
Abstract
Single amino acid mutations in profilin 1 (PFN1) have been found to cause amyotrophic lateral sclerosis (ALS). Recently, we developed a mouse model for ALS using a PFN1 mutation (glycine 118 to valine, G118V), and we are now interested in understanding how PFN1 becomes toxically lethal with only one amino acid substitution. Therefore, we studied mutation-related changes in the PFN1 protein and hypothesized that such changes significantly disturb its structure. Initially, we expressed and studied the purified PFN1WT and PFN1G118V proteins from bacterial culture. We found that the PFN1G118V protein has a different mean residue ellipticity, as measured by far-UV circular dichroism, accompanied by a spectral shift. The intrinsic fluorescence of PFN1G118V showed a small fluctuation in maximum fluorescence absorption and intensity. Moreover, we examined the time course of PFN1 aggregation using SDS-PAGE, western blotting, and MALDI-TOF/TOF and found that compared with PFN1WT, PFN1G118V had an increased tendency to form aggregates. Dynamic light scattering data confirmed this, showing a larger size distribution for PFN1G118V. Our data explain why PFN1G118V tends to aggregate, a phenotype that may be the basis for its neurotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease caused by the progressive degeneration of motor neurons in the brain and spinal cord leading to the loss of voluntary movement and muscle control. It is known as Lou Gehrig’s disease in the United States and as motor neuron disease in Europe. Two different forms of ALS exist: familial (fALS) and sporadic (sALS) (Zarei et al. 2015). Although the etiology of ALS is largely unknown, inheritable genetic mutations have been found to cause fALS in humans (Hardiman et al. 2017; van Es et al. 2017). One such mutation was found in the gene PFN1, which encodes the profilin 1 (PFN1) protein (Wu et al. 2012). Thus far, eight PFN1 mutations (A20T, C71G, T109M, M114T, E117G, G118V, R136W, and Q139L) have been linked to fALS and sALS (Smith et al. 2014; Chen et al. 2013; Ingre et al. 2013; Wu et al. 2012).
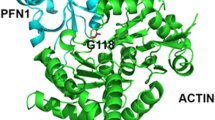
Evolutionarily, PFN1 is a highly conserved small protein, with 139 amino acid residues in humans and 140 amino acid residues in rodents. Among the four known profilin isoforms (PFN 1–4), PFN1 is ubiquitously expressed and abundant in most cell and tissue types (Alkam et al. 2017; Witke 2004). The tertiary structure of PFN1 (Fig. 1) consists of seven anti-parallel β-sheets and five α-helices, two of which are located at the termini (Krishnan et al. 2009; Metzler et al. 1995; Schutt et al. 1993). PFN1 is best known for its actin-binding function and is an essential regulator in converting G-actin to F-actin, a critical step in actin polymerization. Furthermore, multiple other functions were discovered by virtue of its interaction with ligands and cellular signaling partners via its poly-L-proline (PLP)-binding domain (Witke 2004). PFN1 has more than 50 ligands and binding partners, at least two of which are disease-causing proteins: survival of motor neuron protein (SMN), which is implicated in spinal muscular atrophy, and valosin-containing protein (VCP), which is implicated in ALS (Hensel and Claus 2018; Johnson et al. 2010; Fan and Simard 2002). The links between PFN1 and SMN and VCP are of great interest, and further studies are needed to confirm whether their interactions regulate the actin cytoskeleton (Hensel and Claus 2018; Nolle et al. 2011).

Structure of human PFN1WT generated with PyMol (PDB: 1PFL). The G118 residue is depicted in pink, and the remaining ALS-linked residues are shown in yellow and labeled in white. Residues involved in actin binding are shown in blue, and PLP-binding domains are in green. Tryptophan residues (W3 and W31) involved in PLP binding are shown in green and labeled in red. The structure on the left shows the actin-binding site, where the G118V mutation located. The structure on the right is tilted to show the PLP-binding area and other features of PFN1
The recent reports of the work by several groups describe their investigation into the mechanism of mutant PFN1 neurotoxicity in ALS (Boopathy et al. 2015, Freischmidt et al. 2015, Del Poggetto et al. 2015a, b, 2016). The mutations in PFN1 are located in the actin binding domain and in the PLP binding domain. The hypothesis that the neurotoxicity of PFN1 due to loss of actin binding and by loss of interaction with PLP containing domain may not be true. Therefore, there may be different mechanism (s) at play, such as gain-of-toxic function. There is strong evidence that specific mutations thermodynamically destabilize PFN1 and also cause structural changes in its native state that affect its propensity for aggregation, as characterized in vitro (Boopathy et al. 2015; Del Poggetto et al. 2015a, 2016; Wu et al. 2012). A more recent study showed that a C71G mutation causes well-folded PFN1 to co-exist with unfolded PFN1. Unfolded PFN1C71G has accessible amphiphilic and/or hydrophobic regions that might induce a non-native helical conformation, initiating ALS as a result of interacting abnormally with membranes (Lim et al. 2017). On the other hand, the accumulation of TAR DNA-binding protein 43 (TDP-43) aggregates was found to be associated with PFN1 toxicity (Matsukawa et al. 2016; Tanaka and Hasegawa 2016), and this may be an initiating factor in the neuron toxicity and cytoskeletal dysfunction found in ALS.
