
Abstract
Iron dyshomeostasis has been implicated in many diseases, including a number of neurological conditions. Cytosolic NADH cytochrome b5 oxidoreductase (NCB5OR) is ubiquitously expressed in animal tissues and is capable of reducing ferric iron in vitro. We previously reported that global gene ablation of NCB5OR resulted in early-onset diabetes and altered iron homeostasis in mice. To further investigate the specific effects of NCB5OR deficiency on neural tissue without contributions from known phenotypes, we generated a conditional knockout (CKO) mouse that lacks NCB5OR only in the cerebellum and midbrain. Assessment of molecular markers in the cerebellum of CKO mice revealed changes in pathways associated with cellular and mitochondrial iron homeostasis. 59Fe pulse-feeding experiments revealed cerebellum-specific increased or decreased uptake of iron by 7 and 16 weeks of age, respectively. Additionally, we characterized behavioral changes associated with loss of NCB5OR in the cerebellum and midbrain in the context of dietary iron deprivation-evoked generalized iron deficiency. Locomotor activity was reduced and complex motor task execution was altered in CKO mice treated with an iron deficient diet. A sucrose preference test revealed that the reward response was intact in CKO mice, but that iron deficient diet consumption altered sucrose preference in all mice. Detailed gait analysis revealed locomotor changes in CKO mice associated with dysfunctional proprioception and locomotor activation independent of dietary iron deficiency. Finally, we demonstrate that loss of NCB5OR in the cerebellum and midbrain exacerbated harmaline-induced tremor activity. Our findings suggest an essential role for NCB5OR in maintaining both iron homeostasis and the proper functioning of various locomotor pathways in the mouse cerebellum and midbrain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron deficiency is the most prevalent nutrient deficiency worldwide (WHO 2001). Iron plays critical roles in a myriad of cellular pathways and processes, ranging from metabolism to gene transcription. The importance of iron to cellular and systemic processes is evident when considering the wide variety of diseases either directly caused by or associated with iron dyshomeostasis (Ye and Rouault 2010; Fernandez-Real et al. 2015). Maintenance of brain iron homeostasis is relevant to several neurological diseases and the importance of iron in neurocognitive development has been established (Thomas et al. 2009). Alterations in brain-specific or global iron homeostasis have been directly observed or associated with neuropathological diseases like Alzheimer’s disease, Parkinson’s disease, and neurodegeneration with brain iron accumulation, as well as cognitive disorders including autism spectrum disorders, attention deficit hyperactivity disorder, schizophrenia, and many more (Belaidi and Bush 2015; Zucca et al. 2015; Konofal et al. 2007; Insel et al. 2008). A number of studies have evaluated the effects of maternal, neonatal, and adolescent iron deficiency. Whether changes in iron content or homeostasis are a means or an ends has yet to be determined, thus pathways and processes that alter or contribute to iron homeostasis are critical in understanding the underlying etiology and pathology of a number of diseases.
NCB5OR is a highly conserved, ubiquitously expressed oxidoreductase associated with the endoplasmic reticulum whose exact function remains unknown (Zhu et al. 1999, 2004). Naturally occurring polymorphism’s in human NCB5OR have been shown result in increased proteasomal degradation (Kalman et al. 2013). Transgenic mice globally deficient in NCB5OR (GKO) have low birth weight (beginning at 2 weeks of age), petite skeletal structure/build, impaired lipid storage and lipoatrophy, hypermetabolism, mitochondrial dysfunction, and early-onset diabetes from necrotic beta cell loss when compared to normal or wild-type (WT) control mice (Wang et al. 2011; Xie et al. 2004; Larade et al. 2008; Xu et al. 2011). We recently discovered that global loss of NCB5OR results in mild to moderate anemia, significant changes in iron related pathways, and iron deficiency in iron rich tissues such as the spleen and liver (Zhu et al. 2013). Further investigation using a radioactive pulse feeding assay combined with a non-heme iron assay revealed that the brains of GKO mice uptake significantly more circulating 59Fe compared to WT controls, despite showing no apparent change in non-heme iron contentFootnote 1.
Changes in iron homeostasis, mitochondrial dysfunction, and diabetes and their correlation with neurodegenerative diseases (Stroh et al. 2014) led us to ask what specific effects NCB5OR deficiency had on neural tissue. Ataxia with Oculomotor Apraxia (AOA2) and Sideroblastic anemia with ataxia have been shown to be related to changes in iron homeostasis in the cerebellum. Additionally, Friedreich’s ataxia results in mitochondrial iron dyshomeostasis, dorsal root ganglia degeneration, and atrophy of the dentate nuclei without changes in iron content (Solbach et al. 2014). Genes associated with these diseases are ubiquitously expressed; however alleles with mutations or expansions appear to have adverse effects on specific tissue types, namely neural tissue. A microarray study surveying changes in gene expression in Bergmann glia during the postnatal development of the mouse cerebellum revealed NCB5OR to be one of a few that were significantly up-regulated (~120 fold) beginning postnatal day 6 (Koirala and Corfas 2010). This observation coincides with a sequential iron accumulation in chick Bergmann glia beginning postnatal day 2 as well as the localization of transferrin binding protein, ferritin, and iron in Bergmann glia (Kim et al. 2014; Cho et al. 1998). Loss of iron related genes in the cerebellum has been shown to alter locomotion and induce degeneration, ataxia, and developmental issues (Zhao et al. 2015; Capoccia et al. 2015; Maccarinelli et al. 2015; LaVaute et al. 2001). Additionally, cognate motor learning and execution of a complex motor task requires proper nigrostriatal function as well as efficient processing of proprioceptive input by the cerebellum (Salamone 1992). Therefore, we investigated whether loss of NCB5OR in the cerebellum and midbrain would result in locomotor deficits and whether dietary iron deficiency would reveal or exacerbate any deficits.
Materials and methods
Conditional knock out and reporter mice
All mice were of C57BL/6J background and treated according to the University of Kansas Medical Center’s Institutional Animal Care and Use Committee approval protocol. The generation of mice with selective deletion of NCB5OR in the mouse cerebellum and midbrain (conditional knockout (CKO) mice) was achieved by crossing C57BL/6J mice homozygous for a floxed version of NCB5OR’s exon 3 (detailed in Wang et al.)Footnote 2 with En1 tm2(cre)Wrst (The Jackson Laboratory stock no. 007916) mice that are known to excise floxed DNA regions in only the cerebellum and midbrain (Kimmel et al. 2000). To evaluate recombination activity, En1 Cre positive mice were crossed with B6.Cg-Gt(ROSA)26Sor tm14(CAG-tdTomato)Hze/J (stock no. 007914, Jackson Laboratory) which harbor a TdTomato locus with a loxP-flanked STOP cassette that was removed after Cre-mediated recombination, resulting in TdTomato expression. Mice carrying no copies of the Cre gene were considered as normal or wild-type (WT) and all mice used (CKO and WT) were littermates. Evaluation of recombination of NCB5OR was also conducted using qPCR and primer sets targeted to exon 3 and exon 4 of NCB5OR transcripts.
