
Abstract
Traumatic brain injury (TBI) may trigger secondary injury cascades including endoplasmic reticulum stress, oxidative stress, and neuroinflammation. Unfortunately, there are no effective treatments targeting either primary or secondary injuries that result in long-term detrimental consequences. Huperzine A (HupA) is a potent acetylcholinesterase inhibitor (AChEI) that has been used treatment of Alzheimer’s disease (AD). This study aimed to explore the neuroprotective effects of HupA in TBI and its possible mechanisms. Repetitive mild closed head injury (CHI) model was used to mimic concussive TBI. Mice were randomly assigned into three groups including sham, vehicle-treated and HupA-treated injured mice. The HupA was given at dose of 1.0 mg/kg/day and was initiated 30 min after the first injury, then administered daily for a total of 30 days. The neuronal functions including motor functions, emotion-like behaviors, learning and memory were tested. Axonal injury, reactive oxygen species (ROS), and neuroinflammation were examined as well. The results showed that injured mice treated with HupA had significant improvement in Morris water maze performance compared with vehicle-treated injured mice. HupA treatment significantly attenuated markers of neuroinflammation and oxidative stress in the injured mice. Taken together, HupA was effective in reducing neuroinflammation, oxidative stress and behavioral recovery after TBI. HupA is a promising candidate for treatment of TBI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is a major public health issue and a leading cause of death and disability worldwide, especially for those under 45 years old. TBI can lead to acute and potentially long-lasting neurological dysfunction, including the development of chronic traumatic encephalopathy or even Alzheimer’s disease(AD) (Hayes et al. 2017). Both primary and secondary injuries may result in devastating consequences. The primary injury may precede multiple downstream events contributing to secondary injury cascade of cellular, molecular, and metabolic pathological events (Smith et al. 2013). TBI animal model led to significant alterations in the hippocampal excitatory synaptic transmission and plasticity, associated with greater neurodegeneration, neuroinflammation, and cognitive impairments (Schumann et al. 2008). Some of important secondary injury mechanisms involve activation of neuronal cell death pathways, microglial and astrocytic activation, and neurotoxicity. Cholinergic deficits contribute to cognitive impairment after TBI (Kobeissy et al. 2016). TBI may cause hypercholinergic activity early and develops into a chronic hypocholinergic state later. Multiple pharmacological agents for treatment of TBI, to date, fail to show a therapeutic benefits, and some even worsen the outcomes (Kochanek et al. 2016). Therefore, it is urgent to continue searching for effective and safe approaches for TBI treatment. Timing of treatment is a key factor for the efficacy. It is also important to start treatment earlier to prevent activation of downstream cascades after injury (Mei et al. 2017). It is reported that early intervention after TBI, especially the treatment in the early minutes after injury, profoundly influences the outcome of TBI patients (Yang et al. 2017).
The secondary injury is the therapeutic target for TBI. However, the injury mechanisms that have been identified are multifactorial, time dependent, and highly complex (Wheaton et al. 2011). Few therapies have been shown to be successful. Huperzine A (HupA) is an alkaloid isolated from the Chinese herb Huperzia serrata (Qian CengTa), which is now widely used in China to ameliorate mild-to-moderate AD (Zhu et al. 2015). HupA exerts its pharmacological effects primarily by increasing synaptic ACh levels through inhibition of acetylcholinesterase (AChE) activity and has been reported to be an effective cognition enhancer in a broad range of animal models of cognitive impairment in AD and stroke (Scherer et al. 2013). Notably, in addition to its anti-AChE activity, HupA has been shown to possess diverse biological activities such as anti-inflammation, anti-oxidation and anti-apoptosis (Mao et al. 2015). HupA has also been demonstrated to be neuroprotective against glutamate, hydrogen peroxide and amyloid (Wang et al. 2012). These studies suggest the potential therapeutic benefits of HupA in TBI.
To explore the neuroprotective effects of HupA and its possible mechanism of action under TBI condition, repetitive mild closed head injury (CHI) models were established. The effects of HupA on motor and learning function, spatial memory, inflammatory response and oxidative stress after the injury were assessed. The present results show that HupA could be an ideal candidate drug in the treatment of TBI.
