the only GSD with a defect in lysosomal metabolism, and the first GSD which has been identified in 1932 by the Dutch pathologist, C. Pompe (Di Rocco et al. 2007). Contrary to other GSDs, PD and the McArdle disease (GSD V, OMIM # 232600) mostly affect muscles instead of liver (Sun et al. 2015). Based on the difference in the severity of disease and age at onset, two main types of PD are distinguished; Infantile-Onset PD (IOPD) and Late-Onset PD (LOPD). Table 1 summarizes the symptoms and characteristics of PD in infants and children (Milverton et al. 2018; Kishnani and Howell 2004; Bosman et al. 2018; Slonim et al. 2000; Lam et al. 2003). The frequency of PD is estimated to be about 1/40000 in general. The prevalence of IOPD and LOPD are 1/138000 and 1/57000, respectively (Bosman et al. 2018; Benz et al. 2019).
PD has an autosomal recessive inheritance pattern. GAA is located on chromosome 17q25.2-q25.3, spans 18.3 kb and contains 20 exons (https://www.ncbi.nlm.nih.gov/gene(with the first exon being untranslated (Hoefsloot et al. 1990). The corresponding cDNA is 3.6 kb long and encodes a precursor peptide of 952 amino acids with a predicted molecular weight of 105 kD (Peruzzo et al. 2019).
According to the PD Mutation Database (http://www.erasmusmc.nl/klinische_genetica/research/lijnen/pompe_center/moleculaire_aspecten/), to date, 564 sequence variants have been described of which 301 are missense/nonsense mutations, 67 are splicing mutations, 109 are indels mutations, 12 are gross deletions, four are complex rearrangements, and one is a regulatory mutation.
In the present study, we have investigated three Iranian Turkish families with history of PD. Our findings revealed novel mutations in the corresponding gene. This report may expand knowledge about the etiology of the disease and also may be used in future screening or prenatal diagnosis.
Methods and materials
The study population
A total of 7 myopathic cases from three families with average age of 4.6 years (range, 2 month-18.1 years) participated in this study. All patients were referred to the Division of Medical Genetics, Tabriz Children’s Hospital with diagnosis of PD between May 2016 and February 2020 for acid α-glucosidase enzymatic assay using dried blood spot (DBS) analysis. The diagnosis was confirmed through assessment of enzyme activity. Written informed consent forms were obtained from all study participants or their guardians. The study protocol was approved by the ethics committees of Shahid Beheshti Universities of Medical Sciences (IR.SBMU.RETECH.REC.1399.008).
Enzyme activity
Activity of alpha-glucosidase was assessed in whole blood samples using DBS filter paper (Gynzyme, University Medical Center Hamburg, Eppendorf, Germany). Samples were retained at room temperature for 8–10 h to confirm complete drying and were subsequently kept at 4 °C in plastic bags to preclude deterioration from humidity. Enzyme activity was assessed fluorometrically using 4-methylumbelliferone (4-MU) and acarbose as substrate and inhibitor of maltase-glucoamylase activity, respectively.
DNA extraction
Genomic DNA was extracted from peripheral blood samples of patients and their family members using the DNA mini extract kit (Geneall, Korea) according to the manufactures protocol. Concentration and purity of the extracted DNA were determined using a NanoDrop spectrophotometer (Thermo Scientific NanoDrop Instruments, Wilmington, DE, USA).
Polymerase chain reaction (PCR) was performed for 20 GAA gene exons and their flanking regions (exon-intron boundaries). Co-segregation analysis was also performed by investigating the genotype of the detected mutations in other family members to confirm the causative effects of the mutations in patients.
Family A
The proband was a newborn Iranian Azeri Turkish boy, second child of a family, whose parents were consanguineous. Patient was admitted to Children’s Hospital (Tabriz, Iran) with a hoarse voice after birth. The prenatal period was normal. The baby was born at term with a birth weight of 2850 g. He did not show any feeding problems, macroglossia, hypotonia or muscle weakness. No dysmorphic features were observed in the examination. Mental development was normal.