TdTomato reporter and confocal microscopy
TdTomato reporter mice underwent transcardial perfusion with 10 mL of PBS solution immediately followed by 10 mL of 4 % paraformaldehyde. The cerebellum was collected and immediately post-fixed with 4 % paraformaldehyde at room temperature for 1 h. Tissue was then transferred to 20 % sucrose solution and allowed to sit overnight at 4 °C for cryoprotection. Tissues were then embedded in OCT and 12 μm sections were cut, placed on Superfrost Plus slide (Fisher, #12-550-15), and cover slipped. Confocal images of TdTomato were taken using a Nikon Eclipse 90i microscope.
Molecular analysis
Quantitative Reverse Transcription PCR (qRT-PCR) was performed on RNA extracted from WT and CKO whole mouse cerebellum after the mice had been fasted for 4 h. Purity of isolated RNA was assessed by A260/A280 ratio. Reverse transcription of 1 ug total RNA was performed using M-MLV Reverse transcriptase and standard protocol from Life Technologies (Cat # 28025-013). The qPCR reaction was performed on an Applied Biosystems 7900 Fast Real-Time PCR System using SYBR Green master mix from ThermoFisher (Cat. No. 4367659) and all results were normalized to 18S rRNA with the ΔΔCt method. Primer sequences are available upon request. Total protein was isolated from whole mouse cerebellum using a lysis buffer as previously described (Lemaire-Vieille et al. 2013), with the addition of the SigmaFAST protease inhibitor cocktail (s8830). Protein content was determined using a DC assay from Bio-Rad, and total protein was diluted with SDS loading buffer containing beta-mercaptoethanol and boiled briefly at ~95 °C for 5 min. Ninety (90) μg of total protein per sample underwent gel electrophoresis on a mini-protean TGX gel 4–15 % from BioRad (Cat. No. 456-1084). Protein was transferred to a nitrocellulose membrane via cold transfer at 200 mA for 2 h while at 4 °C. Transfer buffer was composed of 25 mM Tris-base (pH 8.8), 192 mM glycine, and 20 % methanol. All antibodies are against mouse antigen: Actin (Sigma, # A1978), APP (Cell Signaling, 2452), ferritin heavy chain/FtH (Santa Cruz Biotechnology, sc-14416), and transferrin receptor 1/TfR1 (Invitrogen, 13-6800). Densitometry was performed using ImageJ software. Percentages represent percentage of signal that is attributed to protein band over the background noise (%Protein or %FtH). Densitometry signal of each protein is normalized against actin (% Actin).
59Fe uptake
Iron uptake in the cerebellum, spleen, and liver was determined by oral 59Fe ingestion. Mice were fasted overnight (14–16 h) but allowed water ad libitum. Each mouse underwent intragastric feeding with a solution containing 10 μCi/mL of 59Fe, 30 μg/mL carrier iron, and 1 M ascorbic acid in phosphate-buffered saline as previously described (Du et al. 2008). Each mouse received 2 μCi 59Fe per 10 g of body weight. One (1) hour post oral gavage, feeding of chow was resumed ad libitum. Approximately 24 h post-gavage the brain, cerebellum, spleen, and liver were collected. Radioactivity was measured (each in a total volume < 1 mL) using a Perkin Elmer 1470 Wizard gamma counter.
Non-heme iron content
Tissue non-heme iron content was measured as previously described (Torrance and Bothwell 1968). Whole cerebellum and spleen from mice were dried at 55 °C for > 48 h, ground into powder, weighed, and then digested in 200 μL or 1 mL of 10 % trichloroacetic acid/10 % HCl at 55 °C for > 48 h, respectively. Samples were then centrifuged at 16,100 rcf for 10 min and 100 μL of the supernatant collected. Ten (10) μL of supernatant was then mixed well in a clear-bottom 96-well plate with 190 μL of chromogen solution, allowed to incubate for 10 min at room temperature, and the absorbance at 535 nm was determined using a Tecan Plate Reader. Chromogen solution consisted of 0.01 % bathophenanthroline-disulfonic acid, 0.1 % thioglycolic acid, and 7 M sodium acetate. Iron concentration was determined by a standard curve dilution from an iron standard (Sigma, St. Louis, MO). All measurements were performed in duplicates.
Histology and iron staining
Sixteen (16) week old male CKO and WT mice were fasted for 4 h and then sacrificed. Whole blood and cerebellum were collected. The cerebellum was immediately placed in 4 % paraformaldehyde and allowed to sit at room temperature for 1–2 h and then placed at 4 °C overnight. The paraformaldehyde was then replaced with 70 % ethanol and the cerebellum was stored at 4 °C until processing. The cerebellum underwent dehydration processing and was embedded in paraffin wax for sectioning. Ten (10) μM sections were taken and placed on Superfrost Plus slide (Fisher, #12-550-15).
Perls’ Prussian blue iron staining with DAB enhancement was performed on sections, as previously described (Smith et al. 1997; Moos and Mollgard 1993; Roschzttardtz et al. 2010; Nguyenlegros et al. 1980). Prior to staining, sections were rehydrated and during the rehydration sequence, a 3 % H2O2 step was added before the final rehydration step for quenching of endogenous peroxidases. Sections were then stained with 3 % HCl / 3 % potassium ferrocyanide / 2 % Triton 100X solution for 30 min and subsequently washed with distilled H2O for 10 min. DAB enhancement was performed for 15 min according to the manufacturer’s protocol (Thermo Fisher cat. No. 34065B). Sections were counterstained with Nissl (cresyl violet). Iron positive and total Purkinje cell counting was performed with a manual counter in real time on a total of 22 sections (10 CKO, 12 WT; 8047 total cells counted). Observer was blinded to genotype during counting.
Dietary treatment
Mice were fed a standard chow, Purina 5053 (LabDiet, Columbia MO), unless otherwise indicated. For dietary iron starvation, pups were given a low-iron diet (TD10210, Teklad Harlan) and allowed to feed ad libitum beginning at weaning (3 weeks of age) and continued for 4 weeks until 7 weeks of age.
Longitudinal behavior assays and gait analysis
Beginning at 3 weeks of age, mice fed either a chow or low-iron diet were screened using a battery of behavioral tests on a weekly basis until 7 weeks of age. The battery of behavioral tests included stationary elevated beam walk, Force-plate Actimetry (BASi), and fore/whole body grip strength. Mice were tested beginning early in the morning (around the beginning of the programmed light cycle of 7 a.m.) and testing was finished prior to noon (12 p.m.) so as to avoid cortisol-associated effects on outcomes. At 7 weeks of age, mice were tested on the Rota-rod and underwent gait analysis using a DigiGait apparatus (Mouse Specifics) prior to euthanasia. Force-plate actimeter measurements were obtained during 10 min recording sessions and DigiGait analysis was conducted at a walking speed of 10 cm/s. Rota-rod data were collected at an accelerating speed of 4 to 40 rpm for a maximum of 5 min.
Sucrose preference test
At 6 weeks of age, mice fed a low-iron or chow diet were individually housed in a two bottle system cage with both bottles containing water. After 1 day of acclimation, one bottle was replaced with a pre-weighed bottle of 4 % sucrose solution and the amount of water in the non-sucrose bottle was measured. The positions of water and sucrose solutions were randomized between cages. After 24 h both the water and sucrose solution weights were recorded and the net consumption for water, sucrose, and total liquid were determined.