Of the fALS PFN1 mutations identified, the G118V mutation is located most proximal to the actin-binding site, according to a model generated with PyMol software and published bovine PFN1 X-ray crystallography data (Alkam et al. 2017; Fil et al. 2017). A recent study showed that in a hPFN1C71G mouse model, motor neuron degeneration preceded mutant protein aggregation and caused ALS phenotypes by a gain of toxicity (Yang et al. 2016). However, the mechanism by which PFN1G118V initiates ALS remains unknown. A recently developed hPFN1G118V mouse model showed pathogenic and behavioral phenotypes of ALS disease, including mutant PFN1 aggregation and abnormally low F/G-actin ratios in lumbar spinal cord sections from fully symptomatic and end-stage animals (Fil et al. 2017). Although some mutations can alter actin binding, the G118V mutation causes PFN1 to lose its ability to bind actin (Freischmidt et al. 2015; Wu et al. 2012). It is possible that this amino acid substitution uniquely alters the biophysical properties of PFN1 to produce a conformational change in the binding site, allowing the valine residue to adapt more energetically favorable dihedral angles (Boopathy et al. 2015).
The misfolding and aggregation of PFN1, due to a single amino acid substitution, could be the underlying cause of neurotoxicity and neurodegeneration in human ALS and in the PFN1 transgenic mouse model (Fil et al. 2017). However, the molecular details behind the misfolding and aggregation of PFN1 are not known; therefore, we sought to investigate the changes that lead to this anomaly. Here, we have investigated the biophysical characteristics of PFN1 containing a G118V mutation. We expressed and purified wild type and mutant PFN1 from E. coli and analyzed the proteins with common biochemical, biophysical, and analytical techniques (e.g., western blotting, MALDI-TOF/TOF, dynamic light scattering [DLS], far-UV circular dichroism [CD], and fluorescence spectroscopy). Our study identified structural changes in PFN1G118V and provides valuable insight into the mechanism of PFN1 toxicity.
Materials and methods
Cloning and expression of recombinant PFN1
Plasmid DNA constructs for human PFN1, both wild type (PFN1WT) and the G118V mutation (PFN1G118V), were obtained from NorClone (Canada). PFN1 cDNA was subcloned into pET21a (Novagen). E. coli BL21 cells transformed with PFN1 constructs were grown in LB medium (Liofilchem, Italy) containing 100 μg/mL ampicillin at 37 °C to an optical density at 600 nm of 0.6 (Fedorov et al. 1994). Subsequently, 1 mM IPTG (SinaClon, Iran) was added to induce PFN1 expression. To allow sufficient protein production, the cells were incubated for either 3 h at 37 °C (wild type) or overnight at 18 °C (G118V) before harvesting by centrifugation.
Purification of PFN1 by ion exchange
Bacterial cells were collected and sonicated in 50 mM Tris-HCl and 100 mM NaCl buffer, pH 7.4, containing protease inhibitor (Roche, Germany), and bacterial cell debris was removed by centrifugation (12,000 g). The supernatant was subjected to a Diethylaminoethyl (DEAE)-Sepharose Fast Flow anion exchange column (GE Healthcare Bio-Sciences AB, Sweden) pre-equilibrated with 50 mM Tris-HCl pH 7.4 (buffer A) with a KNAUER Azura FPLC system coupled to a UV detector (Germany). The protein sample was loaded onto the column at a flow rate of 0.5 ml/min. The column was washed with buffer A (3 column volumes) at a flow rate of 1 ml/min. Then, a linear gradient was established with buffer A and buffer B (1 M NaCl), and the rest of the bound proteins were eluted by increasing the percentage of buffer B from 0 to 100% during 100 min. The UV wavelength was set at 280 nm.
