
Abstract
Sevoflurane, one of the most commonly used volatile anesthetics in clinical treatment, has been shown to induce a widespread increase in brain apoptosis. However, the underlying mechanism is still unknown. Sestrin 2 has been recently shown to regulate intracellular reactive oxygen species (ROS) levels and play a crucial role in p53-dependent antioxidant defenses. In this study, our results indicated that administration of Sevoflurane elevated the gene and protein expression of Sestrin-2 in a dose dependent manner in human neuroblastoma M17 cells. It was shown that silence of Sestrin-2 by small RNA interference (siRNA) ominously exacerbated the increase in intracellular ROS and reduction of SOD activity induced by Sevoflurane treatment. Notably, knockdown of Sestrin-2 in M17 cells significantly increases the number of apoptotic cells after treatment with Sevoflurane. Mechanistically, we also found that Sevoflurane treatment resulted in a reduced amount of the cytosolic anti-apoptotic protein Bcl-2 but an increased amount of Bax, which was exacerbated by knockdown of Sestrin-2. In addition, knockdown of Sestrin-2 remarkably increased the elevated cleaved Caspase-3 expression. Finally, we showed that the induction of Sestrin-2 by Sevoflurane was mediated by p53. These results suggest that the suppressive effects of Sestrin-2 on neuroapoptosis against the Sevoflurane anesthesia in neuronal cells might be associated with modulation of mitochondrial pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sevoflurane, one of the most commonly used volatile anesthetics in clinical treatment, has been displayed to impair long-term emotional memory consolidation in human research (Alkire et al. 2008). In vivo studies have also shown that the long time exposure of Sevoflurane could induce neuronal degeneration and cognitive impairment rats by the Endoplasmic reticulum (ER) stress mediated neurons apoptosis in aging (Chen et al. 2013) and mice (Satomoto et al. 2009). Indeed, multiple lines of evidence have shown a widespread increase in brain apoptosis and long-term impairment of neurologic function shortly after exposure to Sevoflurane in small rodents. A recent study showed that Sevoflurane treatment induced apoptosis in mice by increasing the levels of proapoptotic member Bax, reduced the levels of anti-apoptotic member Bcl-2 and cleaved Caspase-3. Notably, in the developing brain, Sevoflurane exposure lead to wide neuroapoptosis by suppressing certain cell survival signaling (e.g. ERK1/2 MAPK signaling) (Wang et al. 2013).
Sestrins constitute a family of evolutionarily conserved stress-inducible proteins. Increasing evidence has shown that Sestrins protect cells from oxidative stress (Lee et al. 2013). Mammalian cells express three isoforms referred to as Sestrin-1, Sestrin-2, and Sestrin-3. Both Sestrin-1 and Sestrin-2 have been recently shown to regulate intracellular peroxide (ROS) levels. Sestrin-2 has been reported to play a crucial role in p53-dependent antioxidant defenses via regeneration of peroxiredoxins from their hyperoxidized forms (Budanov et al. 2004). Sestrin-2 is implicated in the cellular response to oxidative stress and DNA damage. It exerts its cytoprotective function by regenerating peroxiredoxins, having therefore a major role in the antioxidant defense of the cell (Budanov and Karin 2008). In this study, we speculated that Sevoflurane regulates the gene and protein expression of Sestrin-2. We hypothesized that altered expression of Sestrin-2 has a neuro-protective effect against Sevoflurane induced neurotoxicity. In order to verify these hypothesis, we determined the expression of Sestrin-2 in M17 cells after exposure to Sevoflurane. In addition, we investigated the effects of Sestrin-2 on Sevoflurane induced apoptosis by knocking down the expression of Sestrin-2 using silence of Sestrin-2 with small RNA interference (siRNA). p53 has been reported to play a critical role in regulating the expression of Sestrin-2. Therefore, we also examined the potential roles of p53 in regulating Sevoflurane induced expression of Sestrin-2.
