
Abstract
Mechanosensitive ion channels are widely distributed in the heart, lung, bladder and other tissues, and plays an important role in exercise-induced cardiovascular function promotion. By reviewing the PubMed databases, the results were summarized using the terms “Exercise/Sport”, “Piezo1”, “Transient receptor potential (TRP)” and “Cardiovascular” as the keywords, 124-related papers screened were sorted and reviewed. The results showed that: (1) Piezo1 and TRP channels play an important role in regulating blood pressure and the development of cardiovascular diseases such as atherosclerosis, myocardial infarction, and cardiac fibrosis; (2) Exercise promotes cardiac health, inhibits the development of pathological heart to heart failure, regulating the changes in the characterization of Piezo1 and TRP channels; (3) Piezo1 activates downstream signaling pathways with very broad pathways, such as AKT/eNOS, NF-κB, p38MAPK and HIPPO-YAP signaling pathways. Piezo1 and Irisin regulate nuclear localization of YAP and are hypothesized to act synergistically to regulate tissue mechanical properties of the cardiovascular system and (4) The cardioprotective effects of exercise through the TRP family are mostly accomplished through Ca2+ and involve many signaling pathways. TRP channels exert their important cardioprotective effects by reducing the TRPC3-Nox2 complex and mediating Irisin-induced Ca2+ influx through TRPV4. It is proposed that exercise stimulates the mechanosensitive cation channel Piezo1 and TRP channels, which exerts cardioprotective effects. The activation of Piezo1 and TRP channels and their downstream targets to exert cardioprotective function by exercise may provide a theoretical basis for the prevention of cardiovascular diseases and the rehabilitation of clinical patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A brief introduction of main mechanosensitive ion channels in mammalian
Cells convert mechanical stimuli into electronic and chemical signals through mechanically activated ion channels [1]. Detailed studies of various known mechanosensitive ion channels and active discovery of unknown mechanosensitive ion channels are important for discussing mechanical-signaling mechanisms. Mechanically sensitive ion channels in mammals are mainly tandem-pore-domain potassium channels (K2P), the Piezo family, the transient receptor potential (TRP) channel family, the epithelial Na+ channel/degenerin (ENaC/DEG) superfamily and the acid-sensing ion channels (ASIC) [1]. The K2P family consists of three mechanosensitive ion channels: TREK-1, TREK-2, and TRAAK. Mechanical forces can activate trough currents and stretch currents [2]. K2P is expressed in sensory neurons, promotes cell hyperpolarization and reduces the conductance of non-selective cationic mechanical sensors [3]. Piezo family are very conservative compared to other known channels, Piezo are widely involved in various mechanical conduction processes and in regulating blood pressure and disease. Chemicals, lipid signaling molecules, and physical stimuli can activate TRP channels, causing them to involved in thermal, chemical, mechanical, and permeative processes [4]. TRPV4 is involved in mechanical transduction processes and in the regulation of osmolality, vascular tone, and pain perception [5]. The DEG family was first identified in the mutants MEC-4 and MEC-10 of the Caenorhabditis elegans. In mammals, ENaC and the ASIC channel family share homology with MEC-4 and MEC-10 channels in Caenorhabditis elegans, sensing pain and blood pressure changes [6, 7]. ENaC channels formed by α-, β-, and γ-subunits are typical mechanical sensors in vertebrates with roles in regulating blood pressure and electrolyte/fluid homeostasis [8]. There may be mechanosensitive ion channels that have not yet been identified, and the molecular mechanisms of the mechanical-sensing process need to be further investigated.
The regulation of Piezo1 in cardiovascular function and pathological responses
The role of Piezo1 in regulating the blood pressure
Endothelial cells are continuously undergoing various forms of mechanical stimuli, such as changes in the flow of blood vessels due to changes in blood pressure, causing endothelial cells to sense mechanical forces. Piezo1 is an important mechanosensitive cation channel that occurs in large numbers in vascular endothelial cells and lymphatic vessels. Piezo1 in mice senses mechanical stimuli and generates mechanically activated cationic currents [9, 10]. Endothelial production of nitric oxide (NO) regulates vasoconstriction and maintains blood pressure homeostasis. Mechanical stimuli generated by blood flow activate Piezo1 to induce endothelial NO formation and increase Ca2+ concentration. Piezo1 activates the endothelial nitric oxide synthase (eNOS)/AKT signaling pathway with the assistance of calmodulin, which induces the production of NO and release of ATP, resulting in vasodilation and a reduction in systolic blood pressure [11]. If Piezo1 is absent from endothelial cells, endothelium-dependent responses induced by blood flow shear forces are impaired, eNOS activity is reduced, and vasodilation is impaired, ultimately leading to arterial hypertension. Piezo2 and Piezo1 coactivate pressure receptors in the carotid arteries and aorta to regulate acute blood pressure changes [12]. Double deletion of the Piezo 1/2 gene inhibits pressure receptor reflexes. This leads to a nonbenign increase in blood pressure [13].