Harmaline-induced tremor
At 24 weeks of age, mice were subject to a harmaline-induced tremor assay using subcutaneous injection of harmaline saline solution and assessment of tremor activity on a Force-plate Actimeter, as previously described (Martin et al. 2005). An initial baseline reading of activity for frequencies from 0 to 25 Hz was obtained by allowing mice to explore the Actimeter for 10 min pre-injection. The mice were removed and 4 mL/kg of harmaline stock solution (5 mg/mL harmaline in saline) was injected and the mice were immediately returned to the Actimeter for a 40 min recording session. During this time recordings for signals from 0 to 25 Hz were collected. Subsequently, Fourier transformation of the force-time data facilitated power analysis across frequency and the data was binned into 4 different epochs of 10 min per epoch. The motion power percentage (MPP) for the 10–17 Hz power for each epoch was calculated with the following formula: MPP = [(10–17 Hz) / (0–25 Hz)] × 100. The MPP normalizes the data to give the specific contribution of tremor activity (activity in the 10–17 Hz range) to total motion activity over the 0–25 Hz range, allowing for corrections for intra-animal total movement variation.
Statistical analyses
One (1) way ANOVA was performed for genotype comparison of Western blot quantification and for iron staining quantification of Purkinje cells. Two (2) way ANOVA (sex and genotype) analysis was performed for qPCR, 59Fe uptake, non-heme iron content, and all behavior tests. Three (3) way ANOVA (genotype, sex, and diet) was used to evaluate changes in body weight during the longitudinal studies, sucrose preference test data, and on all metrics from gait analyses. Three (3) way ANOVA (genotype, sex, and age) was used for gait analyses between 7 and 16 week old mice. Student-Newman-Keuls post hoc tests were performed for all ANOVA’s. P-values of <0.05 were considered statistically significant. Values are presented as mean +/- SEM (standard error of mean).
Results
Effective deletion of NCB5OR in the CKO cerebellum
Mice expressing the En-1 driven cre recombinase showed effective recombination of a TdTomato reporter allele in the cerebellum and midbrain (Figure S1). Quantitative PCR (qPCR) analysis of reverse transcribed mRNA with primers targeting exon 4, which is not deleted in CKO tissues, revealed a significantly reduced level of NCB5OR transcripts in the cerebellum of CKO mice relative to WT controls (CKO, 17.6 ± 1.4; WT, 24.9 ± 1.4; p = 0.003, see Table 1 for conditions). In addition, qPCR with primers targeting the floxed exon 3 revealed an absence of functional NCB5OR transcripts in CKO mice relative to WT controls (CKO, 0.33 ± 0.64; WT, 8.33 ± 0.64; p < 0.001).
NCB5OR deficiency results in changes in iron related pathways
To assess whether loss of NCB5OR in the cerebellum facilitated changes in iron related pathways we performed qRT-PCR analysis on mRNA isolated from the cerebellum of CKO mice and WT littermates (Table 1). At 7 weeks of age, CKO mice displayed significantly elevated transcripts for many genes associated with or responsible for proper maintenance of cellular and mitochondrial iron homeostasis. Iron transport and import transcripts transferrin (Tf) and transferrin receptor (TfR1), respectively, were both significantly elevated. Interestingly, transcripts for the only known iron exporter ferroportin (Fpn1) were significantly increased. Transcripts of proteins responsible for iron storage (ferritin heavy chain, FtH, and ferritin light chain, FtL) were also found to be significantly elevated in CKO mice. Additionally, transcripts for amyloid precursor protein (APP) were elevated. APP has been shown to have ferroxidase activity and an iron-responsive element in the 5′ untranslated region of the transcript (Rogers et al. 2002; Duce et al. 2010). Mitochondria-associated iron transcripts frataxin (Fxn) and mitoferrin 2 (MitoFn2) were elevated at 7 weeks as well. By 16 weeks of age, the majority of transcripts returned to levels comparable to WT control mice. However, transcripts for iron regulatory protein 2 (Irp2) and metallothionein 2 (Mt2) were found to be elevated at 16 weeks.
We also performed transcript analysis of cerebellar mRNA at 7 weeks of age after mice had been challenged for 4 weeks with an iron deficient diet for behavioral analysis (Table S1). Transcript levels from male CKO mice showed drastic increases for a number of genes compared to WT controls. Conversely, female CKO mice showed significant decreases in transcripts compared to WT controls and their male counterparts.
Maintenance of iron homeostasis is heavily reliant on both transcriptional and translational regulatory mechanisms. To assess whether the transcript levels accurately reflected the status of TfR1, FtH, and APP protein levels we performed western blot analysis of total protein collected from the cerebellum of male CKO and WT mice at 7 weeks of age (Fig. 1). Levels of TfR1 were significantly elevated (Fig. 1a), while FtH levels were significantly decreased (Fig. 1b). Although CKO mice had elevated APP levels, they did not reach statistical significance when compared to WT (Fig. 1a).

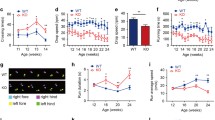
Iron-processing-relevant protein levels in the cerebellum of CKO and WT mice. Specific protein levels were measured using western blot analysis of whole protein extracted from mouse cerebellum at 7 weeks of age. a Amyloid precursor protein (APP) was unchanged in CKO mice, but levels of the iron import protein transferrin receptor (TfR1) were significantly elevated. b Iron storage protein ferritin heavy chain (FtH) was significantly reduced in CKO mice, suggesting an iron-deficient status. Percentages represent percentage of signal that is attributed to protein band over the high background noise. Densitometry signal of each protein is normalized against actin (% Actin). Values are presented as mean +/− SEM (standard error of mean). n = 4:4 (CKO:WT), representative. *, p < 0.05; ***, p < 0.005
NCB5OR ablation alters uptake of circulating 59Fe without changing non-heme iron content
In order to evaluate whether the changes in iron-related pathways were indicative of actual changes in iron uptake we conducted an 59Fe pulse feeding assay on CKO and WT mice at 7 and 16 weeks of age. At 7 weeks of age, CKO mice had significantly greater 59Fe uptake in the cerebellum (Fig. 2a - left), while uptake in control tissues remained unchanged (spleen - Fig. 2b - left, and liver - data not shown). Surprisingly, by 16 weeks CKO mice had significantly lower 59Fe uptake in the cerebellum (Fig. 2a - right) and CKO males exhibited significantly less iron uptake in the spleen (Fig. 2b - right). No recombination in the spleen was seen using PCR (data not shown). There were no changes in liver 59Fe uptake at 16 weeks (data not shown). We tested whether the change in uptake was an indication of altered iron reserves in the form of non-heme iron content; however, measurement of non-heme iron content revealed no changes in the cerebellum (Fig. 2c) or spleen (Fig. 2d) at 7 or 16 weeks of age in CKO mice relative to WT controls.