Material and methods
All experiments were approved by the third Affiliated Hospital of Guangzhou Medical University Institutional Animal Care and Use Committee, and complied with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Mouse model of closed head injury (CHI)
The mouse repetitive mild closed head injury (CHI) model was used as previously described (Mannix et al. 2017). Briefly, C57BL/6 male mice (8 weeks old) were anesthetized for 45 s using 3% isoflurane in a 70:30 nitrous oxide/oxygen mixture. Anesthetized mice were placed on a delicate task wiper and positioned such that the head was placed directly under a hollow guide tube 28 in. in length. The mice were grasped by the tail. A 54 g metal bolt was used to deliver an impact to the dorsal aspect of the skull. Injured mice underwent 7 concussive injuries over 9 days. Sham operated mice underwent anesthesia without trauma. After injury, mice were returned to their cages, with food and water.
Treatment protocol with HupA
Mice were randomized divided into 3 group: sham (n = 10), vehicle (n = 10) and HupA group (n = 10). Sham group mice underwent anesthesia but no concussive injury. HupA and vehicle group mice were underwent 7 concussive injuries over 9 days and were administered daily intraperitoneally injection of HupA or normal saline. HupA dose was selected by targeting clinically significant concentrations as described previously (Wang et al. 2011). HupA (98% pure; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in normal saline solution to a concentration of 1.0 ml/kg. HupA (1.0 mg/kg) or an equal volume of saline was injected intraperitoneally (IP) 30 min after TBI for up to 30 days. Animals were sacrificed 30 days after the first injury and the brains were removed from the skull for further experiments.
mRNA extraction and quantitative real-time polymerase chain reaction analysis
Total RNA were extracted from hippocampus or cortex and purified using iScript™ RT-qPCR Kit (BIO-RAD, Hercules, CA). One microgram of total RNA was reverse-transcribed using the Applied Biosystems High-Capacity cDNA reverse transcription kit. Quantitative real-time polymerase chain reaction analysis was performed on an Applied Biosystems Prism 7900 System using Power SYBR Green PCR Master Mix. The thermal cycler conditions were as follows: hold for 10 min at 95 °C, followed by 45 cycles of a 2-step PCR consisting of a 95 °C step for 15 s followed by a 60 °C step for 25 s. Sequences of primer used are given in Table 1. Amplifications were carried out in triplicates and the relative expression of target genes was determined by the ∆∆CT method.
Western blotting
Western blotting was performed to determine the expression of APP, NF-κB(p65), and β-actin. Proteins (30-40 μg) from each sample supernatant were separated by electrophoresis on 4–12% SDS polyacrylamide gels (Invitrogen, Carlsbad, CA), then transferred to a nitrocellulose membrane. The membranes were blocked with 5% fat free milk for 1 h and incubated overnight at 4 °C with antibodies against APP (Abcam, Cambridge, MA, USA, 1:1000), NF-κB(Abcam, Cambridge, MA, USA,1:2500), or a mouse anti-β-actin monoclonal antibody (1:5000; Abcam, Cambridge, MA, USA,). The blots were washed in TBS-Tween and further incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies (Abcam, Cambridge, MA, USA,1:5000). Immunoreactivity was detected using a chemiluminescence system according to the manufacturer’s protocol. Band signal intensity was quantified using Image J software. The density of each sample was normalized to its own density of β-actin.
Immunohistochemistry
Animals were anesthetized and then transcardially perfused with phosphate buffered saline (PBS) followed by 4% formaldehyde for 15 min at 30 days after the first injury. Brains were removed and fixed in 4% formaldehyde for 48 h, and incubated in 30% sucrose for an additional 48 h at 4 °C. Coronal sections (20 μm) were cut on a cryostat and mounted onto glass slides. To assess inflammation after injury, coronal sections were first blocked with 3% normal goat serum, then incubated overnight at 4 °C with anti-Iba1(Novus Biologicals, Littleton, CO, USA, 1:100) antibody. The sections were then reacted for 1 h at room temperature with a biotinylated secondary antibody, followed by ABC elite kit (Vector, USA) and diaminobenzydine (DAB) reaction (Vector, USA). The positive staining cell was counted in hippocampus area. Cell counts were performed with ImageTool software.
Measurement of malondialdehyde (MDA) level
The level of MDA was measured according to the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The level of MDA was expressed as nmol/mg protein.
Measurement of superoxide dismutase (SOD) activities
The enzymatic activities of SOD in the cortex tissue homogenates were measured by the commercial assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturers’ instructions. The enzymatic activity of the protein were expressed as U/mg protein.