A cardiac ultrasound was considered to examine the heart, which indicated the presence of cardiac hypertrophy. Electrocardiogram (ECG) and echocardiography showed features of concentric LV hypertrophy with GradeIdiastolic dysfunction. Serum muscle enzymes, including lactate dehydrogenase, aspartate aminotransferase and creatine kinase were found to be elevated. The results revealed that the GAA activity was 0.45 nmol/spot*21 h (normal reference range, 1.5–10 nmol/spot*21 h) at pH 3.8. Although the family members had no history of this disorder, they were advised to undergo genetic testing.
Family B
The subject was a 2-year-old Iranian Azeri Turkish female who had diagnosed with features of Ross Class III heart failure (HF) at age of 9 months. She was under treatment with diuretics and angiotensin-converting enzyme inhibitors when referred to our hospital. She had history of muscle weakness, floppiness, and head lag. On examination, her pulse rate was 110/min, respiratory rate was 30/min, with BP of 106/86 in the upper limbs and 120/86 in the lower limbs. Her cardiovascular system examination was normal. She had mild hepatomegaly with increased liver enzymes. Her serum muscle enzymes, including lactate dehydrogenase, aspartate aminotransferase and creatine kinase were high. The assay for GAA activity from whole blood using DBSs was done. The activity of the alpha-glucosidase in the patient was 0.21 nmol/spot*21 h (normal reference range, 1.5–10 nmol/spot*21 h) at pH 3.8 consistent with the diagnosis of PD.
Family C
The proband was referred to our hospital with a diagnosis of hypertrophic cardiomyopathy in the fourth month of his life. The case was the first child of a consanguineous Azari Turkish parents and had no family history of disease. A Grade II ejection systolic murmurs was detected during the first hours of hospitalization, and an ECG showed severe biventricular hypertrophy. Wet lung signs were present. Moderate hepatosplenomegaly and elevated liver enzymes were dteceted. The patients had muscle weakness and floppiness.
A cardiac assessment showed cardiomegaly. The 2-dimensional ECG showed severe biventricular hypertrophy. The assay for GAA activity from whole blood using DBS filter paper was done. The activity of the alpha-glucosidase in the patient was 0.2 nmol/spot*21 h (normal reference range, 1.5–10 nmol/spot*21 h) at pH 3.8, consistent with the diagnosis of PD.
Bioinformatics analysis
To assess the conservation of the GAA sequence in DNA and protein in humans and near species, and compare the mutated sequence with these sequences, we used MUSCLE software (version 3.8.31) (Edgar 2004a; Edgar 2004b) and clustal omega (Chojnacki et al. 2017). DNA and protein sequences of other species (Chimpanzee, Angola colobus, Bonobo, Capuchin, Gorilla, Orangutan, Macaque and Mouse) were gained from the Ensemble genome browser 99. We used Mutation Taster (http://mutationtaster.org) (Schwarz et al. 2014) and Franklin (https://franklin.genoox.com/) for prediction of functional effect mutations. Also, for analysis of protein domain of GAA, we used the NCBI Conserved Domains (Lu et al. 2020). In order to illustrate the effect of the recognized mutations on the structure of normal protein and the mutant forms, we used the SWISS-MODEL software (https://swissmodel.expasy.org/) (Waterhouse et al. 2018).