Purification of PFN1 by affinity column
Cyanogen bromide (CNBr)-activated sepharose was prepared based on Kohn’s method (Kohn and Wilchek 1984). Poly-L-proline, with molecular weight 1–10 kDa (Sigma-Aldrich, USA), was dissolved in the coupling buffer (0.1 M NaHCO3 and 0.5 M NaCl, pH 8.5), and added to CNBr-activated resin. The resin was stirred for 20 h at 4 °C then washed with the coupling buffer and blocked with 0.1 M Tris-HCl (pH 8.0) plus 0.5 M NaCl for 2 h. Then, the lysate was washed 3 times with 0.1 M Tris buffer (pH 8.0) and 0.1 M acetate buffer (pH 4.0) plus 0.5 M NaCl. The lysate was diluted with wash buffer (10 mM Tris, 100 mM NaCl, 1 mM EDTA, and 2 mM DTT, pH 7.4) and incubated with PLP-sepharose resin, pre-equilibrated with wash buffer, in an ice bath for 2 h with gentle shaking. PFN1 was eluted from the resin with wash buffer plus 8 M urea and then dialyzed against wash buffer without urea for 48 h.
The purified proteins were concentrated using Vivaspin® centrifugal concentrators with a 5 kDa molecular-weight cutoff. The total protein concentration was determined with a Pierce BCA Protein Assay Kit (Thermo-Fisher Scientific, USA), according to the manufacturer’s protocol.
SDS-PAGE analysis and western blotting
Samples were mixed with SDS-PAGE sample buffer (0.5 M Tris-HCl pH 6.8, 10% SDS, 3 M glycerol, 0.05% bromophenol blue) and separated with 15% tris-glycine SDS-PAGE according to the Laemmli procedure (Laemmli 1970). SDS-PAGE gels were run with or without reducing agent. If added reducing agent (10X, ThermoFisher, Waltham, MA), it was diluted to 0.25% and added to the buffer in the inner chamber of gel tank to maintain the protein samples in a reduced state during electrophoresis. Reducing agent (1%) added to the protein samples prior to loading. The gels were stained with Coomassie Brilliant Blue G-250 or silver nitrate. Separated proteins on the SDS gel were transferred to a PVDF membrane using the Invitrogen iBlot device. The membranes were blocked with 5% skim milk in PBS-T (0.1% Tween 20) buffer for 1 h, incubated overnight at 4 °C with anti-PFN1 primary antibody (Sigma-Aldrich #P7749) diluted in PBS-T 5% milk (1:1000), and rinsed 3 times with PBS-T buffer. Then, proteins were visualized with an HRP-conjugated secondary antibody (Amersham Corp., USA) diluted in PBS-T 5% milk (1:5000) and detected with the ECL system (Amersham).
Protein identification by mass spectrometry
Intact PFN1 proteins and in-gel digested PFN1 peptides were analyzed by MALDI mass spectrometry (Applied Biosystems 4800 MALDI-TOF/TOF). In-gel trypsin digestion was performed on the protein bands of interest excised from the Coomassie blue-stained gels, as described previously (Shevchenko et al. 2007). The tryptic peptide mixtures were mixed with an equal volume of α-cyano-4-hydroxycinnamic acid matrix solution in 50% acetonitrile containing 0.1% trifluoroacetic acid, spotted (0.7 μL) onto a MALDI plate, air dried, and analyzed with a MALDI-TOF/TOF operated in reflector positive mode. The monoisotopic peptide mass values of digested PFN1 were searched in the Swiss-Prot and NCBI databases using the MASCOT peptide mass fingerprinting search program (www.matrixscience.com). For intact mass analysis, sinapinic acid was used as a matrix, and equine cytochrome c (average mass 12,362 Da) and apomyoglobin (average mass 16,952 Da) were used as internal and external standards for calibration. All were analyzed with MALDI-TOF/TOF in linear positive mode. Unless otherwise stated, all materials in this experimental section were purchased from Sigma-Aldrich.
Far-UV CD
Far-UV CD (190–260 nm) measurements were carried out on a JASCO J-715 spectropolarimeter (Tokyo, Japan) at a protein concentration of 0.1 mg/ml in 50 mM Tris-HCl (PH 7.4) at 25 °C. The blank-subtracted data were then converted to mean residue ellipticity [Ө]. A quartz cuvette with a 1-mm path length was used for analysis.
Intrinsic fluorescence
Intrinsic fluorescence spectra were recorded at a protein concentration of 0.4 mg/ml in 50 mM Tris-HCl (PH 7.4) using the Cary Eclipse spectrofluorometer (Varian Ltd., Spain) and a 10-mm quartz cuvette with excitation at 280 nm and emission in the 280–500 nm range. The slit widths were set at 5 nm.
Dynamic light scattering
A DLS device (Nanophox Sympatech GmbH, Germany) was used to assess the size distribution of proteins in 0.4 mg/ml protein samples in 50 mM Tris-HCl (PH 7.4) at 25 °C (refraction index of 1.33 and a viscosity of 0.88 cP in a 10-mm plastic cell).
All samples used in the far-UV CD, intrinsic fluorescence, and DLS experiments were purified by anion-exchange chromatography, and all analyses were performed immediately after centrifugation (15,700 g, 10 min) and filtration (0.22 μm).