Measurement of TNF-α and IL-1β levels
The levels of TNF-α and IL-1β on the cerebral hippocampus were quantified using specific ELISA kits according to the manufacturers’ instructions (Biosource Europe SA, Nivelles, Belgium). Brain tissues were harvested and frozen 3 days after last injury. Tissues were then homogenized in a lysis buffer. Tissue homogenates were centrifuged at 13,000 rpm for 20 min at 4 °C. Supernatants were transferred to new tubes and used for analysis. Values were expressed as pg/mg protein.
Behavioral analysis
Morris water maze test
The cognitive function of the mice was assessed using Morris water maze test, as previously described (Mannix et al. 2013). During training, the platform was placed in one quadrant of the pool. The animal was gently and randomly placed in the water facing the wall at one of the remaining quadrants, and allowed to swim to find the platform. Mice were underwent four training trials per day from day 14 to day 18 after the first injury. Each animal was given 60 s to find the platform, and could remain on the platform for 15 s to become familiar with visual cues from the surroundings before being removed from the platform. One hour after the last training session on day 18, the platform was removed and animals were tested in a probe trial to measure quadrant preference and platform localization. On the sixth day, the hidden platform was removed, and the mice were released in the quadrant opposite the target quadrant (original platform quadrant).The time spent in the target quadrant and the percentage of swimming distance spent within the target quadrant within a 30s period were used to evaluate the spatial memory ability of the mice.
Elevated plus maze
The elevated plus maze is used to test anxiety-like behavior in rodents. The test was performed on day 24 after the first injury. Mice were placed on the center platform of the maze, facing a closed arm, and were allowed to explore the apparatus for 5 min. Basic locomotor activity and percent of time spent in open versus closed arms were recorded as described.
Rotarod test
Motor ability and function were assessed on Days 8–10 and after the first injury using a rotarod. The rotarod test has been described previously(Tu et al. 2017). The time between placement on the rotarod and fall off from the rotarod was recorded as a measure of motor function. The first day of rotarod comprised training. During training mice learned to walk on the rotating rod for 5 min. The 2nd and 3rd days comprised testing. On testing days, mice were placed on the rod at 4 rpm for 10 s to acclimate to the rod speed. After the 10-s acclimation period, the rod accelerated at 0.1 rpm/s. Each mouse completed 4 trials on the testing days with a minimum of 5 min rest between each trial.
Statistical analyses
Data are presented as mean ± standard error of the mean. Continuous variables were compared between injured and sham injured mice and HupA treated using analysis of variance (ANOVA) or Kruskal-Wallis for univariate testing as appropriate. To account for repeated measures over time, MWM and rotorod latencies were analyzed by linear regression with clustered robust standard errors. Statistical significance was considered p < 0.05. All analyses were performed using Stata 11.2.
Results
HupA decreased TBI-induced cognitive deficits
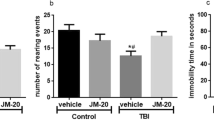
Persistent deficits in memory and learning are a commonality among TBI survivors. To determine whether HupA could improve learning and memory, Morris water maze test was used to evaluate spatial learning impairment. The less time it takes for animals to find the platform, the better the spatial learning function. Mice were tested on day 14 after the first injury. Compared with sham group, TBI animals took more time to find the platform. However, relative to the vehicle treatment group, HupA treatment spent significantly less time to find the platform (Fig. 1). There were no statistically significant difference of performance in elevated plus maze and rotarod test between vehicle treated and HupA treated injury groups.

Effects of HupA treatment on CHI-induced spatial memory performance. a Morris water maze test were performed on day 14 after the first injury. Mean daily latencies to escape from start point to the hidden platform were measured. Animals received HupA exhibited less escape latencies compare to vehicle animals. b Elevated plus maze test. c Rotarod task demonstrated deficits of motor balance in injured compare to sham, *P<0.05 compare to sham
HupA attenuated TBI-induced axonal damage
Amyloid precursor protein (APP) is a membrane spanning glycoprotein of nerve cells which is transported by fast axoplasmic flow and accumulates after axonal damage. The upregulation of APP has been shown a marker for axonal damage. We examined APP expression in acute time point after injury. The brain cortex tissues were collected 3 days after the last injury and analyzed by western blot. The result showed that after injury APP expression was increased and HupA significantly attenuated the expression of APP compared with vehicle-treated group (Fig. 2).