Piezo1 is involved in the remodeling of blood vessels in atherosclerosis
Regular beating of the heart results in increased blood flow and mechanical pressure stimulation of vascular endothelial cells and vascular smooth muscle cells. Mechanical stress is determined by vascular pressure. Circulation is the main source of blood flow shear. Shear affects the structure and function of the vascular intima and is involved in the development of atherosclerosis [14]. Piezo1 is an important mechanosensitive cation channel that regulates angiogenesis, development, and regeneration [15,16,17]. The activation of Piezo1 by mechanical stimulation increases Ca2+ inflow, which activates calpsin-2 (CAPN2), matrix metalloproteinase-2 (MMP2) and membrane type 1 matrix metalloproteinase (MT1-MMP), resulting in corresponding biological functions, including endothelial cell alignment and migration, vascular development, maturation, regeneration and remodeling18. Piezo1 contributes to early vascular development [18]. Endothelium-specific knockout of Piezo1 impairs vascular maturation and leads to embryonic death [19], suggesting that Piezo1 is essential for early vascular development. In endothelial cells, blood flow activates Piezo1. This leads to the phosphorylation of AKT and eNOS through P2Y2/Gq/G11 signaling, accompanied by the release of ATP [11]. Both laminar and turbulent flow can generate mechanical stress and activate Piezo1 [20]. Piezo1 activation by laminar flow mediates eNOS activation and NO release, which can ameliorate atherosclerosis. Conversely, Piezo1 activation by turbulence increases endothelial inflammatory responses and promotes atherosclerosis [21] (Fig. 1).

Different blood flow characteristics affect Piezo1 regulation of cardiovascular function and pathological changes
Electrophysiologic and ionic changes during ischemia
Ischemia causes cellular depolarization, resulting in an increase in extracellular potassium concentration [K+]o, ATP-sensitive inward-rectifying potassium current (IKATP) peak and acetylcholine-activated potassium current (IKACh), as well as a decrease in action potential duration, thereby accelerating the repolarization process. After ischemia, there was a slight increase in cellular excitability. Within 2–3 min, the negative cellular resting voltage decreased from approximately − 85 to − 60 mV, resulting in the inhibition of Na+ channel activity and the action potential (AP) conduction, which decreased the conduction velocity to approximately 0.03 cm/ms and emergence of slower inactivating sodium current, leading to a decreased cellular excitability and prolonged effective refractory period [22]. Elevated [K+]o leads to a lengthening of the post-repolarization refractoriness. This effect is most pronounced when the IKATP reaches its peak. Additionally, both the maximum rate of rise of the AP and the maximum transmembrane voltage are reduced [23]. The ventricular activation time is prolonged from 80–100 to 200–300 ms [22].
Ischemia shifts cellular metabolism towards anaerobic conditions, increases NADH, promotes Na+/Ca2+ Exchangers (NCX) activities, inhibits sarcoplasmic reticulum Ca-ATPase (SERCA) activities, decreases calcium transient (CaT), increases diastolic intracellular calcium concentration [Ca2+]i and disrupts [Ca2+]i homeostasis [24, 25]. The use of ORM-10103, a specific inhibitor of NCX, was found to reduce [Ca2+]i, alleviate the negative effects of ischemia/reperfusion on cells, and ameliorate the reduction in AP and CaT after reperfusion, but still caused arrhythmias [25].
The electrocardiogram pattern of ischemia is characterized by reduced R-wave amplitude, ST-segment elevation, T-wave inversion, prominent Q-wave, shortened RR interval, prolonged QT interval, etc. [26]. Additionally, cardiac output, stroke volume, fractional shortening, and ejection fraction are all impaired, and there is a delay in ventricular depolarization and repolarization, which disturbs the activity of the autonomic nervous system and leads to arrhythmia [26].