Materials and methods
Cell culture
The human neuroblastoma M17 cells were maintained in Opti-MEM medium (Invitrogen, Eugene, OR, USA), supplemented with 5 % fetal bovine serum (FBS) and 1 % penicillin–streptomycin (P/S), in a humid incubator with 5 % CO2 at 37 °C.
Intracellular Reactive Oxygen Species (ROS) determination
Intracellular ROS production was determined by using Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay. Briefly, upon completion of indicated treatment and transfection, cells were gently washed with PBS for three times, followed by loaded with 10 μM DCFH-DA and incubated at 37 °C for 1 h. Fluorescence signals were examined by a microscope and the average fluorescence intensity was used to reflect the level of intracellular ROS.
Determination of Superoxide Dismutase (SOD) activity
Upon completion of indicated treatment and transfection, SOD activity was determined by a commercial SOD Assay Kit (Cayman Chemical) according to manufacturer’s instructions. Protein concentration was measured by the bicinchoninic acid (BCA) assay. Optical density (OD) values were recorded and normalized to protein concentration to reflect SOD activity.
Real-time Polymerase Chain Reaction (PCR)
Upon completion of indicated treatment, total RNA was isolated from M17 cells using Qiazol reagent (Qiagene, USA) according to the manufacturer’s protocol. 2 μg of total isolated RNA was then used as template for reverse transcription PCR to synthesize cDNA. Synthesized cDNA was used for quantitative real-time PCR analysis using the step one plus real time PCR system (Applied Biosystems, USA) with the SYBR Green detection methods. GAPDH was selected as stable reference gene under the experimental conditions of this study. The cycle threshold (Ct) value of each gene was normalized to GAPDH using the comparative Ct method.
Western blot analysis
Protein was isolated from cells using cell lysis buffer (Cell Signaling Technology, USA). Protein concentration was measured by the bicinchoninic acid (BCA) assay. Equal amount of protein was subject to 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto high quality polyvinylidene difluoride membrane (Invitrogen, USA) (Zhou et al. 2008). Membranes were blocked with 10 % non-fat dry milk in TBST buffer. Then membranes were sequentially incubated with primary antibodies overnight at 4 °C and horse radish peroxidase conjugated secondary antibodies for 2 h at room temperature (RT). Blots were developed by the Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA). Band density was measured by the software Image J (NIH, USA).
Morphological analysis of apoptosis
Upon completion of indicated treatment, the nuclear morphology and chromatin condensation in human neuroblastoma M17 cells were investigated by Hoechst staining. Fluorescent dye Hoechst 33258 is a non-toxic, water-soluble double benzene imidazole compounds and can be used to bind to the DNA molecule as a fluorescent probe. Briefly, cells were loaded with 50 μg/ml Hoechst 33258 and incubated for 15 min at RT in darkness (Sheng et al. 2009). The blue fluorescence of nuclear morphology and chromatin condensation was observed by a confocal fluorescence microscope (FV500, Olymplus) equipped with an argon.
Cells are scored as apoptotic if their nuclei present chromatin condensation and marginalization or nuclear beading. Often, apoptotic nuclei fragment into smaller structures. Fluorescence signals were examined by Apoptotic cells showed blue nuclear condensation and fragmentation. The fractions of apoptotic cells were determined relative to total cells. A blind study with random group selection was performed. At least 200 cells were counted in each experiment.
Statistical analysis
All experimental data are shown as mean ± SD. Statistical analysis was performed by using one-way analysis of variance (ANOVA) followed by Dunnett’s significant post-hoc test, and differences between means were considered statistically significant at P < 0.05.
Results
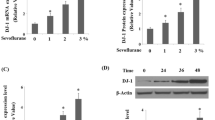
We firstly examined the alteration in levels of Sestrin-2 in response to Sevoflurane treatment. As shown in Fig. 1a, real time PCR results indicated that treatment with Sevoflurane evidently increased the expression of Sestrin-2 at mRNA levels in a dose dependent manner. Consistently, western blot results revealed that administration of Sevoflurane elevated the expression of Sestrin-2 at protein levels (Fig. 1b).