HupA ameliorated APP overexpression. a The expression of APP in cortex from sham and rmTBI mice treated with HupA or normal saline was assessed by western blots. β-actin was used as a loading control. b Quantification of immunoblot bands from three independent experiments for APP. Data are presented as mean ± SEM. *P < 0.05 by Student’s t-test compared to sham group. #P < 0.05 by Student’s t-test compare to vehicle group
HupA reduced oxidative stress in TBI animals
Oxidative stress has been associated with secondary injury after TBI. To determine whether the neuroprotective effect of HupA was derived from its ability to alleviate the oxidative stress caused by TBI, MDA and SOD were analyzed in brain tissue. Mice subjected to TBI exhibited a marked increase in MDA levels, compared with sham group (P < 0.05). The administration of HupA to CHI animal significantly reduced TBI-mediated lipid peroxidation in comparison with the vehicle-treated group (P < 0.05). Furthermore, the activity of antioxidative enzymes were measured in the cortex homogenates from the animals. The activity of SOD were reduced in vehicle-treated group compared with sham group, and HupA increased activity of SOD in cortex (Fig. 3, P < 0.05).

HupA alleviated oxidative stress caused by TBI. a Measurement of malondialdehyde (MDA) levels. b The activities of SOD. *P < 0.05 by Student’s t-test compared to sham group. #P < 0.05 by Student’s t-test compare to vehicle group
HupA reduced neuroinflammation in TBI animals
To determine the effect of HupA on inflammation, we performed immunohistochemical staining of Iba-1 for detection of microglia. There was a significant difference in the number of Iba-1-positive cells in cortex among different group. As shown in Fig.4, a significantly reduced Iba-1 immunolabeling cells were observed in HupA-treated group compared to vehicle treated group (p < 0.05). These findings suggest that HupA treatment attenuated CHI-induced inflammation.

HupA attenuated increase of microglial cell after TBI. Representative immunostaining images of Iba-1 in cortex of 3 group mice on day 30 post first injury
HupA attenuates the elevated level of inflammatory factors in TBI animals
IL-1β is released by activated microglia after stimulation. Following the injury, there was a significant increase of expression of both TNF-α and IL-1β in hippocampus on day 3 after TBI in comparison with sham-operated animals. This elevation of TNF-α and IL-1β expression in hippocampus was significantly attenuated in HupA-treated mice in comparison with the vehicle group (Fig. 5a, b). Figure 5c, d showed that TNF-α and IL-1β mRNA levels were increased in the hippocampus 1 d after TBI and expression of these markers was attenuated by HupA intervention after TBI.

HupA inhibited the increase of TNF-α and IL-1β after TBI. The relative protein content of TNF-α and IL-1β in per milligram of total protein was assessed. TNF-α and IL-1β expressions examined by real time PCR. a The TNF-α concentration. b The IL-1β concentration. c The mRNA expression of TNF-α. d The mRNA expression of IL-1β. The data are expressed as means ± SEM. * p < 0.05 compare to sham group; # p < 0.05 compare to vehicle group
To further explore the molecular mechanisms underlying the inhibitory effect of HupA on expressions of IL-1β and TNF-α after injury, the expressions of NFκB p65 were determined by western blot analysis. The well-established, classic activation pattern of NFκB is translocation of p65 from the cytoplasm to the nucleus. We investigated the expression of cytoplasmic NFκB p65 and nuclear NFκB p65. There was a significant difference in the NF-κB p65 expression of the hippocampus among different group. Nuclear NFκB p65 was significantly enhanced in TBI group. Furthermore, treatment with HupA caused a significant decrease in the expression of nuclear NF-κB p65 (Fig. 6) compared to vehicle treated group.