Arrhythmias are primarily caused by two types of activity: early after depolarizations (EAD) and delayed after depolarizations (DAD), which are related to calcium overload and catecholamine secretion. EAD occur in and out of the sinoatrial node through activation of ion channels such as transient outward potassium current. Ventricular tachycardia typically occurs within 2–10 min after ischemia, while ventricular fibrillation usually occurs within 20–30 min [22]. Studies of electrophysiological and ionic changes during cardiac ischemia have contributed to the important role of mechanosensitive ion channels in the cardiovascular system.
The role of Piezo1 in ischemic heart injury
Ischemic heart injury initiates with changes in the mechanical environment of the heart. Piezo1 is expressed in the heart and is involved in mechanical transduction processes [27, 28]. Piezo1 mediates pathological cardiac hypertrophy by regulating the expression levels of calmodulin and calpain through a positive feedback mechanism [27]. Piezo1 is upregulated in the ischemic heart and regulates Ca2+ production and reactive oxygen species (ROS) signaling in the cardiomyocyte through a positive feedback mechanism. This allows the heart to adapt to pathological mechanical load [29]. Piezo1 mediates pathological cardiac hypertrophy by regulating the expression levels of calmodulin and calpain through a positive feedback mechanism. Cardiac Piezo1 expression increases after myocardial infarction, associated with ventricular remodeling and the development of myocardial hypertrophy. Inhibition of Piezo1 expression improves cardiac function and alleviates myocardial hypertrophy. Ischemic injury-induced myocardial fibrosis is triggered by an increase in extracellular matrix (ECM) stiffness [30]. Piezo1, in conjunction with integrin β1, acts as an initial signaling molecule in the activation of cardiac fibroblasts. Piezo1 and integrin β1 form a mutually reinforcing positive feedback loop in which increased ECM hardness promotes expression of both. As the expression levels of Piezo1 and integrin β1 increased, the stiffness of the ECM and the feedback strength of the loops increased, further accelerating the process of cardiac fibrosis [30]. Piezo1 in cardiac fibroblasts activates the p38MAPK signaling pathway, linking Ca2+ entry to interleukin-6 (IL-6) secretion, promoting cardiac hypertrophy and remodeling [31] (Table 1).
The role of TRP channels in regulating the cardiovascular function and pathological processes
TRP channels play a role in the development of cardiovascular disease and regulate cardiovascular function.
TRPP1 channels sense blood flow in the vasculature, and blood flow stimulates TRPP1 in endothelial cells, activating small conductance and intermediate Ca2+-activated potassium channels (SK/IK), as well as activating eNOS, resulting in vasodilation and reduced blood pressure [39]. TRPP1/2 is expressed on the endothelial membrane of cardiomyocytes, and cardiac-specific knockout of TRPP1 resulted in abnormally increased blood pressure in mice [40]. In the heart, simultaneous expression of both forms large conductance non-selective cation channels. TRPP2-deficient mice are embryonic lethal and exhibit cardiac septal defects [41]. Mutations in TRPP2 lead to autosomal dominant polycystic kidney disease (ADPKD), and patients with ADPKD suffer from cardiac dysplasia manifested by septal morphology or mitral valve prolapse, suggesting a certain role for TRPP1/2 in cardiac development [42].