Altered iron uptake in CKO mice. 59Fe pulse feeding assay revealed (a) significantly elevated 59Fe uptake in the cerebellum of 7 week old CKO mice and significantly decreased uptake by 16 weeks of age. (b) Serving as a control, at 7 weeks the spleen showed no change in iron uptake. Sixteen week male CKO mice acquired significantly less 59Fe. Non-heme iron content assays of the (c) cerebellum and (d) spleen from 7 to 16 week old CKO and WT mice showed no changes in non-heme iron content. Values are presented as mean +/− SEM (standard error of mean). 7 week; n = 10 CKO (5 Male, 5 Female), n = 10 WT (5 Male, 5 Female). 16 week; n = 10 CKO (5 Male, 5 Female), n = 10 WT (5 Male, 5 Female). *, p < 0.05; **, p < 0.01
NCB5OR deficiency results in less ferric iron staining in Purkinje cells
The altered state of 59Fe uptake combined with no observable difference in non-heme iron content at 16 weeks of age in CKO mice led us to question whether there was a shift in the ratio of free to stored iron content. A shift of this nature would result in different amounts of iron in the ferrous state (Fe2+) and stored iron in the ferric state (Fe3+). In order to test this hypothesis we needed a test that would differentiate between the two oxidative states of iron. Therefore, we performed histological analysis of 16 week old male cerebellum sections combined with Perls’ Prussian blue iron staining, which uses ferrocyanide to specifically stain for ferric iron. Perls iron staining in combination with DAB enhancer revealed distinct staining in Purkinje cells of the cerebellum (Fig. 3a). Iron positive and negative Purkinje cell counts revealed that CKO mice possessed a significantly reduced number of iron-positive Purkinje cells when compared to WT counterparts (Fig. 3b). Control sections treated with only DAB (enhancer) and Nissl (counterstain) in the absence of Perls’ iron stain revealed no positive staining, indicating iron-specific staining (data not shown).

Reduced iron-positive Purkinje cells in male CKO mice. (Left) Perls iron staining in the CKO cerebellum with Nissl counterstaining. Note the positive staining (red arrows) and negative staining (black arrow) of Purkinje cells. (Right) CKO mice at 16 weeks of age have a reduced number of iron-positive Purkinje cells as a percentage of total Purkinje cells counted. Values are presented as mean +/− SEM (standard error of mean). Male mouse n = 8 (4 CKO, 4 WT), sections used for quantification n = 22 (10 CKO, 12 WT). *, p < 0.05
Iron deficiency alters execution of complex motor tasks in CKO mice
Based on the changes in iron uptake and related pathways, we hypothesized that loss of NCB5OR in the cerebellum and midbrain would result in locomotor defects. Additionally, we investigated whether generalized iron deficiency brought about by chronic dietary iron deprivation would exacerbate any underlying deficits. It is important to note that gross differences in body weight can contribute to significant changes in certain behavior measures. There were no body weight differences between CKO and WT mice in either the chow or low-iron diet fed groups, but we did note a significant difference in body weight of all mice between the chow and low-iron-diet treated groups during ages 4–7 weeks (Fig. 4a)Footnote 3. All CKO and WT mice were used for behavioral studies below since no gender difference was observed.

Diet-dependent effects of NCB5OR deficiency during a complex motor task. (a) There were no genotypic differences in weight within chow or low-iron diet treated sets, however low-iron diet treated animals weighed significantly less than chow fed controls. CKO mice on a low iron diet challenged with a complex motor task (elevated beam walk) showed (b) increased beam traversal time accompanied by (c) increased slips/errors during traversal. Dietary iron deficiency also resulted in (d) a decreased latency to fall on an accelerating Rota-rod task. Values are presented as mean +/− SEM (standard error of mean). Chow; n = 8:10 (CKO: WT). Low iron; n = 8:8 (CKO:WT). *, p < 0.05; **, p < 0.01; ***, p < 0.005
While there were no apparent differences in performance of the beam walk task between chow-fed CKO and WT mice (Figures S2A and S2B), CKO mice on an iron-deficient diet took significantly longer to traverse an elevated beam at weeks 1,2, and 4 of low-iron-diet treatment (Fig. 4b). CKO mice also committed an increased number of paw slips (Fig. 4c) compared to WT mice at weeks 2 and 3 of low-iron-diet treatment. Additionally, Rota-rod tests revealed that 7 week old CKO mice treated with an iron deficient diet for 4 weeks had a decreased latency to fall compared to WT mice (Fig. 4d), indicating a deficit in coordination. When we compared the dietary effects within genotypes, we observed that the increased number of slips during the coordinated motor task was the result of the genotype differences between CKO and WT mice (Figure S3A and S3B). However, the increased beam traversal time in the low-iron diet group was due to a hyperactive response in WT mice compared to no response in CKO mice (Figures S3C and S3D).
An iron-deficient diet decreases exploratory locomotor activity in CKO mice
The resistance to a low-iron-diet-induced hyperactive response in CKO mice led us to investigate whether CKO mice had differences in locomotor activity when fed a low-iron diet. Force-plate actimeter data revealed no differences between CKO and WT mice on a chow diet (Figures S4A-D), with the exception of a difference in total distance traveled and spatial statistic during the initial 3 week age time point. However, treatment with a low-iron diet resulted in significantly reduced locomotor activity in CKO mice, yielding reduced total distance traveled (Fig. 5a) and area measure (Fig. 5b) compared to WT mice after 1–3 weeks of dietary iron deprivation. Spatial statistics also indicate that the reduced locomotor output correlated with lowered exploratory activity during weeks 1 and 2 of dietary iron deprivation, with CKO mice spending more time in one area, generating a higher spatial statistic (Fig. 5c). It is possible that changes in total distance traveled and area measures are indicative of slower locomotion rather than reduced overall locomotor activity and exploratory behavior, however CKO mice displayed significantly more bouts of low mobility during weeks 1 and 2 of low-iron-diet treatment (Fig. 5d), suggesting that they spent more time being immobile than their WT counterparts.

Diet-dependent effects of NCB5OR deficiency on locomotion. CKO mice on an iron-deficient diet had decreased total locomotor output as indicated by (a) significant decreased in distance traveled and (b) area measure. CKO mice also exhibited reduced exploratory activity resulting in (c) a higher spatial statistic and (d) an increased number of low mobility bouts compared to WT. Spatial statistic scores: 100 = All time spent in one place, 1 = time spent evenly distributed across force plate. Bout of low mobility: 1 bout of low mobility = no change in distance or area (no movement) within a 10 s frame. Values are presented as mean +/− SEM (standard error of mean). n = 8:8 (CKO:WT). *, p < 0.05; **, p < 0.01; ***, p < 0.005
CKO mice display changes in locomotion independent of dietary treatment
To our surprise, hind-paw gait analyses revealed a number of significant changes in 7 week old CKO mouse locomotion that were present in both chow- and low-iron-fed mice and were ultimately unaffected by the dietary iron deficiency. Compared to WT mice, CKO mice had a wider stance width (Fig. 6b), decreased step angle (Fig. 6c), and increased distance to fore limb-hind limb paw overlap (decreased occurrence of superimposed tracks; Fig. 6d). Additionally, CKO mice displayed changes in the rate in which they reach full stance (increased max ∆A/∆T, rate of change of paw area over time; Fig. 6f) as well as an increase in the amount of paw drag (Fig. 6g) from the time of full stance to liftoff.