HupA inhibited NFκB activation. The hippocampus from sham, injured vehicle or injured HupA treated mice were lysed in RIPA buffer. Nuclear NFκB p65 and cytoplasmic NFκB p65 expressions were examined by western blot. a Western blots of nuclear NFκB p65. b Western blots of cytoplasmic NFκB p65. * p < 0.05 compare to sham group; # p < 0.05 compared to vehicle group
Discussion
TBI is a leading cause of mortality and morbidity around the world. Repetitive mild brain injuries can result in cumulative brain damage and neurodegeneration that have been linked to neuroinflammation and oxidative stress. To date, there is no approved drug for treatment of TBI, despite a broad number of drug classes may potentially target a range of different mechanisms pertinent to TBI (Wheaton et al. 2011). In this study, we determined HupA reduced neuroinflammation, oxidative stress and neurological impairments in experimental rmTBI. The results demonstrate that HupA is neuroprotective in TBI mice. The main findings in the present study were as following: 1) TBI caused spatial cognitive impairment and HupA treatment improved cognitive functional outcomes; 2) TNF-α and IL-1β were increased remarkably following TBI; 3) TBI induced lipid peroxidation and 4) HupA treatment significantly ameliorated neuroinflammation, oxidative stress and axonal injury.
Cognitive impairment is one of the most prevalent chronic TBI symptoms (Lee et al. 2015). Spatial cognition, as assessed in the water maze, was shown to be impaired in rmTBI mice through the measure of time spending in finding platform, consistent with previous reports (Velosky et al. 2017). HupA treated mice spent less time finding the platform compared to vehicle group, suggesting that HupA was able to preserve memory after TBI. There was also a significant improvement of hidden trail in HupA treated mice compared to vehicle-treated mice, which further supporting the notion that HupA preserved cognitive function in the water maze. Motor function was assessed by rotarod test and the results showed that HupA did not improve motor function in TBI mice. Nonetheless, these findings indicate that HupA treatment is able to attenuate cognitive dysfunction after TBI.
Accumulation of APP in axons has been described in trauma. APP upregulation is a universally accepted marker of traumatic axonal injury (Johnson et al. 2011). APP immunostaining was performed to detect axonal injury in the traumatic injured brain (Johnson et al. 2013). An anti-inflammatory therapy may decrease inflammation and APP upregulation in TBI whereby primary and secondary damage are followed by neuroinflammatory responses (Acosta et al. 2017). Our study suggested that HupA markedly reduced post-traumatic APP accumulation in the cortex and prevented TBI- induced cognitive impairment. This indicates that axonal number and function are protected, which also prevents cognitive impairments.
The pathogenesis of TBI likely involves a number of secondary injury processes. Growing evidence, supported by our findings here, implicates neuroinflammation and oxidative stress as a key factors (Ding et al. 2014). Significantly increased free radical production is one of the main causes of secondary injury in TBI, leading to further damage to neuronal tissue. After brain injury, infiltrating neutrophils produce oxidative stress, which contributes to neurodegeneration (Shu et al. 2016). TBI models demonstrated a rapid increase in brain reactive oxygen species (ROS) levels. Several oxidants and their derivatives are generated after TBI, including superoxide anions and hydroxylradicals. This enhanced production of ROS along with exhausted antioxidant defense enzymes, such as superoxide dismutase, catalases, and GPx, causes oxidative stress. MDA formation in the brain tissue is widely used as the index of lipid peroxidation and it increased after TBI(Cheng et al. 2016). SOD is a well-known intracellular antioxidant enzyme, We found that its activity was decreased after TBI. The level of MDA in brain of TBI mice was consistently increased compared with sham group in our study. Furthermore, HupA inhibited lipid peroxidation (MDA production) and increased the activities of endogenous antioxidant enzymes (SOD). Neuroprotective effects of HupA against traumatic brain injury are associated with its antioxidant properties. In agreement with our findings, HupA has been reported to reduce oxidative damage in D-galactose-treated rats (Ruan et al. 2014). These results suggest that HupA can keep balance between oxidants and antioxidants in different brain injury model. Some pathways including lipid peroxidation is changed after TBI by gene expression microarray analysis (Kobeissy et al. 2016). The neuroprotective mechanisms of HupA might be associated with signaling pathways involved in lipid peroxidation. Further studies are required to define HupA mediated which pathways involved in lipid peroxidation.
Neuroinflammation has been shown to be responsible for many of cognitive deficits after injury (Tabas and Glass 2013). Excessive neuroinflammation can exacerbate the functional deficits, and moderate inhibition to the inflammatory response may be neuroprotective. Post-traumatic neuroinflammation is characterized by glial cell activation, leukocyte recruitment, and upregulation of inflammatory mediators (Aungst et al. 2014). Microglial cells are the master regulators of the neuroinflammatory response associated with brain disease and the activated microglia has been demonstrated in models of TBI (Lin et al. 2017). Activated microglia release a number of pro-inflammatory mediators including TNF-α and IL-1β that contribute to secondary damage (Perry et al. 2010). The current study found that TBI induced abundant microglia accumulation in hippocampus, and HupA administration significantly inhibited this accumulation.