Expression of Sestrin-2 in M17 cells after Sevoflurane treatment. M17 cells were incubated with Sevoflurane at concentrations of 1, 2, 3 % for 24 h. a mRNA levels of Sestrin-2 in M17 cells were measured by real time PCR. b Protein levels of Sevoflurane in M17 cells were measured by western blot analysis (*p < 0.01 vs. non-treated control; n = 3–5)
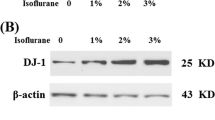
Elevated expression of Sestrin-2 might indicate a contributive role of Sestrin-2 in neurotoxicity of Sevoflurane or act as a compensatory response for cell survival. In order to illuminate the role of Sestrin-2 in Sevoflurane-induced neurotoxicity, we investigated the effects of Sestrin-2 in Sevoflurane neurotoxicity by silencing the expression of Sestrin-2 using Sestrin-2 small RNA interference in human M17 cells. Western blot analysis revealed the successful inhibition of Sestrin-2 at protein levels (Fig. 2).

Knockdown of Sestrin-2 in M17 cells by transfection with small RNA interference. siSestrin-2, Sestrin-2 siRNA group; NS, nonspecific group. Western blot analysis result revealed the successful knockdown of DJ-1 (*p < 0.01 vs. NS group)
Intracellular levels of ROS were determined by the DCFH-DA method. And the results indicated that silence of Sestrin-2 by siRNA ominously exacerbated the increase in intracellular ROS induced by Sevoflurane treatment (Fig. 3a). Furthermore, Sevoflurane treatment significantly decreased SOD activity, which was exacerbated by knockdown of Sestrin-2 (Fig. 3b).

Inhibition of Sestrin-2 by transfection with small RNA interference exacerbates Sevoflurane-induced oxidative stress. a Intracellular ROS in M17 cells were determined by the DCFH-DA assay; Scale bar, 50 μm; b SOD activity was determined by a commercial kit (*p < 0.01 vs. control group; #p < 0.01 vs. Sevoflurane treated group; n = 4)
Cell apoptosis of M17 cells were measured by the Hoechst staining assay. And the results indicated that treatment with Sevoflurane significantly increased the nuclear condensation characteristic of apoptosis in M17 cells. Notably, knockdown of Sestrin-2 in M17 cells significantly increases the number of apoptotic cells after treatment with Sevoflurane (Fig. 4). Mitochondria apoptotic pathway has been associated with the neurotoxicity of Sevoflurane (Zhang et al. 2010). Bax and Bcl-2 are the two important mitochondrial proteins regulating apoptosis. Thus, we speculated that alterations of Bax and Bcl-2 might be involved in the neuroprotective effects of Sestrin-2 on Sevoflurane neurotoxicity. And our results indicated that treatment with Sevoflurane resulted in a reduced amount of the cytosolic anti-apoptotic protein Bcl-2 but an increased amount of Bax, which was exacerbated by knockdown of Sestrin-2 (Fig. 5a). In addition, knockdown of Sestrin-2 remarkably increased the elevated cleaved Caspase-3 expression, which was induced by 3 % Sevoflurane (Fig. 5b).

Silence of Sestrin-2 promotes Sevoflurane-induced apoptosis. siSestrin-2, Sestrin-2 siRNA group; NS, nonspecific group. After indicated treatment, cells were subjected to Hoechst 33258 staining for morphological assessment of apoptosis. 50 μm; Apoptotic cells were recognized by condensed/fragmented nuclei and used for statistical analysis (*p < 0.01 vs. control group; #p < 0.01 vs. nonspecific siRNA-transfected group; n = 4)

Silence of Sestrin-2 regulates mitochondria related apoptotic pathway. siSestrin-2, Sestrin-2 siRNA group; NS, nonspecific group. a The expression of Bax and Bcl-2 as measured by western blot analysis (*p < 0.01 vs. control group; #p < 0.01 vs. Sevoflurane treated group; n = 5); b The levels of cleaved Caspase-3 as measured by western blot analysis (*p < 0.01 vs. control group; #p < 0.01 vs. Sevoflurane treated group; n = 5)
p53 is a tumor suppressor gene, which initiates the process of programmed cell death, or apoptosis. The p53 protein is upregulated in response to a diverse array of cellular stresses. Importantly, it has been shown that Sestrin-2 is a target of the tumor suppressor gene p53 (Budanov and Karin 2008). Indeed, our results indicated that Sevoflurane significantly increased the expression level of p53 (Fig. 6a). To further assess whether p53 is involved in the induction of Sestrin-2 by Sevoflurane, we down-regulated the expression of p53 in M17 cells by using siRNA. Successful knockdown of p53 was shown in Fig. 6b. Importantly, the elevated expression of Sestrin-2 induced by Sevoflurane was abolished by silence of p53 (Fig. 6c).