Diet-independent effects of NCB5OR deficiency on gait. a Posture plots of WT and CKO mice at 7 weeks. DigiGait analysis at 7 weeks revealed that CKO mice had b a wider stance width, c decreased step angle, and d a greater distance to paw overlap compared to WT mice. e A visual representation of change in paw area over time. CKO mice show significant changes indicative of hind limb paresis with increased f max ΔA/ΔT and g paw drag. Max ΔA/ΔT is the rate of change of paw area during paw placement and full stance. Paw drag is the paw area under the curve from full stance to liftoff. CKO mice were quick to load their paw and spent more time initiating the liftoff stage (flopping and dragging their paws). Values are presented as mean +/− SEM (standard error of mean). n = 16:18 (CKO:WT) *, p < 0.05; **, p < 0.01
In order to confirm locomotor changes independent of dietary treatment and assess whether age was in important factor, we performed gait analysis on 16 week old CKO and WT mice fed a chow diet. Comparative gait analysis between 7 and 16 week old CKO mice revealed that CKO mice had significantly increased Max ∆A/∆T (Figure S5A), midline distance (Figure S5B), and distance to paw overlap (Figure S5C) regardless of age. In addition, CKO mice had decreased swing duration (Figure S5D), however this significance was not seen when comparing dietary influence at 7 weeks of age (data not shown).
Reward response remains intact in CKO mice
Upon analysis of locomotor activity and Rota-rod performance, it became apparent that the effects of an iron-deficient diet not only had negative effects on CKO mice, but the effects appeared to be opposite that of WT mice exposed to a low-iron diet when compared to the chow diet. For example, CKO mice on a low-iron diet displayed decreased performance on the Rota-rod task compared to CKO mice on chow, while Rota-rod performance was greater in WT mice on a low-iron diet compared to WT mice on chow. The increased performance of the WT mice under iron-deficient conditions led us to question whether the ventral tegmental area (VTA) was affected in both WT and CKO mice on low-iron diets. Lesions in the tail of the VTA have been reported to result in increased Rota-rod performance in rats (Bourdy et al. 2014) and deletion of the dopamine D2 receptors in the VTA results in increased cocaine-induced locomotion. Moreover, loss of dopamine D2 receptors in the VTA results in increased motivation for sucrose (de Jong et al. 2015).
Therefore, we used a sucrose preference test to determine whether the loss of NCB5OR and the low-iron diet altered behaviors associated with the VTA. In both chow-fed and low-iron- diet treated groups there were no significant differences between 6 week old CKO and WT mice for sucrose preference based on net consumption over a 24 h period (Fig. 7a and b). Interestingly, when comparing all mice treated with either a low-iron or chow diet, mice treated with a low-iron diet showed a significant shift away from sucrose preference compared to those on a chow diet (Fig. 7c). This suggests that dietary iron deficiency alters the reward response such that there is less preference for sucrose without affecting total liquid consumption.

Intact reward response and increased peak harmaline tremor in CKO mice. Sucrose preference testing for CKO and WT mice at 6 weeks of age fed a (a) chow diet and (b) low-iron diet. (c) A reduction in sucrose preference is seen when comparing the effects of a low-iron diet on all mice, suggesting that generalized iron deficiency significantly alters reward behavior in mice. (d) Treatment with subcutaneous harmaline results in rhythmic burst firing in the inferior olive and subsequent activation of rubrospinal and reticulospinal tracts via cerebellar efferents, generating a high frequency tremor. (e) Actimeter signals show a power strike in the 10–17 Hz range, indicative of a high frequency tremor. (f) CKO and WT mice at 24 weeks of age had comparable motion power percentages (MPP) in the 10–17 Hz range during baseline and harmaline tremor onset (10 min post injection), However, by 20 and 30 minuted post injection CKO mice had significantly elevated responses to harmaline induced tremor over WT. Values are presented as mean +/− significantly elevated responses to harmaline induced tremor over WT. Values are presented as mean +/− SEM (standard error of mean). DCN deep cerebellar nuclei, IO inferior olive, PC Purkinje cell, RF reticular formation, RN red nucleus. For figures A–C: Chow; n = 8:10 (CKO:WT). Low iron; n = 8:8 (CKO:WT). For figure F: n = 10:10 (CKO:WT). *, p < 0.05; ***, p < 0.005. MPP = [(10–17 Hz) / (0–25 Hz)] × 100
NCB5OR ablation in the cerebellum exacerbates peak harmaline-induced tremor
Since CKO mice did not show signs of gait asymmetry (data not shown) or overt ataxia like those seen in mice with cerebellar or dorsal root ganglia lesion or degeneration, we hypothesized that pathways responsible for rhythmic, central pattern generation as well as gross limb movement were being affected by loss of NCB5OR in the cerebellum. These include the cerebelloreticular and cerebellorubral tracts (Fig. 7d). In order to test these, we investigated subcutaneous harmaline tremor response, a commonly known pharmacological method of induced essential/cerebellar tremor that utilizes the suspect afferent and efferent pathways. Twenty-four (24) week old CKO and WT mice were placed in an actimeter that allowed for spectral analysis of power for vertical forces between 0-25Hz (Fig. 7e). Motion power percentages (MPP) for initial baseline session readings revealed no differences between CKO and WT mice. MPP’s for the initial onset of tremor activity (10 min post injection) in the 10–17 Hz range did not differ between CKO and WT mice. By 20 min post injection, CKO mice exhibited significantly greater MPP’s than WT mice (Fig. 7f). The significant difference continued through 30 min post injection.
Discussion and summary
Our study aimed to characterize iron-related and behavioral effects due to absence of a reductase, NCB5OR, in the mouse cerebellum and midbrain. To avoid complications of previously described phenotypes and changes associated with global loss of NCB5OR, we generated a cerebellum/midbrain specific conditional knockout (CKO) mouse. Strategic ablation of NCB5OR in the cerebellum and the midbrain allowed us to investigate the specific molecular effects of NCB5OR deficiency on neural tissue as well as evaluate changes in locomotive behavior due to loss of NCB5OR in areas known to affect locomotion. We demonstrated that ablation of NCB5OR in the cerebellum resulted in altered profiles of gene transcripts and proteins critical to iron metabolism and proper maintenance of cellular iron homeostasis. Normally, pathways responsible for iron uptake and export respond in accordance with cellular iron status. During times of iron sufficiency, import and export of iron are regulated such that there is not significant uptake or release of iron, since cytosolic iron stores should compensate for any immediate increase in demand. However, in times of iron deficiency, export is inhibited and uptake is increased, allowing for the accumulation of more iron without significant loss. In our model, loss of NCB5OR resulted in an abnormal response to normal iron bioavailability. At 7 weeks of age, neural tissue devoid of NCB5OR simultaneously acquired and exported available iron, consistent with observed changes in iron uptake (Tf and TfR) and export-related transcripts (Fpn1, APP, CP). At 16 weeks no differences in levels of these transcripts were observed while less iron was acquired by NCB5OR deficient tissue. It is important to note that we did observe elevated IRP2 and MT2 transcripts in the CKO mouse cerebellum at 7 and 16 weeks of age. IRP2 is a regulator of iron-related transcript translation and therefore plays an important role in the abundance of proteins directly responsible for iron homeostatic maintenance (Samaniego et al. 1994; Guo et al. 1994). Levels of IRP2 protein are inversely correlated with iron content and are therefore increased in iron-depleted cells (Iwai et al. 1995). Increased IRP2 transcript levels could indicate a shift from a transcriptional to a protein regulatory response to NCB5OR deficiency. MT2 also plays a role in metal homeostasis by regulating everything from heavy metal scavenging to zinc (Zn) acquisition and signaling. A role for Zn and metallothionein’s in autism spectrum disorders has been of growing interest (Russo 2009; Vela et al. 2015). Faber et al. reported that the Zn/Cu ratio in children with autism spectrum disorder’s was abnormally low, suggesting alterations in metallothionein related pathways (Faber et al. 2009).