Previous studies have suggested that the inflammatory response participates in the pathogenesis of TBI, and NF-κB is regarded as an important factor. The activation of NF-κB leads to the release of pro-inflammatory cytokines, such as TNF-α and IL-1β (Kumar and Loane 2012). We therefore evaluated the possible impact of the HupA treatment on TNF-α and IL-1β. We found that both TNF-α and IL-1β levels were significantly reduced in the brains of TBI mice treated with HupA compared to sham animals, further illustrating the mitigation of neuroinflammation by HupA. Our study revealed that HupA inhibited the excessive activation of NF-κB p65 and reduced the release of TNF-α and IL-1β in mice closed head injury model. Preventing the inflammatory response is considered a high potential target for neuroprotection after TBI. In addition, accumulating evidence indicated that HupA could inhibit neuroinfalmmation. HupA has been reported to suppress D-gal-induced neurovascular damage and BBB dysfunction, partly by preventing NF-κB nuclear translocation (Ruan et al. 2014). HupA blocked D-gal-induced chronic sterile inflammation by inhibiting cytoplasmic IκBα degradation and NF-κB activation(Ruan et al. 2013). The finding suggested that HupA has central anti-inflammatory effects plausibly via suppressing NF-κB pathway and interleukin release from neurons and microglial cells. Overall, we found that HupA attenuated neuroinflammation and oxidative stress and that these changes were associated with improved long-term cognitive outcomes in mice with TBI. The neuroprotective effects of HupA may involve multiple mechanisms.
However, there are also several limitations of this study. First, only one dose was attempted. Second, the neuroprotective effects of HupA need to be affirmed in other TBI models. Third, the time windows for treatment after TBI need to be further determined.
In conclusion, our data show that treatment with HupA can inhibit oxidative stress, reduce inflammation and alleviate some behavioral impairments in a TBI animal model. This findings suggest that HupA may be an effective neuroprotectant as an early pharmacological intervention for TBI.
References
Acosta SA, Tajiri N, Sanberg PR, Kaneko Y, Borlongan CV (2017) Increased amyloid precursor protein and tau expression manifests as key secondary cell death in chronic traumatic brain injury. J Cell Physiol 232:665–677
Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI (2014) Repeated mild traumatic brain injury causes chronic Neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab 34:1223–1232
Cheng T et al (2016) Cerebroprotection of flavanol (−)-epicatechin after traumatic brain injury via Nrf2-dependent and -independent pathways. Free Radic Biol Med 92:15–28
Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M (2014) Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2–ARE signaling pathway as a potential mechanism. Free Radic Biol Med 73:1–11
Hayes JP et al (2017) Mild traumatic brain injury is associated with reduced cortical thickness in those at risk for Alzheimer’s disease. Brain. doi:10.1093/brain/aww344
Johnson MW, Stoll L, Rubio A, Troncoso J, Pletnikova O, Fowler DR, Li L (2011) Axonal injury in young pediatric head trauma: a comparison study of β-amyloid precursor protein (β-APP) Immunohistochemical staining in traumatic and nontraumatic deaths*. J Forensic Sci 56:1198–1205
Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43
Kobeissy FH et al (2016) Cognitive impairments induced by concussive mild traumatic brain injury in mouse are ameliorated by treatment with Phenserine via multiple non-cholinergic and cholinergic mechanisms. PLoS One 11:e0156493
Kochanek PM et al (2016) Approach to modeling, therapy evaluation, drug selection, and biomarker assessments for a multicenter pre-clinical drug screening consortium for acute therapies in severe traumatic brain injury: operation brain trauma therapy. J Neurotrauma 33:513–522
Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26:1191–1201
Lee DJ et al (2015) Septohippocampal neuromodulation improves cognition after traumatic brain injury. J Neurotrauma 32:1822–1832