During cardiac remodeling in ischemic cardiomyopathy, the Ca2+ response is dramatically altered, and some mechanosensitive cation channels are acting to mediate the response. TRPC3 channels are overexpressed and mediate Ca2+ influx into cardiomyocytes in hypertrophied hearts [43]. Angiotensin II (Ang II) induces cardiac hypertrophy and activates TRPC1/3/4/6 [44]. Dual inhibition of TRPC1 and TRPC4 expression ameliorates cardiac hypertrophy [45]. Inhibition of TRPC3/6 expression in myocardial infarction or transverse aortic constriction (TAC)-induced cardiac hypertrophy results in reduced myocardial infarction, decreased levels of cardiac fibrosis, and improved cardiac function [46]. TRPC3 interacts with nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (Nox2) to promote ROS generation and increase levels of oxidative stress in the heart. Inhibition of TRPC3 reduces Nox2 expression and ROS production, thereby improving cardiac function [47]. Studies have shown that cardiac TRPC6 is activated by stretch, which increases myocardial Ca2+ concentration and endothelin-1 expression, leading to the onset and development of arrhythmias [48]. In mice with MLP (-/-)-induced dilated cardiomyopathy, TRPC3 activates Ca2+/calmodulin-dependent kinase II (CaMKII). Inhibition of TRPC3 attenuates this process and reduces Rac1-mediated increase in ROS production, ameliorating cardiomyopathy [49]. Inhibition of TRPC3 can also reduce apoptosis in endothelial cells and has atherosclerosis-protective effects. TRPC channels are involved in regulating pathological processes such as angiogenesis, arterial atherosclerosis, inflammatory responses, and hypertension [50]. TRPC1 is overexpressed in patients who have undergone coronary artery bypass graft surgery and in pigs with coronary artery injury, and the over-proliferation of smooth muscle cells promotes Angiogenesis [51]. Hypertension is associated with changes in TRPC channel activity. In hypertensive patients, there is an increase in vascular expression of TRPC1/3/5, which disrupts Ca2+ homeostasis [52,53,54]. Additionally, TRPC6 knockout mice exhibit a hypertensive phenotype, and it is hypothesized that TRPC3 negatively regulates TRPC6 expression through a feedback mechanism [50]. In myocardial hypertrophic and spontaneously hypertensive rats, the expression of TRPM4 is increased, causing a dramatic increase in Ca2+ release from atrial cardiomyocytes and an imbalance in Ca2+ homeostasis, followed by delayed depolarization and changes in ECG signals, leading to myocardial hypertrophy-associated arrhythmias [55].
TRPV channels are critical in the regulation of cardiac structure and function.TRPV1 is expressed in cardiomyocytes, vascular endothelial cells, and smooth muscle cells and is involved in the regulation of calcium in cardiomyocyte differentiation during early heart development [56]. The expression of TRPV1 is increased in TAC-induced myocardial hypertrophy, leading to elevated levels of cardiac inflammatory markers TNFα and IL-6, and activation of the NF-κB signaling pathway, which promotes cardiac hypertrophy [57]. Cardiac fibrosis is related to TRPV1/2/3, and TRPV1 deletion leads to an increase in cardiac collagen fibers and increased expression levels of the fibrosis-related genes TGFβ1, Col1a1, and Col3a1 [58]. TRPV2 and TRPV3 are ion channels that play important roles in the heart. TRPV2 converts mechanical signals into electrochemical signals that perform biological functions in response to mechanical stimuli [59]. Knockout of TRPV2 in cardiomyocytes results in embryonic death. TRPV3 increases intracellular Ca2+ concentration, activates CaMKII, Calcineurin (CaN), and NFATc3, and is involved in the development of pathological cardiac hypertrophy [60]. In cardiac hypertrophy induced by abdominal aorta coarctation (AAC), rather than physiological cardiac hypertrophy induced by regular exercise, the expression of TRPV3 and hypertrophic markers brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC) has increased [61]. In an in vitro model of fibrosis induced by Ang II, inhibition of TRPV3 resulted in a downregulation of Collagen I and Collagen III, as well as the expression of cell cycle protein E and cell cycle protein-dependent kinase 2 (CDK2) [62]. Similarly, TRPV4 activates eNOS in the same way as TRPP1 to regulate vascular tone [63]. Knockout of TRPV4 results in loss of vasodilator capacity by impairing the endothelium-dependent hyperpolarization (EDH) process [64] (Fig. 2).