Nutritional iron deficiency helped confirm our initial observations and led to interesting observations regarding sexual dimorphism in the iron-deprivation response. Comparison of transcript data from 7 week old iron-diet-treated mice revealed an opposite response between male and female CKO mice, but these trends were not reflected in behavior metrics. The presence of sexual dimorphism in the NCB5OR model has been in a delayed onset of diabetes in female global knockout mice (W.F. Wang and H. Zhu, unpublished data), decreased voluntary exercise in male CKO mice, and altered responses to thirst and refeeding after fasting in CKO mice (unpublished data)Footnote 4.
The stark contrast in transcript and iron uptake response to NCB5OR deficiency at 7 and 16 weeks of age may indicate a pattern of compensatory response and a subsequent compensated state, respectively. That is to say that by 16 weeks the loss of NCB5OR led to a shift in major iron homeostatic pathways as well as pathways dependent on iron. This resulted in the establishment of new homeostatic criteria, negating a need for a compensatory cellular response. This suggests not only an abnormal iron homeostatic response, but also abnormal iron flux, sensing, and pathway crosstalk. We used spleen and liver 59Fe levels as positive and negative controls, respectively, during our uptake experiments due to previous findings showing significantly elevated 59Fe uptake in the spleen and unchanged uptake in the liver of NCB5OR global knockout mice. As expected, at 7 weeks of age we observed no differences in 59Fe uptake in either the spleen or the liver. However, at 16 weeks we were surprised to find significantly decreased uptake in the spleens of male CKO mice while PCR confirmed no loss of NCB5OR in the spleen (data not shown). Direct communication and regulation from brain to the spleen moderated by cholinergic innervation has been identified and may contribute to this observation, and there is significant evidence demonstrating the existence of ‘brain-spleen inflammatory coupling’ (For review see (Rasouli et al. 2011)). Histological analysis of iron staining in the cerebellum revealed further evidence of an altered homeostatic state in 16-week-old CKO mice. The results, along with the observation of no alteration in total non-heme iron content, suggest that CKO mice may possess altered homeostatic criteria that allow for more ferrous (labile) iron. The latter is known to potentiate oxidative stress via the classical Fenton reaction mechanism and is part of ferroptosis, a regulated cell death pathway that has recently been implicated in a variety of diseases (for review, see (Xie et al. 2016)) The potential role of ferroptosis in the pathological changes of NCB5OR-deficient mice is currently under investigation.
Brain iron homeostasis has been the focus of numerous studies, especially those focusing on the effects of neonatal/adolescent iron deficiency. In fact, iron deficiency, dietary or otherwise, has been linked to a number of neurological and psychiatric conditions including restless leg syndrome and attention deficit hyperactivity disorder (Konofal et al. 2004, 2007, 2008; Capoccia et al. 2015; Maccarinelli et al. 2015; Qu et al. 2007; Earley et al. 2005; Connor et al. 2003; Percinel et al. 2015; Provini and Chiaro 2015). An investigation into the effects of iron deficiency and iron deficiency anemia in young Zanzibari children demonstrated lower motor activity scores and less time in locomotion in affected children (Olney et al. 2007). However, there is evidence that early dietary iron deficiency can result in either reduced or increased locomotion (Fiset et al. 2015; Bourque et al. 2008). To date, NCB5OR (or Cyb5R4) gene mutations have not been reported in patients with neurological diseases. However, all previous studies have been centered on diabetic cases (Andersen et al. 2004; Brehm et al. 2012), leaving the possibility that NCB5OR’s contribution to human neurological disease has yet to be investigated and uncovered. Targeted deletion of NCB5OR in the cerebellum and midbrain in the present study did not result in overt changes in locomotion or coordination when mice were fed a chow diet with sufficient iron content. However, NCB5OR deficiency combined with dietary iron deficiency significantly altered locomotion and coordination. Interestingly, when compared to chow fed controls, the effects of dietary iron deficiency appeared to be opposite for CKO and WT mice, with CKO mice becoming hypoactive (or unaffected, as seen in the beam walk time to traverse) and WT mice becoming hyperactive. Given that multiple studies have reported opposing effects of dietary iron deficiency on locomotion, it is interesting to note that ablation of NCB5OR in the cerebellum and midbrain resulted in a resistant or hypoactive response to iron deficiency while WT counterparts responded in a hyperactive manner. Additionally, CKO mice fed an iron-deficient diet displayed decreased coordination and movement execution during complex motor tasks. These changes indicate possible detrimental effects of NCB5OR deficiency on nigrostriatal pathways in the context of dietary iron deficiency. Genetic variations resulting in altered iron content in ventral midbrain nuclei have been linked to changes in locomotor activity (Unger et al. 2008). Also, interactions between iron, dopamine, and neuromelanin-related pathways have been indicated in Parkinson’s disease (Zucca et al. 2015).
Changes in gait and ataxia are commonly associated with dysfunction in the cerebellum or proprioceptive pathways (Miyajima et al. 2001). However CKO mice did not display overt changes in gait symmetry or exhibit progressive ataxia (as is common with cerebellar lesion, degeneration, or altered proprioceptive input) compared to WT controls. Additionally, when compared to WT controls, CKO mice did not display changes in fore limb or whole body grip strength (data not shown), ruling out confounding factors of muscle weakness. It is important to note that while there were no genotype differences in the chow and low-iron diet groups, the low-iron diet did induce a mildly ataxic phenotype in all mice.
The VTA contributes both to reward response and locomotor integration in reward-associated tasks. Since CKO mice exhibited locomotor effects opposite those of WT mice when treated with a low-iron diet, we evaluated whether CKO mice in the low-iron diet treated group harbored changes in reward response compared to WT mice. Results indicate that reward response was intact in CKO mice and was comparable to that of WT mice in both chow and low-iron diet groups. This suggests that the diet-dependent effects of NCB5OR deficiency are likely not originating from the VTA or VTA control over substantia nigral pathways. However, a low-iron diet significantly reduced sucrose preference in all mice, indicating that iron dyshomeostasis or deficiency does alter reward response in mice. Dopaminergic neurons in the VTA are thought to be the largest contributors to VTA mediated reward response. Since we suspect that dopaminergic pathways in the substantia nigra are at the root of the diet dependent effects of NCB5OR ablation, the lack of genotype-diet interaction in VTA-dopaminergic-neuron- mediated behavior might indicate genetic susceptibility of certain dopaminergic neuron populations over others.