Lin C et al (2017) Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp Neurol 290:115–122
Mannix R et al (2013) Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol 74:65–75
Mannix R, Berkner J, Mei Z, Alcon S, Hashim J, Robinson S, Jantzie L, Meehan WP, Qiu J (2017) Adolescent mice demonstrate a distinct pattern of injury after repetitive mild traumatic brain injury. J Neurotrauma 34:495–504
Mao X-Y, Zhou H-H, Li X, Liu Z-Q (2015) Huperzine a alleviates oxidative glutamate toxicity in hippocampal HT22 cells via activating BDNF/TrkB-dependent PI3K/Akt/mTOR signaling pathway. Cell Mol Neurobiol 36:915–925
Mei Z, Qiu J, Alcon S, Hashim J, Rotenberg A, Sun Y, Meehan WP 3rd, Mannix R (2017) Memantine improves outcomes after repetitive traumatic brain injury. Behav Brain Res. doi:10.1016/j.bbr.2017.04.017
Perry VH, Nicoll JAR, Holmes C (2010) Microglia in neurodegenerative disease. Nat Rev Neurol 6:193–201
Ruan Q, Liu F, Gao Z, Kong D, Hu X, Shi D, Bao Z, Yu Z (2013) The anti-inflamm-aging and hepatoprotective effects of huperzine A in d-galactose-treated rats. Mech Ageing Dev 134:89–97
Ruan Q, Hu X, Ao H, Ma H, Gao Z, Liu F, Kong D, Bao Z, Yu Z (2014) The neurovascular protective effects of Huperzine a on D-galactose-induced inflammatory damage in the rat hippocampus. Gerontology 60:424–439
Scherer RW, Yang G, Wang Y, Tian J, Liu J-P (2013) Huperzine A for Alzheimer’s disease: a systematic review and meta-analysis of randomized clinical trials. PLoS One 8:e74916
Schumann J, Alexandrovich GA, Biegon A, Yaka R (2008) Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J Neurotrauma 25:945–957
Shu L, Wang C, Wang J, Zhang Y, Zhang X, Yang Y, Zhuo J, Liu J (2016) The neuroprotection of hypoxic preconditioning on rat brain against traumatic brain injury by up-regulated transcription factor Nrf2 and HO-1 expression. Neurosci Lett 611:74–80
Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221
Tabas I, Glass CK (2013) Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339:166–172
Tu TM, Kolls BJ, Soderblom EJ, Cantillana V, Ferrell PD, Moseley MA, Wang H, Dawson HN, Laskowitz DT (2017) Apolipoprotein E mimetic peptide, CN-105, improves outcomes in ischemic stroke. Ann Clin Transl Neurol 4:246–265
Velosky AG, Tucker LB, Fu AH, Liu J, McCabe JT (2017) Cognitive performance of male and female C57BL/6J mice after repetitive concussive brain injuries. Behav Brain Res 324:115–124
Wang C-Y, Zheng W, Wang T, Xie J-W, Wang S-L, Zhao B-L, Teng W-P, Wang Z-Y (2011) Huperzine A activates Wnt/β-catenin signaling and enhances the Nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 36:1073–1089
Wang Y, Tang XC, Zhang HY (2012) Huperzine a alleviates synaptic deficits and modulates amyloidogenic and nonamyloidogenic pathways in APPswe/PS1dE9 transgenic mice. J Neurosci Res 90:508–517
Wheaton P, Mathias JL, Vink R (2011) Impact of pharmacological treatments on cognitive and behavioral outcome in the Postacute stages of adult traumatic brain injury. J Clin Psychopharmacol 31:745–757
Yang L et al (2017) DRα1-MOG-35-55 treatment reduces lesion volumes and improves neurological deficits after traumatic brain injury. Metab Brain Dis. doi:10.1007/s11011-017-9991-6
Zhu D, Lei Y, Yang L, Ye CY, Qin MY, Yang HY, Jiang HL, Tang XC, Zhang HY (2015) Involvement of intracellular and mitochondrial Aβ in the ameliorative effects of Huperzine A against oligomeric Aβ42-induced injury in primary rat neurons. PLoS One 10:e0128366
Acknowledgements
The authors would like to thank Dr. Jianhua Qiu for his assistance with the revision English language.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work is supported by a grant from Guangzhou Education Bureau (1201421151).
Conflict of interest
No competing financial interests exist.
Rights and permissions
About this article

Cite this article
Mei, Z., Zheng, P., Tan, X. et al. Huperzine A alleviates neuroinflammation, oxidative stress and improves cognitive function after repetitive traumatic brain injury. Metab Brain Dis 32, 1861–1869 (2017). https://doi.org/10.1007/s11011-017-0075-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-017-0075-4