Sevoflurane-induced elevation of Sestrin-2 is mediated by p53. a. M17 cells were treated with 3 % Sevoflurane for 24 h, the level of p53 was determined by immunoblot and quantification analysis (*p < 0.01 vs. nontreated control; n = 5); b Western blot analysis revealed successful knockdown of p53; c Western blot analysis revealed that Sevoflurane-induced induction of Sestrin-2 could be attenuated by knockdown of p53 (*p < 0.01 vs. nontreatment; #p < 0.01 vs. Sevoflurane treatment; n = 3–5)
Discussion
General anaesthetic exposure to Sevoflurane induced widespread cerebral neuronal apoptosis and subsequent cognitive dysfunction (Jevtovic-Todorovic et al. 2003; Brambrink et al. 2010). It was also showed that developmental neurotoxicity of Sevoflurane is correlated with hippocampal injury (Fanselow 2000). In addition, in vitro studies displayed that neuronal cultures exposed to 3 % Sevoflurane for 6 h induced about 36 % cell apoptosis (Wang et al. 2013). Similarly, it was also demonstrated that administration of Sevoflurane induced significant inhibition of cell proliferation and apoptosis in A549 cells (Liang et al. 2011). Another in vitro study showed that Sevoflurane had an effect of inhibiting growth and promoting apoptosis of colonic and laryngeal cancer cells (Kvolik et al. 2005). However, the mechanisms need to be elucidated. In this study, we found that Sestrin-2 expression is increased after Sevoflurane treatment. In order to elucidate whether Sestrin-2 plays a contributive role in Sevoflurane neurotoxicity or as a compensatory regulator for cellular survival, we examined the effects of Sestrin-2knockdown in M17 cells after Sevoflurane treatment. Our data show that silence of Sestrin-2 exacerbates Sevoflurane-induced oxidative stress and apoptosis. These findings suggest that upregulation of Sestrin-2 expression after Sevoflurane treatment might act as a compensatory response for cell survival. A schematic representation of the underlying mechanism is shown in Fig. 7.

Schematic drawing of the effects of the mechanisms underlying protective action of Sestrin-2 against Sevoflurane neurotoxicity
Sestrin-2, also known as Hi95, is induced by stress signals and plays a vital role in maintaining redox homeostasis (Budanov et al. 2002). Sestrin-2 has been shown to be a target gene of p53 and play a key role in p53-dependent antioxidant defenses through inhibiting the mTOR signaling pathway (Budanov and Karin 2008). Interestingly, Sevoflurane treatment has been reported to induce p53-dependent apoptosis in the Caco-2 cells (Kvolik et al. 2009). In this study, our data demonstrates that p53 mediates the induction of Sestrin-2 in response to Sevoflurane treatment, suggesting the critical roles of p53 in Sevoflurane neurotoxicity. Intrinsic antioxidant defenses are important for neuronal longevity. Notably, Sestrin-2 has been reported to be expressed in neurons and acts as an antioxidant factor in the brain (Papadia et al. 2008).