Aggregation analysis
Purified recombinant PFN1WT and PFN1G118V samples (0.7 mg/ml, purified with anion-exchange chromatography and stored at −20 °C) were thawed, centrifuged at 15,700 g for 10 min, and then filtered (0.22 μm filters). Samples in aggregation buffer (100 mM Tris-HCl and 11.2 mM NaCl, pH 7.4) were incubated at 37 °C with shaking on an orbital shaker at 200 rpm. Aliquots were removed from each sample over a time course of 96 h at 0, 4, 24, 48, 72, and 96 h, and the samples were analyzed by MALDI-TOF/TOF and SDS-PAGE.
Results
Expression, purification, and validation of recombinant PFN1WT and PFN1G118V
Using E. coli as a simple and robust system to overexpress recombinant protein, we obtained high quality and reasonable yields of both wild type (PFN1WT) and mutant (PFN1G118V) PFN1 protein. Prominent bands at a molecular weight corresponding to PFN1 (14–15 kDa) were visible after SDS-PAGE of the recombinant protein homogenates suggesting that PFN1 proteins were expressed (Fig. SI1). Moreover, the proteins from the excised bands were analyzed with MALDI-TOF/TOF to validate the gel-based identification of PFN1 (Fig. SI1). The peptide sequences were matched with the Swiss-Prot database and rated by a scoring system. MALDI-TOF/TOF analysis identified 21 peptides from the complete digestion of PFN1WT (81% sequence coverage) and 11 matched peptides from the partial digestion of PFN1G118V (47% sequence coverage) (Table 1). This bottom-up proteomic analysis resulted in 100% confidence with human PFN1 for both expressed recombinant proteins (Tables SI1 and SI2).
To purify recombinant PFN1WT and PFN1G118V, we performed PLP-sepharose affinity chromatography with bacterial cell lysates and obtained high quality protein, as indicated by western blots and mass spectrometry peaks. We found that samples of PFN1G118V contained multiple bands (higher molecular weights) by western blotting, suggesting that PFN1G118V oligomerized (Fig. 2a). As shown by western blotting, the presence of higher molecular-weight bands recognized by anti-PFN1 (Fig. 2b, arrows) confirmed that the bands seen with silver staining (Fig. 2a, arrows) were SDS-resistant PFN1 oligomers. The arrows in Fig. 2b point to two bands that are dimers and trimers. These bands are much more prominent in the PFN1G118V sample; however, the 30–35 kDa band is less prominent in the PFN1WT sample, and the 45 kDa trimer is absent. Furthermore, PFN1WT and PFN1G118V were purified with DEAE-Sepharose Fast Flow anion-exchange resin to avoid the use of urea. As shown in Figure SI2, an aliquot of each fraction (labeled 1–4) was subjected to SDS-PAGE, and PFN1 bands (arrows) were processed for sequencing with MALDI-TOF/TOF. Fraction 1 contained most of the PFN1WT or PFN1G118V proteins, while fractions 2–4 contained very little of either protein.

Silver-stained SDS-PAGE and western blot of purified PFN1WT and PFN1G118V. a Samples purified with PLP-sepharose were subjected to SDS-PAGE and stained with silver nitrate. These gels were ran without any reducing agent. The G118V sample contains higher molecular weight bands (arrows), evidence for PFN1 oligomerization. b Western blots of purified wild type (WT) and mutant samples were probed with anti-PFN1, and oligomers are confirmed to be PFN1 with specific antibody that only recognize PFN1. M, pre-stained protein ladder; molecular weights are shown in kDa
Biophysical characterization of PFN1WT and PFN1G118V
The conformational properties of the purified recombinant PFN1WT and PFN1G118V were assessed with far-UV CD, intrinsic fluorescence, and DLS techniques.
Far-UV CD
The far-UV CD spectrum of PFN1WT revealed a typical α + β structure (Fig. 3a) that is consistent with observations reported by others (Boopathy et al. 2015; Del Poggetto et al. 2015b; Lambrechts et al. 2002). PFN1WT exhibited negative ellipticity over the range of approximately 204–240 nm and positive ellipticity over the range of approximately 192–204 nm, with a maximum at 195 nm. In contrast, the CD plot of PFN1G118V showed lower values of positive and negative mean residue ellipticity [Ө] (deg cm2 dmol−1). Additionally, this spectrum showed a maximal positive CD signal at 193 nm and a small shift from approximately 204 nm to a lower wavelength (200 nm) corresponding to a “blue shift” compared to PFN1WT (Fig. 3a).