We observed a number of changes in CKO mouse locomotion that were independent of and unaffected by dietary iron deficiency, suggesting that regions outside of the nigrostriatal and mesolimbic pathways may be affected. Gait analyses revealed that CKO mice had gait alterations indicating altered proprioception and central pattern-generated locomotion, resulting in hypometric hindlimb movements. In addition, these changes were found to occur in chow fed CKO mice regardless of age, confirming previous observations. However, the absence of ataxia and gait asymmetry in CKO mice suggests that changes may be due to physiological rather than pathological mechanisms. We suspected the origins of diet-independent changes to be of lower, non-cognate motor task-associated pathways. We investigated whether pathways responsible for central pattern-generated locomotion (reticulospinal, rubrospinal, and associated tracts) were being affected by altered cerebellar input by using a harmaline-induced tremor assay. Harmaline treatment results in rhythmic burst firing in the medial accessory olive (MAO) and dorsal accessory olive (DAO) nuclei that excites vermis and paravermis Purkinje cells in the cerebellum. The Purkinje cells project to the deep cerebellar nuclei (fastigial and interposed nuclei). Climbing fibers from the MAO and DAO also directly project to deep cerebellar nuclei. The deep cerebellar nuclei stimulate reticulospinal and rubrospinal fibers which play a role in control of posture, locomotion, and central pattern generation of locomotion (Martin et al. 2005; Mori et al. 1998). At the cellular level, harmaline administration results in increased intraneuronal Ca2+ spikes in oscillating olivary neurons, leading to greater excitability (Martin et al. 2005). The increased response to harmaline tremor in CKO mice could indicate changes in other metal ion (i.e. Ca2+, Zn2+, Na+) homeostatic maintenance pathways in the absence of NCB5OR. Increased serotonin has also been shown to potentiate a greater response to harmaline-induced tremor. These are important considerations when exploring further explanation of the changes seen in CKO mice.
In summary, our study has shown that genetic alterations leading to iron dyshomeostasis can affect a range of movement-related neural pathways, especially when combined with environmental factors such as generalized iron deficiency. These data suggest that NCB5OR deficiency promotes iron dyshomeostasis, but do not prove that NCB5OR directly contributes to iron homeostasis. In addition, our data suggest that NCB5OR deficiency might serve as a model of iron dyshomeostasis resulting from altered iron flux rather than iron deposition or deficiency, per se. The nature of NCB5OR’s contribution to iron pathways and other iron-related genes is currently unknown and further experimentation is needed to address how NCB5OR’s function is necessary for proper maintenance of cellular iron. In all, our current findings demonstrate that NCB5OR-related iron dyshomeostasis, and pathways and processes affected by iron, are of significance to the proper functioning of multiple neural pathways, specifically those contributing to quantitative and qualitative locomotor output.
Notes
Wang WF, et al., NCB5OR deficiency causes anemia, iron dyshomeostasis and increased susceptibility to mitochondrial dysfunction. Manuscript in preparation;
Wang WF, et al., Monogenic NCB5OR diabetes is a result of impaired iron homeostasis and mitochondrial dysfunction in beta-cells. Manuscript in preparation;
Low-Fe is used in lieu of low-iron for figures.
Stroh MA, et al., NCB5OR deficiency in the cerebellum and midbrain affects fasted feeding behavior, thirst response, and voluntary exercise in mice. Publication in process.
Abbreviations
- NCB5OR:
-
NADH Cytochrome b5 Oxidoreductase
- CKO:
-
Conditional knockout
- WT:
-
Wild-type
- MPP:
-
Motion power percentage
- VTA:
-
Ventral tegmental area
References
Andersen G et al (2004) Variation in NCB5OR: studies of relationships to type 2 diabetes, maturity-onset diabetes of the young, and gestational diabetes mellitus. Diabetes 53(11):2992–2997
Belaidi AA, Bush AI (2015) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem. doi:10.1111/jnc.13425
Bourdy R et al (2014) Control of the nigrostriatal dopamine neuron activity and motor function by the tail of the ventral tegmental area. Neuropsychopharmacol 39(12):2788–2798
Bourque SL, Iqbal U, Reynolds JN, Adams MA, Nakatsu K (2008) Perinatal iron deficiency affects locomotor behavior and water maze performance in adult male and female rats. J Nutr 138(5):931–937
Brehm MA, Powers AC, Shultz LD, Greiner DL (2012) Advancing animal models of human type 1 diabetes by engraftment of functional human tissues in immunodeficient mice. Cold Spring Harb Perspect Med 2(5):a007757
Capoccia S et al (2015) Behavioral characterization of mouse models of neuroferritinopathy. PloS one 10(2):e0118990
Cho SS, Shin DH, Lee KH, Hwang DH, Chang KY (1998) Localization of transferrin binding protein in relation to iron, ferritin, and transferrin receptors in the chicken cerebellum. Brain Res 794(1):174–178
Connor JR et al (2003) Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 61(3):304–309
de Jong JW et al (2015) Reducing ventral tegmental dopamine D2 receptor expression selectively boosts incentive motivation. Neuropsychopharmacol 40(9):2085–2095
Du X et al (2008) The serine protease TMPRSS6 is required to sense iron deficiency. Science 320(5879):1088–1092
Duce JA et al (2010) Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142(6):857–867
Earley CJ, Connor JR, Beard JL, Clardy SL, Allen RP (2005) Ferritin levels in the cerebrospinal fluid and restless legs syndrome: effects of different clinical phenotypes. Sleep 28(9):1069–1075
Faber S, Zinn GM, Kern JC, Kingston HMS (2009) The plasma zinc/serum copper ratio as a biomarker in children with autism spectrum disorders. Biomarkers 14(3):171–180
Fernandez-Real JM, McClain D, Manco M (2015) Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care 38(11):2169–2176
Fiset C, Rioux FM, Surette ME, Fiset S (2015) Prenatal iron deficiency in guinea pigs increases locomotor activity but does not influence learning and memory. PloS one 10(7):e0133168
Guo B, Yu Y, Leibold EA (1994) Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J Biol Chem 269(39):24252–24260
Insel BJ, Schaefer CA, McKeague IW, Susser ES, Brown AS (2008) Maternal iron deficiency and the risk of schizophrenia in offspring. Arch Gen Psychiatry 65(10):1136–1144
Iwai K, Klausner RD, Rouault TA (1995) Requirements for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J 14(21):5350–5357
Kalman FS et al (2013) Natural mutations lead to enhanced proteasomal degradation of human Ncb5or, a novel flavoheme reductase. Biochimie 95(7):1403–1410
Kim JY et al (2014) Sequential accumulation of iron in glial cells during chicken cerebellar development. Acta Histochem 116(4):570–576
Kimmel RA et al (2000) Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev 14(11):1377–1389
Koirala S, Corfas G (2010) Identification of novel glial genes by single-cell transcriptional profiling of Bergmann glial cells from mouse cerebellum. PLoS One 5(2), e9198