Results
Molecular genetics results
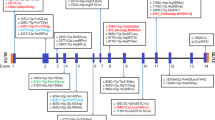
In the proband from the family A, we detected deletion of GAG nucleotides at position 1966–1968 (located in exon 14) which leads to one amino acid deletion at the mentioned position (Fig. 1). In the probanf from family B, we detected deletion of AT nucleotides at position 2011–2012 (located in exon 14). The mutation results in frameshift, creation of a premature stop codon at codon 65 downstream of the mutation and production of a truncated protein with 216 amino acids missing. Finally, in the patient from family C, duplication of seven nucleotide (c.1475-1481dup ACCCCAC) in exon 10 of GAA gene was detected which was predicted to cause a frameshift mutation and premature stop codon in 13 codons downstream of mutation resulting in 444 amino acids missing in the truncated protein. Assessment of in–house genotypic data showed absence of these variants in more than 100 tested cases. Figure 2 shows the location of mutations in the DNA and Protein sequences of GAA gene.

Pedigrees of studied family and their recognized mutations. The suffering family members and their linked mutations are provided. N indicates normal allele, M1 indicates c.1966_1968delGAG mutation and M2 indicates c.1828G˃A mutation in part A. N indicates normal allele and M1 indicates c.2011-2012del AT mutation in part B. N indicates normal allele and M1 indicates c.1475-1481dupACCCCAC mutation in part C

Position of the mutations in DNA (A) and protein (B) sequence. The mutations are located in exon 10 and exon 14 of the GAA gene which affect the same domain in the protein product
Bioinformatics evaluation results
Table 2 shows the results of prediction by bioinformatics tools. As indicated in Fig. 3, protein structure prediction was performed for evaluating mutant and normal shapes of these proteins using SWISS-MODEL software. As shown in Fig. 2, frameshift mutations lead to production of truncated proteins which may result in functional and structural changing. According on Mutation Taster, all three mutations [c.1966-1968del GAG, c.2011-2012del AT and c.1475-1481dup] are disease causing. Franklin software also predicted all of the established mutations to be likely pathogenic. Based on the results of DNA and protein sequence alignments, the locations of mutations in DNA and protein are strongly conserved between the near species (Figs. 4 and 5).

Predicted structure of the GAA protein (A, B, C) carrying the recognized mutations in comparison with the normal protein

Protein sequence alignment for detected mutations

DNA sequence alignment result for detected mutations
Discussion
PD comprises a group of glycogen storage disorder with variable ages of onset. This disease has been characterized by accumulation of glycogen in lysosomes resulting in severe hypotonia, eating trouble, recurrent respiratory infection and death which might occur during the first year of life (Milverton et al. 2019; Kohler et al. 2018). In the present study, we have reported the clinical and mutational characteristics of three patients with PD. Here, we identified three mutations in the GAA gene, including one duplication mutation and two deletion mutations. These mutations have not been reported in ClinVar. A total of 564 various mutations have been described in the GAA gene (www.pompecenter.nl). Six previous studies have reported GAA mutations in Iranian families all of them being novel mutations (c.693insA, c.317-318insCCC, c.1650del G, c.730C > T, c.1824-1828dupATACG, c.2040 + 2dup, c.1650delG, c. 1837 T > G and c.2596del G) (Emami 2013; Aryani et al. 2014; Nazari et al. 2017; Ebrahimi et al. 2017; Bahreini et al. 2016; Moravej et al. 2018). The identified mutations in the current study are located in exon 14 and 10. This region is an important region regarding the high incidence of detected mutations (from c.1039 to c.2454) (Kroos et al. 2012). Kroos et al. have shown relationship between genotype and phenotype in PD patients. They also showed a direct relationship between glycogen build-up in lysosomes and the remaining protein activity (Kroos et al. 2012). However, Wan et al. demonstrated no correlation between GAA activity and clinically course (Wan et al. 2008). Lack of genotype-phenotype relationship might be due to the presence of modifying genetic factors, environmental factors or diversity in specificity of diagnostic laboratory procedures (De Filippi et al. 2014).