Structural characterization of PFN1WT and PFN1G118V with different probes. a Far-UV circular dichroism spectra and (b) intrinsic fluorescence spectra of PFN1WT and PFN1G118V
Intrinsic fluorescence
The intrinsic tryptophan fluorescence of PFN1, due to tryptophan residues 3 and 31 in the PLP-binding site located on a solvent-exposed hydrophobic surface, was used to determine structural fluctuations in the protein by comparing the fluorescence intensities and maximum wavelength (McLachlan et al. 2007). As shown in Fig. 3b, the intrinsic fluorescence spectrum of PFN1WT showed a peak from 325 to 350 nm. Although this peak, with a maximum absorption of 335 nm, represented the compact folded structure of PFN1WT in its native form, small changes in maximum fluorescence absorption to approximately 340 nm (a “red shift”) and in fluorescence intensity were apparent in PFN1G118V, indicating that the change from glycine to valine caused this shift (Fig. 3b). This suggests that a structural change within a more flexible hydrophobic surface, although the exposure of the tryptophan indole groups to solvent is similar to the native protein.
Dynamic light scattering
The hydrodynamic diameter of protein samples were measured by DLS, where the intensity of light scattering is proportional to the sixth power of the particle diameter, causing the scattered light from small species to be very small and undetectable (Lorber et al. 2012). Our DLS analysis of PFN1WT showed an average peak for particle sizes of 4–5 nm, which was consistent with the size of a native PFN1 monomer (Fig. 4a). On the other hand, DLS analysis of PFN1G118V showed an average peak for size distribution in the range of 230–320 nm. This rather large hydrodynamic diameter for mutant PFN1G118V (40 to 60 times larger than for PFN1WT) suggested that aggregated proteins were present (Fig. 4b).

Size distribution profile of PFN1WT and PFN1G118V. Dynamic light scattering graph showing the average size distribution of (a) PFN1WT and (b) PFN1G118V
Aggregation analysis of PFN1WT and PFN1G118V in vitro
To gain further insight into the characteristics and mechanism of PFN1G118V aggregation, we subjected PFN1WT and PFN1G118V to a time-course condition that might influence the aggregation process. As shown in Fig. 5, MALDI-TOF/TOF and SDS-PAGE assays showed a decrease in soluble PFN1 during the time-course study. At time zero, intact mass analysis of PFN1WT and PFN1G118V showed the most abundant peak to be the PFN1 monomer in both samples (Fig. 5a and c). Moreover, we observed a weak signal consistent with the mass-to-charge (m/z) ratio of a PFN1 dimer in these MALDI mass spectra (Fig. 5a and c, arrows). When PFN1 protein samples were incubated at 37 °C with constant shaking and analyzed with MALDI, we observed a peak for monomeric PFN1WT, consistent with a corresponding band on the gel, suggesting that PFN1WT did not aggregate into larger protein complexes during the 96 h of incubation as shown in Fig. 5b. On the other hand, the band corresponding to monomeric PFN1G118V began to diminish after four hours and was undetectable after two days of incubation (Fig. 5d). Likewise, MALDI analysis of samples incubated for more than 48 h no longer showed the monomeric spectra of PFN1G118V that was present in the baseline samples (data not shown). We also confirmed that PFN1G118V was present in the sample by re-solubilizing the insoluble fraction (that formed during incubation) in loading buffer with 2-mercaptoethanol (to reduce the samples) and performing SDS-PAGE. Coomassie-blue staining indicated a band corresponding to monomeric PFN1 (~15 kDa). We also noticed that some aggregated PFN1G118V (high molecular-weight protein aggregates) was not solubilized in the presence of reducing agent and did not migrate on the SDS gel, but remained in the stacking gel. As shown in Fig. 5d, a Coomassie-stained band that is likely PFN1G118V was noticeably reduced, and there was no detectable Coomassie-stained band in the 72-h lane (Fig. 5d).

Intact mass and time-course aggregation analysis of PFN1WT and PFN1G118V. MALDI-TOF/TOF spectra of (a) PFN1WT and (c) PFN1G118V proteins at time zero before aggregation analysis. SDS-PAGE of aliquots from (b) PFN1WT samples were incubated at 37 C under constant shaking for 0, 24, 48, 72 and 96 h and (d) PFN1G118V samples incubated at 37 °C under constant shaking for 0, 24, 48, and 72 h. Insoluble (Ins) fraction from PFN1G118V sample solubilized in sample buffer containing 2-mercapthoethanol; arrowhead pointing to the Coomassie-stained band in the top of the Ins lane (d) indicates PFN1 aggregates. Other fractions were solubilized in sample buffer without reducing agent
Discussion and conclusion
PFN1 has attracted attention because of its significant role in many cellular activities and its contribution to various diseases. Mutations in its actin- and PLP-binding domains (Fig. 1) potentially interfere with its de novo activities, resulting in the toxicity associated with disorders like ALS. Here, we expressed, purified, validated, and investigated the physical properties and differences between wild type and mutant recombinant human PFN1. These types of analyses require the appropriate methodology, a sufficient amount of quality protein, and tools like MALDI-TOF/TOF and SDS-PAGE for manipulation and analysis. First, we ensured that we had isolated pure (90–95%) wild type and mutant human PFN1, as confirmed by MALDI-TOF/TOF (Tables 1, SI1, and SI2).