Konofal E, Lecendreux M, Arnulf I, Mouren MC (2004) Iron deficiency in children with attention-deficit/hyperactivity disorder. Arch Pediatr Adolesc Med 158(12):1113–1115
Konofal E et al (2007) Impact of restless legs syndrome and iron deficiency on attention-deficit/hyperactivity disorder in children. Sleep Med 8(7–8):711–715
Konofal E et al (2008) Effects of iron supplementation on attention deficit hyperactivity disorder in children. Pediatr Neurol 38(1):20–26
Larade K et al (2008) Loss of Ncb5or results in impaired fatty acid desaturation, lipoatrophy, and diabetes. J Biol Chem 283(43):29285–29291
LaVaute T et al (2001) Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet 27(2):209–214
Lemaire-Vieille C et al (2013) Ataxia with cerebellar lesions in mice expressing chimeric PrP-Dpl protein. J Neurosci 33(4):1391–1399
Maccarinelli F et al (2015) A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiol Dis 81:119–133
Martin FC, Le AT, Handforth A (2005) Harmaline-induced tremor as a potential preclinical screening method for essential tremor medications. Mov Disord 20(3):298–305
Miyajima H et al (2001) Cerebellar ataxia associated with heteroallelic ceruloplasmin gene mutation. Neurology 57(12):2205–2210
Moos T, Mollgard K (1993) A sensitive post-DAB enhancement technique for demonstration of iron in the central nervous system. Histochemistry 99(6):471–475
Mori S et al (1998) Cerebellar-induced locomotion: reticulospinal control of spinal rhythm generating mechanism in cats. Ann N Y Acad Sci 860:94–105
Nguyenlegros J, Bizot J, Bolesse M, Pulicani JP (1980) Diaminobenzidine black as a new histochemical-demonstration of exogenous iron. Histochemistry 66(3):239–244
Olney DK et al (2007) Young Zanzibari children with iron deficiency, iron deficiency anemia, stunting, or malaria have lower motor activity scores and spend less time in locomotion. J Nutr 137(12):2756–2762
Percinel I, Yazici KU, Ustundag B (2015) Iron deficiency parameters in children and adolescents with attention-deficit/hyperactivity disorder. Child Psychiatry Hum Dev 47(2):259–269
Provini F, Chiaro G (2015) Neuroimaging in restless legs syndrome. Sleep Med Clin 10(3):215–226
Qu S et al (2007) Locomotion is increased in A11-lesioned mice with iron deprivation: a possible animal model for restless legs syndrome. J Neuropathol Exp Neurol 66(5):383–388
Rasouli J, Lekhraj R, Ozbalik M, Lalezari P, Casper D (2011) Brain-spleen inflammatory coupling: a literature review. Einstein J Biol Med 27(2):74–77
Rogers JT et al (2002) An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem 277(47):45518–45528
Roschzttardtz H, Conejero G, Curie C, Mari S (2010) Straightforward histochemical staining of Fe by the adaptation of an old-school technique: identification of the endodermal vacuole as the site of Fe storage in Arabidopsis embryos. Plant Signal Behav 5(1):56–57
Russo AJ (2009) Anti-metallothionein IgG and levels of metallothionein in autistic children with GI disease. Drug Healthcare Patient Saf 1:1–8
Salamone JD (1992) Complex motor and sensorimotor functions of striatal and accumbens dopamine: involvement in instrumental behavior processes. Psychopharmacology 107(2–3):160–174
Samaniego F, Chin J, Iwai K, Rouault TA, Klausner RD (1994) Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. Structure, function, and post-translational regulation. J Biol Chem 269(49):30904–30910
Smith MA, Harris PLR, Sayre LM, Perry G (1997) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94(18):9866–9868
Solbach K et al (2014) Cerebellar pathology in Friedreich’s ataxia: atrophied dentate nuclei with normal iron content. Neuroimage Clin 6:93–99
Stroh M, Swerdlow RH, Zhu H (2014) Common defects of mitochondria and iron in neurodegeneration and diabetes (MIND): a paradigm worth exploring. Biochem Pharmacol 88(4):573–583
Thomas DG, Grant SL, Aubuchon-Endsley NL (2009) The role of iron in neurocognitive development. Dev Neuropsychol 34(2):196–222
Torrance JD, Bothwell TH (1968) A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci 33(1):9–11
Unger EL, Sterling ME, Jones BC, Beard JL (2008) Genetic variations in ventral midbrain iron influence diurnal locomotor activity and body temperature in BXD mice. FASEB J 22:694.10
Vela G et al (2015) Zinc in gut-brain interaction in autism and neurological disorders. Neural Plast 2015:972791
Wang WF et al (2011) Development of diabetes in lean Ncb5or-null mice is associated with manifestations of endoplasmic reticulum and oxidative stress in beta cells. BBA Mol Basis Dis 1812(11):1532–1541
WHO (2001) Iron deficiency anaemia: assessment, prevention and control
Xie J et al (2004) Absence of a reductase, NCB5OR, causes insulin-deficient diabetes. Proc Natl Acad Sci U S A 101(29):10750–10755
Xie Y et al (2016) Ferroptosis: process and function. Cell Death Differ 23(3):369–379
Xu M et al (2011) Ncb5or deficiency increases fatty acid catabolism and oxidative stress. J Biol Chem 286(13):11141–11154
Ye H, Rouault TA (2010) Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 49(24):4945–56
Zhao L, Hadziahmetovic M, Wang C, Xu X, Song Y, Jinnah HA, Wodzinska J, Iacovelli J, Wolkow N, Krajacic P, Weissberger AC, Connelly J, Spino M, Lee MK, Connor J, Giasson B, Leah Harris Z, Dunaief JL (2015) Cp/Heph mutant mice have iron-induced neurodegeneration diminished by deferiprone. J Neurochem 135(5):958–974
Zhu H, Qiu H, Yoon HW, Huang S, Bunn HF (1999) Identification of a cytochrome b-type NAD(P)H oxidoreductase ubiquitously expressed in human cells. Proc Natl Acad Sci U S A 96(26):14742–14747
Zhu H et al (2004) NCB5OR is a novel soluble NAD(P)H reductase localized in the endoplasmic reticulum. J Biol Chem 279(29):30316–30325
Zhu H, Wang WF, Wang HP (2013) Impaired iron metabolism in monogenic Ncb5or diabetes. Diabetes 62:A566–A566
Zucca FA et al (2015) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog Neurobiol. doi:10.1016/j.pneurobio.2015.09.012
Acknowledgments
Authors thank Dr. John Stanford (University of Kansas Medical Center, KUMC) for providing critiques and comments for the manuscript. Authors acknowledge Dr. WenFang Wang (KUMC) for preparing the NCB5OR-floxed line for crossing and Dr. Alexandra Joyner at Memorial Sloan-Kettering Cancer Center for providing the En1-cre driver. This study was supported by the KUMC School of Health Professions research funds (H.Z.) and a Ruth L. Kirschstein National Research Service Award (T32 HD057850, PI: R. Nudo) supporting M.A.S. Access to the KUMC Rodent Behavior Facility and shared equipment core facilities was provided by the Kansas Intellectual and Developmental Disabilities Research Center (P30 HD02528).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to report.
Rights and permissions
About this article

Cite this article
Stroh, M.A., Winter, M.K., Swerdlow, R.H. et al. Loss of NCB5OR in the cerebellum disturbs iron pathways, potentiates behavioral abnormalities, and exacerbates harmaline-induced tremor in mice. Metab Brain Dis 31, 951–964 (2016). https://doi.org/10.1007/s11011-016-9834-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-016-9834-x