In patient A, a deletion mutation has been detected in exon 14 (c.1966-1968delGAA). The mutations deletes a negative charged amino acid (Glutamic) which might effectively change the structure of the protein. Exon 14 has 152 nucleotides which encode 51 amino acids (Ensemble Genome Browser 99). Currently, 34 mutations have been reported in exon 14, four of them exerting very severe impact on GAA activity (www.hgmd.cf.ac.uk/ac/index.php). In patient B, a deletion mutation has been detected in exon 14 (c.2011-2012delAT) which alters the amino acid methionine to alanine causing a frameshift and creating a premature stop codon at 65 codons downstream of the mutation that might lead to nonsense-mediated RNA decay (NMD) in the cell. NMD is a regulatory process during which mRNAs with premature termination codons are degraded to avoid non-functional or detrimental effects on proteins. Deficiency in the NMD has been shown in some brain disorders (Jaffrey and Wilkinson 2018). As mentioned before, 34 different mutations have been reported in the exon 14of them being very severe. In patient C, a duplication mutation located in the exon 10 (c. 1475-1481dup ACCCCAC) changes alanine to proline, resulting in a frameshift and creation of a premature termination codon at 13 codons downstream of the mutation. Exon 10 has 114 nucleotides and encodes for 38 amino acids. To date, 22 mutations have been reported in exon 10, six of them having very severe impact on GAA activity. As we can see in fig. 2, GAA protein have four functional domains. The most important domain is GH31-MGAM-SI-GAA domain (starting from exon 7 to exon 15). Approximately all active sites and catalytic site of the GAA protein have been located in this domain. These mutations lead to destruction of the GH31 domain and may result in creation of truncated protein or even complete loss of GAA enzyme activity. With respect to the in-silico analysis, all described novel mutations of this study have a harmful effect on the GAA protein structure and enzymatic activity. As predicted by Mutation Taster and Franklin tools, these mutations are damaging. In addition, alignment results have this region of GAA is highly conserved among species, indicating its vital function in the cell. In brief, the identified mutations in the current study have expanded the data regarding GAA mutations and might be beneficial in future functional analysis, genetic counseling and prenatal diagnosis.
References
Aryani O, Manshadi MD, Tondar M, Khalili E, Kamalidehghan B, Ahmadipour F, Fani S, Houshmand M (2014) A newly identified c. 1824_1828dupATACG mutation in exon 13 of the GAA gene in infantile-onset glycogen storage disease type II (Pompe disease). Mol Biol Rep 41:6211–6214
Bahreini F, Houshmand M, Modaresi MH, Tonekaboni H, Nafissi S, Nazari F, Akrami SM (2016) Mitochondrial copy number and D-loop variants in pompe patients. Cell J (Yakhteh) 18:405
Benz k, Hahn p, Hanisch m, Lücke k, Lücke t, Jackowski j (2019) Systematic review of oral and craniofacial findings in patients with Fabry disease or Pompe disease. Br J Oral Maxillofac Surg 57:831–838
Bosman L, Hoeks SE, González Candel A, Van Den Hout HJ, Van Der Ploeg AT, Staals LM (2018) Perioperative management of children with glycogen storage disease type II—Pompe disease. Pediatr Anesth 28:428–435
Chojnacki S, Cowley A, Lee J, Foix A, Lopez R (2017) Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res 45:W550–W553
De Filippi P, Saeidi K, Ravaglia S, Dardis A, Angelini C, Mongini T, Morandi L, Moggio M, Di Muzio A, Filosto M (2014) Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet J Rare Dis 9:102
Di Rocco M, Buzzi D, Taro M (2007) Glycogen storage disease type II: clinical overview. Acta Myologica 26:42
Ebrahimi, M., Behnam, M., Behranvand-Jazi, N., Yari, L., Sheikh-Kanlomilan, S., Salehi, M., Tahmasebi, P., Amini, M., Behjati, M. & Hosseini, N. 2017. Identification a novel mononucleotide deletion mutation in GAA in pompe disease patients. J Res Med Sci: Off J Isfahan Univ Med Sci, 22