We used appropriate techniques such as DLS, far-UV CD, and intrinsic fluorescence that were able to reveal the molecular dynamics and characteristics of PFN1WT and PFN1G118V proteins, from size distribution profiles to secondary and tertiary structures. Our data on PFN1WT (Figs. 3 and 4a) were consistent with data on the native, folded PFN1 protein published by others (Boopathy et al. 2015; Del Poggetto et al. 2015b). When we analyzed our DLS data, the average hydrodynamic diameter for PFN1G118V was 265 nm. However, there was no evidence of aggregation by PFN1WT, as the average hydrodynamic diameter was less than 5 nm (Fig. 4a and b). Furthermore, differences in the far-UV CD spectrum for PFN1G118V (Fig. 3a), along with a red shift in fluorescence and a slight decrease in fluorescence intensity (Fig. 3b), indicated changes in the secondary and tertiary structure of PFN1G118V compared to wild type. These results are in agreement with those previously published by Del Poggetto et al. (2015a), but they differ from those published by Boopathy et al. (2015), who reported that PFN1G118V has a native-like CD spectrum. Given the phi and psi angles for Gly118 according to the Ramachandran plot, the substitution of glycine with valine induces conformational changes that form more energetically favorable dihedral angles. The far-UV CD values for PFN1G118V suggest that the glycine to valine mutation may produce a significant intermolecular force, resulting in an abnormally different secondary structure and affecting the fifth alpha helix. This is consistent with the results of our own in silico study (Kiaei et al., 2018, manuscript under review). The small blue shift at approximately 204 nm to 200 nm, where the molar ellipticity is 0, and lower positive and negative ellipticity values for PFN1G118V in the CD spectrum relative to PFN1WT (Fig. 3a) can also be attributed to high molecular-weight species. Likewise, with western blot of purified PFN1G118V, we observed bands corresponding to the molecular weights of PFN1 dimers and trimers (approximately 30 kDa and 45 kDa) (Fig. 2).
In addition, the intrinsic tryptophan fluorescence assay indicated a folded structure for both PFN1WT and PFN1G118V, as the maximum absorption at 350–357 nm indicating solvent-exposed tryptophan residues in the unfolded protein was not observed here (Del Poggetto et al. 2015b). Given the minor red shift in maximum fluorescence presented here and considering that the G118V mutation has a modest effect on the interaction between PFN1 and PLP (Boopathy et al. 2015), we propose that the environment of tryptophan residues is very similar in the mutant and native state. One may speculate that, altogether, the structural changes that occur when PFN1G118V is folded may make the hydrophobic core somewhat solvent accessible.
This type of structural change following a single, critical amino acid substitution may favor the oligomerization and aggregation of PFN1G118V. Consistent with our observations, in vitro and in vivo studies by others support our evidence for the tendency of PFN1G118V to aggregate (Fil et al. 2017; Tanaka and Hasegawa 2016; Tanaka et al. 2016; Yang et al. 2016; Smith et al. 2014). There are also reports of a correlation between changes in how mutant PFN1 variants fold and their tendency for aggregation (Del Poggetto et al. 2015a, 2016). Indeed, our evidence that PFN1G118V has a larger hydrodynamic diameter than PFN1WT further supports the conclusion that PFN1G118V forms aggregates (Fig. 4b). Looking back at the challenges we encountered when purifying human PFN1G118V from our bacterial system, the fact that a majority of the mutant protein was recovered from the pellet instead of the supernatant suggests that the protein forms aggregates (data not shown). Additionally, in our time-course study with SDS-PAGE and MALDI-TOF/TOF, we found a significant decrease in the band and peak of soluble PFN1G118V monomer after 24 h of incubation at 37 °C, and we observed the formation of larger insoluble protein complexes that were stable under denaturing conditions (Fig. 5d). However, we observed bands of SDS-soluble, monomeric PFN1WT on the gel after 96 h of incubation, indicating that PFN1WT is less prone to aggregating (Fig. 5b). The work of other colleagues supports the idea that SDS-resistant oligomers of PFN1G118V (see Fig. 2b) may seed the formation of inclusions and accelerate the recruitment of monomeric PFN1G118V into aggregates (Tanaka et al. 2016). Previous studies demonstrated that the G118V mutation, which is proximal to the actin-binding site (Fig. 1), causes PFN1 to lose its ability to bind actin, sequester G-actin, and regulate the F/G-actin ratio, thus affecting the dynamics of actin polymerization and de-polymerization (Fil et al. 2017; Freischmidt et al. 2015). Our data suggest that the G118V mutation causes structural alterations in PFN1 that may impair its interaction with actin as a consequence of abnormal binding-site conformation and/or PFN1 aggregation.