Both Bcl-2 and Bax are two members of the Bcl-2 family, which regulates the mitochondria pathway related apoptosis. Bcl-2 is an anti-apoptotic protein while Bax is a proapoptotic member. Bcl-2 prevents apoptosis by inhibiting the release of mitochondrial apoptogenic factors into the cytoplasm. In contrast, Bax promotes apoptosis by activating caspases. We observed an up-regulation in Bax and a down-regulation in Bcl-2 in M17 cells after the Sevoflurane exposure, which were exacerbated by silence of Sestrin-2. These results suggest that the suppressive effects of Sestrin-2 on neuroapoptosis against the Sevoflurane anesthesia in neuronal cells might be associated with modulation of mitochondrial pathway.
References
Alkire MT, Gruver R, Miller J, McReynolds JR, Hahn EL et al (2008) Neuroimaging analysis of an anesthetic gas that blocks human emotional memory. Proc Natl Acad Sci U S A 105:1722–1727
Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X et al (2010) Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology 112(4):834–841
Budanov AV, Karin M (2008) p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134:451–460
Budanov AV, Shoshani T, Faerman A et al (2002) Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 21:6017–6031
Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM (2004) Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 304(5670):596–600
Chen G, Gong M, Yan M, Zhang X (2013) Sevoflurane induces endoplasmic reticulum stress mediated apoptosis in hippocampal neurons of aging rats. PLoS One 8(2), e57870
Fanselow MS (2000) Contextual fear, gestalt memories, and the hippocampus. Behav Brain Res 110(1–2):73–81
Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF et al (2003) Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 23(3):876–882
Kvolik S, Glavas-Obrovac L, Bares V, Karner I (2005) Effects of inhalation anesthetics halothane, sevoflurane, and isoflurane on human cell lines. Life Sci 77:2369–2383
Kvolik S, Dobrosevic B, Marczi S, Prlic L, Glavas-Obrovac L (2009) Different apoptosis ratios and gene expressions in two human cell lines after sevoflurane anaesthesia. Acta Anaesthesiol Scand 53(9):1192–1199
Lee JH, Budanov AV, Karin M (2013) Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab 18(6):792–801
Liang H, Gu MN, Yang CX, Wang HB, Wen XJ, Zhou QL (2011) Sevoflurane inhibits proliferation, induces apoptosis, and blocks cell cycle progression of lung carcinoma cells. Asian Pac J Cancer Prev 12(12):3415–3420
Papadia S, Soriano FX, Léveillé F et al (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11(4):476–487
Satomoto M, Satoh Y, Terui K et al (2009) Neonatal exposure to Sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology 110:628–637
Sheng B, Gong K, Niu Y, Liu L, Yan Y, Lu G, Zhang L, Hu M, Zhao N, Zhang X, Tang P, Gong Y (2009) Inhibition of gamma-secretase activity reduces Abeta production, reduces oxidative stress, increases mitochondrial activity and leads to reduced vulnerability to apoptosis: implications for the treatment of Alzheimer’s disease. Free Radic Biol Med 46(10):1362–1375
Wang WY, Yang R, Hu SF, Wang H, Ma ZW, Lu Y (2013) N-stearoyl-L-tyrosine ameliorates sevoflurane induced neuroapoptosis via MEK/ERK1/2 MAPK signaling pathway in the developing brain. Neurosci Lett 541:167–172
Zhang Y, Dong Y, Wu X, Lu Y, Xu Z, Knapp A, Yue Y, Xu T, Xie Z (2010) The mitochondrial pathway of anesthetic isoflurane-induced apoptosis. J Biol Chem 285:4025–4037
Zhou F, Zhang L, Gong K, Lu G, Sheng B, Wang A, Zhao N, Zhang X, Gong Y (2008) LEF-1 activates the transcription of E2F1. Biochem Biophys Res Commun 365(1):149–153
Acknowledgements
Natural Science Foundation of Shandong Province (Studies on Anesthesia Expert Decision Support System, ZR2009CM098).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yi, W., Zhang, Y., Guo, Y. et al. Elevation of Sestrin-2 expression attenuates Sevoflurane induced neurotoxicity. Metab Brain Dis 30, 1161–1166 (2015). https://doi.org/10.1007/s11011-015-9673-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-015-9673-1