Edgar RC (2004a) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Edgar RC (2004b) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Emami M (2013) Detection of a novel mutation in the GAA gene in an Iranian child with glycogen storage disease type II. Archives of Iranian medicine 16:126
Hirschhorm, R. 2001. Glycogen storage disease type II. Acid alpha-glucosidase (acid maltase) deficiency. Metabolic Mole Bases Inherited Dis 3389-3420
Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra B (1990) Characterization of the human lysosomal α-glucosidase gene. Biochem J 272:493–497
Jaffrey SR, Wilkinson MF (2018) Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat Rev Neurosci 19:715–728
Kishnani PS, Howell RR (2004) Pompe disease in infants and children. J Pediatr 144:S35–S43
Kohler L, Puertollano R, Raben N (2018) Pompe disease: from basic science to therapy. Neurotherapeutics 15:928–942
Kroos, M., Hoogeveen-Westerveld, M., Van Der Ploeg, A. & Reuser, A. J. The genotype–phenotype correlation in Pompe disease. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 2012. Wiley Online Library, 59–68
Lam C, Yuen Y, Chan K, Tong S, Lai C, Chow T, Lee K, Chan Y, Martiniuk F (2003) Juvenile-onset glycogen storage disease type II with novel mutations in acid α-glucosidase gene. Neurology 60:715–717
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS (2020) CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48:D265–D268
Milverton, J., Newton, S. & Merlin, T. 2018. The effectiveness of enzyme replacement therapy for juvenile-onset Pompe disease: a systematic review. J Inherited Metab Dis, 1-8
Milverton J, Newton S, Merlin T (2019) The effectiveness of enzyme replacement therapy for juvenile-onset Pompe disease: A systematic review. J Inherit Metab Dis 42:57–65
Moravej H, Amirhakimi A, Showraki A, Amoozgar H, Hadipour Z, NIKFAR G (2018) A new mutation causing severe infantile-onset pompe disease responsive to enzyme replacement therapy. Iran J Med Sci 43:218
Nazari F, Sinaei F, Nilipour Y, Fatehi F, Streubel B, Ashrafi MR, Aryani O, Nafissi S (2017) Late-onset pompe disease in Iran: A clinical and genetic report. Muscle Nerve 55:835–840
Peruzzo P, Pavan E, Dardis A (2019) Molecular genetics of Pompe disease: a comprehensive overview. Ann Trans Med 7:278
Schoser, B., Hahn, A., James, E., Gupta, D., Gitlin, M. & Prasad, S. 2019. A systematic review of the health economics of Pompe disease. Pharmacoecon Open, A Systematic Review of the Health Economics of Pompe Disease
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362
Slonim AE, Bulone L, Ritz S, Goldberg T, Chen A, Martiniuk F (2000) Identification of two subtypes of infantile acid maltase deficiency. J Pediatr 137:283–285
Sun B, Brooks ED, Koeberl DD (2015) Preclinical development of new therapy for glycogen storage diseases. Current gene therapy 15:338–347
Wan L, Lee C-C, HSU C-M, Hwu W-L, Yang C-C, Tsai C-H, Tsai F-J (2008) Identification of eight novel mutations of the acid α-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J Neurol 255:831–838
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, De Beer TAP, rempfer c, bordoli l (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303
Yang CF, Liu HC, Hsu TR, Tsai f C, Chiang SF, Chiang CC, Ho HC, Lai CJ, Yang TF, Chuang SY (2014) A large-scale nationwide newborn screening program for pompe disease in Taiwan: towards effective diagnosis and treatment. Am J Med Genet A 164:54–61
Acknowledgments
This study is financially supported by Shahid Beheshti University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gharesouran, J., Jalaiei, A., Hosseinzadeh, A. et al. GAA gene mutation detection following clinical evaluation and enzyme activity analysis in Azeri Turkish patients with Pompe disease. Metab Brain Dis 35, 1127–1134 (2020). https://doi.org/10.1007/s11011-020-00586-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-020-00586-3