TRP channel regulate cardiovascular function and pathological processes
Exercise plays an important physiological function in cardiovascular by regulating Piezo1 and TRP channels
The cardiovascular benefits of regular moderate exercise are well-documented, regardless of the type of exercise [65]. However, overtraining can lead to a decrease in cardiovascular function. The exercise done by competitive athletes causes serious cardiac remodeling leading to increase arrhythmia development. Dogs trained to exercise at a high level for extended periods of time are at a greater risk of developing arrhythmias compared to dogs that are sedentary [66]. They had prolonged cardiac repolarization times, increased repolarization instability, and slight ventricular fibrosis compared to sedentary dogs [66]. Competitive athletes commonly develop various heart rate arrhythmias such as sinus bradycardia, premature beats, and ventricular tachycardia, called “athlete’s heart” [67]. A study found that after inducing physiologic cardiac hypertrophy through 12 weeks of swimming training, rats showed increased cardiac output, decreased long-term QT variability, and increased abnormal ventricular beats, suggesting arrhythmic events. This may be due to increased sarcoplasmic reticulum Ca2+ content disrupting cardiac calcium homeostasis [68]. Therefore, excessive exercise may be detrimental to the improvement of cardiovascular function and the rehabilitation of clinical patients. The amount, duration, and mode of exercise should be properly adjusted accordingly for different cardiovascular risk factors. However, there is currently no personalized exercise prescription available and there is an urgent need for an in-depth study of the relevant molecular mechanisms. We have summarized the role of different exercise modes in regulating the physiological functions of Piezo1 and TRP in Table 2. Hypertrophic cardiomyopathy is a significant factor affecting the careers of athletes, and further research is needed to develop exercise training programs for competitive athletes and to consider how to train for positive cardiac remodeling. Additionally, accurate risk assessment and the development of individualized exercise plans for sedentary individuals with hypertrophic cardiomyopathy is an issue that needs to be further examined [69]. Research into the mechanisms of exercise cardio-protection provides a molecular basis for the selection of appropriate exercise programs for different groups.
Exercise activates vascular endothelial cell Piezo1 to exert its physiological function
Exercise has been shown to be beneficial for cardiovascular disease. Piezo1 acts as an “exercise sensor” that regulates cardiovascular homeostasis by improving vasoconstriction and diastolic function, improving blood redistribution in the body during exercise, and increasing physical fitness in adults [70]. During whole body physical activity, Piezo1 links communication between endothelial cells and mesenteric vascular smooth muscle cells, inducing vasoconstriction, inhibiting EDH, and increasing blood pressure. Endothelium-specific depletion of Piezo1 inhibits the increase in blood pressure during physical activity and reduces exercise capacity [19, 70]. Exercise increases blood flow, thereby increasing blood flow shear, activating Piezo1 and paracrine signaling of eNOS/platelet reactive protein-2 (TSP2), and maintaining capillary density and endothelial microcirculatory stability. These findings suggest a link between Piezo1 and muscle work capacity and physical performance. The study found that endothelial-specific knockdown of Piezo1 inhibits the eNOS/TSP2 signaling pathway, leading to increased endothelial cell apoptosis in muscle micro vessels, reduced capillaries, and decreased physical capacity [71].
Exercise modulates cardiac Piezo1 activity and its physiological function
Exercise can increase the rate of myocardial contraction and stroke volume, and stretch and shear stress can strongly stimulate myocardial and endothelial tissue. At the molecular level of the cardiac system, Piezo1 senses pressure and stretch stimuli, generating appropriate mechanical signals that regulate cell contractility and other physiological properties [72]. Piezo1 responds differentially to varying degrees of mechanical stimulation, and appropriate mechanical forces activate Piezo1 to upregulate the expression of SERCA2 and eNOS, thereby increasing cardiac contractility [72, 73]. Exercise leads to increased cardiac activity, and the mechanosensory system senses cardiac activity and participates in cardiac mechanical transduction activities that regulate changes in cellular function. At the molecular level of the cardiac system, Piezo1 senses pressure and stretch stimuli, generating appropriate mechanical signals that regulate cell contractility and other physiological properties [74]. A study demonstrated that Piezo1 is expressed in myotubes. In cultured mouse myotubes, Yoda1 chemically activates Piezo1 channels to stimulate IL-6 expression. Inhibition of Piezo1 reduced IL-6 secretion. It is believed that Piezo1 activation is responsible for the increased secretion of muscle factors during myocardial diastole during exercise [75].