In conclusion, we investigated the substitution of glycine 118 in PFN1, a solvent-exposed residue within a flexible loop, with valine, a residue with a larger side chain. Our findings support our hypothesis that this mutation significantly alters the secondary and tertiary structures of PFN1. Therefore, the toxicity of mutant PFN1 could result from the placement of two valines side by side, thus changing the structure and leading to misfolding, oligomerization, and aggregation. Our results present a possible mechanism by which mutant PFN1 aggregates, with important implications for the development of drugs that may block this process.
References
Alkam D, Feldman EZ, Singh A, Kiaei M (2017) Profilin1 biology and its mutation, actin (g) in disease. Cell Mol Life Sci 74:967–981
Boopathy S, Silvas TV, Tischbein M, Jansen S, Shandilya SM, Zitzewitz JA, Landers JE, Goode BL, Schiffer CA, Bosco DA (2015) Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc Natl Acad Sci USA 112:7984–7989
Chen Y, Zheng ZZ, Huang R, Chen K, Song W, Zhao B, Chen X, Yang Y, Yuan L, Shang HF (2013) PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol Aging 34(7):1922 e1–e5
Del Poggetto E, Bemporad F, Tatini F, Chiti F (2015a) Mutations of Profilin-1 associated with amyotrophic lateral sclerosis promote aggregation due to structural changes of its native state. ACS Chem Biol 10:2553–2563
Del Poggetto E, Chiti F, Bemporad F (2015b) The folding process of human Profilin-1, a novel protein associated with familial amyotrophic lateral sclerosis. Sci Report 5:–12332
Del Poggetto E, Gori L, Chiti F (2016) Biophysical analysis of three novel profilin-1 variants associated with amyotrophic lateral sclerosis indicates a correlation between their aggregation propensity and the structural features of their globular state. Biol Chem 397:927–937
Fan L, Simard LR (2002) Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet 11:1605–1614
Fedorov A, Pollard T, Almo S (1994) Purification, characterization and crystallization of human platelet profilin expressed in Escherichia coli. J Mol Biol 241:480–482
Fil D, DeLoach A, Yadav S, Alkam D, MacNicol M, Singh A, Compadre CM, Goellner JJ, O'Brien CA, Fahmi T, Basnakian AG, Calingasan NY, Klessner JL, Beal FM, Peters OM, Metterville J, Brown RH Jr, Ling KKY, Rigo F, Ozdinler PH, Kiaei M (2017) Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum Mol Genet 26:686–701
Freischmidt A, Schopflin M, Feiler MS, Fleck AK, Ludolph AC, Weishaupt JH (2015) Profilin 1 with the amyotrophic lateral sclerosis associated mutation T109M displays unaltered actin binding and does not affect the actin cytoskeleton. BMC Neurosci 16:77
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17071
Hensel N, Claus P (2018) The actin cytoskeleton in SMA and ALS: how does it contribute to Motoneuron degeneration? Neuroscientist 24(1):54–72
Ingre C, Landers JE, Rizik N, Volk AE, Akimoto C, Birve A, Hübers A, Keagle PJ, Piotrowska K, Press R, Andersen PM, Ludolph AC, Weishaupt JHIngre C, Landers JE, Rizik N, Volk AE, Akimoto C, Birve A, Hübers A, Keagle PJ, Piotrowska K, Press R, Andersen PM, Ludolph AC, Weishaupt JH (2013) A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging 34(6):1708 e1–e6
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, van Deerlin V, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurrò MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, ITALSGEN Consortium, Galassi G, Scholz SW, Taylor JP, Restagno G, Chiò A, Traynor BJ (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857–864
Kohn J, Wilchek M (1984) The use of cyanogen bromide and other novel cyanylating agents for the activation of polysaccharide resins. Appl. Biochem. Biotechnol. 9:285–305
Krishnan K, Holub O, Gratton E, Clayton AH, Cody S, Moens PD (2009) Profilin interaction with phosphatidylinositol (4,5)-bisphosphate destabilizes the membrane of giant unilamellar vesicles. Biophys J 96:5112–5121
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lambrechts A, Jonckheere V, Dewitte D, Vandekerckhove J, Ampe C (2002) Mutational analysis of human profilin I reveals a second PI(4,5)-P2 binding site neighbouring the poly (L-proline) binding site. BMC Biochem 3:12
Lim L, Kang J, Song J (2017) ALS-causing profilin-1-mutant forms a non-native helical structure in membrane environments. Biochim Biophys Acta 1859(11):2161–2170
Lorber B, Fischer F, Bailly M, Roy H, Kern D (2012) Protein analysis by dynamic light scattering: methods and techniques for students. Biochem Mol Biol Educ 40:372–382