Exercise regulates TRP channels expression and its physiological function
Cardiorespiratory responses are regulated by exercise muscle afferent feedback during exercise. Studies have shown that TRPV1 and COX-2 expression is upregulated and exercise muscle afferent feedback is enhanced during 5 min of cycling exercise at 65% peak workload in heart failure patients with reduced ejection fraction (HFrEF) [76]. Additionally, TRPV2 plays a role in regulating the cardiac stress response, maintaining cardiac structural integrity, and regulating cardiac Ca2+ flow in both healthy and pathological conditions. TRPV2-deficient mice lose the ability to perform aerobic exercise, and voluntary wheel running activities impairs cardiac function by reducing cardiomyocyte volume, stroke volume and ejection fraction compared to wild mice [77]. Furthermore, the cardioprotective effects of exercise were nullified in knockout mice, and the heart’s adaptive response to exercise stimuli was diminished [78]. TRPC3 activates downstream signaling molecules such as Ca2+-activated protein kinase C (PKC) and regulates Nox2 activity in the heart in response to mechanical stimuli. Knockdown of TRPC3 inhibits Adriamycin-induced increase in Nox2, reduces ROS production, and protects cardiac function. Physical exercise reduced TRPC3 and Nox2 in mouse hearts and similarly improved cardiac function [47]. Exercise is widely recognized as a preventative measure and treatment for diet-related diseases, reducing the risk of illness. In mice fed a high-fat diet, Voluntary wheel running exercise was found to reduce levels of TRPA1 and AKAP150. AKAP150 was found to regulate the membrane movement of TRPV1, and TRPA1 was found to co-localize with and act through TRPV1. These findings suggest that membrane movement of TRP channels may be involved in the mechanism by which exercise benefits high-fat-fed mice [79]. A link between exercise-stimulated mechanosensitive ion channels and motility factors has been found. In endothelial cells, activation of TRPV4 increases intracellular calcium concentration, resulting in vasodilation. TRPV4 mediates Irisin-induced Ca2+ influx and endothelium-dependent vasodilation, which protects vascular function [80] (Table 2, Fig. 3).

Exercise modulates Piezo1 and TRP channels to exert cardioprotective effects in normal and pathological hearts
Advances in mechanism research of exercise modulation of Piezo1 and TRP channels to improve cardiovascular function
Exercise and Piezo1 downstream signaling pathway mechanisms
Piezo1 plays a wide range of roles in cardiovascular biology, where it generates clear connections with multiple signaling pathways involved in the regulation of cardiovascular activity.
AKT/eNOS signaling pathway
Piezo1 activates eNOS at Ser1177 and Ser635, resulting in increased NO production and leading to vasodilation. At Ser635, shear-activated Piezo1 stimulates adrenomedullin to bind to Gαs-coupled receptors, which activates protein kinase A (PKA) and subsequently eNOS via the second messenger cAMP [87]; At Ser1177, mechanical stimulation activates Piezo1 to couple with P2Y2 receptors via Gq/G11 on the cell membrane. This increases levels of the endothelial cell adhesion proteins PECAM-1 and VE-cadherin, activates the AKT signaling pathway, and leads to NO production through eNOS phosphorylation. Piezo1 is activated by shear in blood flow, leading to an increase in Ca2+ influx and ATP release via pannexin [11]. This process has been confirmed by studies conducted by Cinar [88]. The sudden increase in blood flow causes an increase in cellular Ca2+ concentration, which activates calmodulin and eNOS, resulting in vasodilation and constriction [89].
NF-κB signaling pathway
Activation of Piezo1 by flow shear in laminar and turbulent flow has different effects on vascular physiology. Laminar flow stimulates endothelial cells to align and grow in the direction of blood flow and exert biological functions through the eNOS signaling pathway [90]. In contrast, turbulent flow generates oscillatory shear, leading to the production of atherosclerosis-associated endothelial inflammatory signaling molecules and activation of the NF-κB signaling pathway [91]. Recent studies have demonstrated the distinction between these two biological processes [32]. Turbulence-generated blood flow shear activates Piezo1, which promotes Ca2+ release. It binds to the Gq/G11-coupled P2Y2 receptor and activates the focal adhesion kinase (FAK) and NF-κB signaling pathways in the presence of PI-3 kinase (PI3K), thereby promoting the development of atherosclerosis. Additionally, integrin activation triggers phosphodiesterase 4D (PDE4D), which exerts an inhibitory effect on cAMP, leading to a reduction in cAMP-dependent kinase activation by eNOS and inhibition of vasodilation [92].
p38MAPK signaling pathway
Fibroblast-secreted IL-6 promotes cardiac hypertrophy and plays a crucial role in damaged cardiac tissues. The p38MAPK signaling pathway is activated in the pathological heart and is involved in cardiac signaling [93]. Activation of Piezo1 in cardiac fibroblasts, through pharmacological agonists or stretch stimulation, promotes an increase in Ca2+ influx. This leads to downstream activation of p38MAPK, which results in increased expression and secretion of IL-6 [31]. The cascade of Piezo1 and p38MAPK pathways provides new ideas for elucidating the interaction between signaling molecules involved in cardiac fibroblast differentiation and cardiac remodeling.