Matsukawa K, Hashimoto T, Matsumoto T, Ihara R, Chihara T, Miura M, Wakabayashi T, Iwatsubo T (2016) Familial amyotrophic lateral sclerosis-linked mutations in profilin 1 exacerbate TDP-43-induced degeneration in the retina of drosophila melanogaster through an increase in the cytoplasmic localization of TDP-43. J Biol Chem 291:23464–23476
McLachlan GD, Cahill SM, Girvin ME, Almo SC (2007) Acid-induced equilibrium folding intermediate of human platelet profilin. Biochemistry 46:6931–6943
Metzler WJ, Farmer BT, Constantine KL, Friedrichs MS, Mueller L, Lavoie T (1995) Refined solution structure of human profilin I. Protein Sci 4:450–459
Nölle A, Zeug A, van Bergeijk J, Tönges L, Gerhard R, Brinkmann H, Al Rayes S, Hensel N, Schill Y, Apkhazava D, Jablonka S, O'mer J, Srivastav RK, Baasner A, Lingor P, Wirth B, Ponimaskin E, Niedenthal R, Grothe C, Claus P (2011) The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum Mol Genet 20(24):4865–4878
Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U (1993) The structure of crystalline profilin-beta-actin. Nature 365:810–816
Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2007) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1:2856–2860
Smith BN, Vance C, Scotter EL, Troakes C, Wong CH, Topp S, Maekawa S, King A, Mitchell JC, Lund K, Al-Chalabi A, Ticozzi N, Silani V, Sapp P, Brown Jr. RH, Landers JE, Al-Sarraj S, Shaw CE (2014) Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging 36:1602.e1617–1602.e1627
Tanaka Y, Hasegawa M (2016) Profilin 1 mutants form aggregates that induce accumulation of prion-like TDP-43. Prion 10:283–289
Tanaka Y, Nonaka T, Suzuki G, Kametani F, Hasegawa M (2016) Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum Mol Genet 25:1420–1433
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, van den Berg LH (2017) Amyotrophic lateral sclerosis. Lancet 390(10107):2084-2098
Witke W (2004) The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol 14:461–469
Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, Kost JE, Gonzalez-Perez P, Fox AD, Adams J, Taroni F, Tiloca C, Leclerc AL, Chafe SC, Mangroo D, Moore MJ, Zitzewitz JA, Xu ZS, van den Berg LH, Glass JD, Siciliano G, Cirulli ET, Goldstein DB, Salachas F, Meininger V, Rossoll W, Ratti A, Gellera C, Bosco DA, Bassell GJ, Silani V, Drory VE, Brown Jr RH, Landers JE (2012) Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488:499–503
Yang C, Danielson EW, Qiao T, Metterville J, Brown RH Jr, Landers JE, Xu Z (2016) Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc Natl Acad Sci U S A 113:E6209–E6218
Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano OF, Pagani W, Lodin D, Orozco G, Chinea A (2015) A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 6:171
Acknowledgements
We gratefully acknowledge Dr. Seyed Omid Ranaei Siadat from the Protein Research Center (Shahid Beheshti University) for allowing us to access his laboratory, which focuses on protein expression. Financial support by the Shahid Beheshti University Research Council is also gratefully acknowledged. This manuscript was edited by the Science Communication Group at the University of Arkansas for Medical Sciences (UAMS); we also thank Caroline Barham (UAMS) for proofreading the manuscript. The authors acknowledge funding from the UAMS startup fund, UAMS Center for Translational Neurosciences, NIGMS IDeA Program Award P30 GM110702, P20GM109005, and NINDS NS088653 and NS101334.
Author information
Authors and Affiliations
Contributions
M.K. introduced the idea, designed the study, analyzed the data, and wrote the manuscript. A.G. designed the study, analyzed the data, and wrote the manuscript. M.N. designed and performed experiments, analyzed data, and wrote the manuscript. P.G. assisted with MALDI analysis and helped prepare the manuscript. A.A. assisted with the expression of recombinant PFN1, and S.A. assisted with sub-cloning experiments.
Corresponding authors
Ethics declarations
Conflict of interest
Dr. Kiaei and UAMS have a financial interest in the technology discussed in this publication. These financial interests have been reviewed and approved in accordance with the UAMS conflict of interest policies. Other authors have no conflict to disclose.
Electronic supplementary material
ESM 1
(DOCX 1242 kb)
Rights and permissions
About this article

Cite this article
Nekouei, M., Ghezellou, P., Aliahmadi, A. et al. Changes in biophysical characteristics of PFN1 due to mutation causing amyotrophic lateral sclerosis. Metab Brain Dis 33, 1975–1984 (2018). https://doi.org/10.1007/s11011-018-0305-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-018-0305-4