Piezo1 and integrins
The interaction of Piezo1 and integrins play an important role in the regulation of cardiovascular function: On the one hand, Piezo1 activation regulates integrin expression, and turbulence-generated blood flow shear activates Piezo1 to upregulate integrin α5 expression, with atherogenic phenotypes; On the other hand, integrins modulate the response to mechanical force stimulation of Piezo1, and different shear force stimulus intensities activate signaling pathways with different Piezo1/integrin interactions. High shear stress stimulates Piezo1 to regulate the interaction of fibronectin with integrin α5β1, and Piezo1 promotes increased Ca2+ influx, activation of calpain causes protein tyrosine phosphatase 1B (PTP1B) to bind to the integrin α5 subunit after dephosphorylating annexin A2 (ANXA2) at Y24, thereby activating integrin α5β1; Low shear stress stimulates the involvement of Piezo1 in regulating the interaction between collagen type IV and integrin αvβ3 through PI3K [94]. The process of cardiac fibrosis is associated with activation and phenotypic transformation of cardiac fibroblasts, and the conversion of cardiac fibroblasts into myofibroblasts has a pro-fibrotic effect. In cardiac fibroblasts, Piezo1 and integrins are involved in regulating the phenotypic transformation of cardiac fibroblasts through a mutually reinforcing positive feedback loop, which provides a new target for the treatment of myocardial fibrosis [30].
Exercise protects cardiac function through the HIPPO-YAP signaling pathway
The Hippo/YAP signaling pathway consists of three main components: upstream signaling inputs, core kinase cascades and downstream transcription factors. YAP/TAZ, as an essential component of the core kinase cascade, is important for the regulation of downstream transcription factors [95], involved in the regulation of cell proliferation and cell growth and development. Inhibition of YAP/TAZ expression reduces endothelial inflammation and delays atherogenesis and progression of atherosclerosis, which is closely related to atherogenic inflammation [95]. The regulatory effect of Piezo1 on YAP has been validated in cardiac fibroblasts [96], endothelial cells [97], cancer cells [98], osteoblasts [99], and human neural stem cells [100] in validated in a variety of tissues. In drug-induced endothelial cell inflammatory response, the expression of both Piezo1 and YAP/TAZ was found to be significantly increased. Activated Piezo1 increased YAP nuclear translocation, and inhibition of Piezo1 expression inhibited the endothelial cell inflammatory response and reduced or even eliminated YAP expression [97], suggesting that Piezo1 has the function of regulating the nuclear localization of YAP. A similar regulatory relationship between Piezo1 and YAP was shown in Liu et al. [101]. Piezo1 mediates YAP/TAZ activation and nuclear localization in cascade with the JNK signaling pathway and the MAPK classical signaling pathway to increase endothelial inflammation and contribute to the process of atherosclerosis development [102, 103]. Piezo1 is involved in the regulation of organismal valve generation and development, and plays an important role in cell differentiation during the embryonic period [104], and Piezo1 may act as a regulator of tissue mechanical properties in the zebrafish heart valve outflow tracts and flowing vascular system by regulating YAP expression and localization [105, 106]. Piezo1 regulation of YAP nuclear localization acts as a modulator of the macrophage inflammatory response [107], ultimately promoting angiogenesis and producing osteogenesis [108].
After myocardial infarction, a large number of cardiomyocytes die, and the dead cardiomyocytes have irreversible damage, which is a serious danger to cardiac function [109]. Cardiomyocyte proliferation and regeneration as potential pathways to improve cardiac function, pointing to new directions in cardiac regeneration [110]. Irisin plays an important role in improving tendinopathy by promoting the proliferation and differentiation of tendon-derived stem cells through up-regulation of YAP/TAZ expression, a process associated with ubiquitin proteasome hydrolysis [111]. Recombinant Irisin upregulates the expression of ECM proteins and angiogenic factors and promotes osteogenesis, showing its potential to improve adverse remodeling in response to mechanical stimulation [112]. Therefore, it is proposed whether Irisin can also target endogenous cardiac stem cells to induce cardiomyocyte proliferation in the heart. Although current research evidence and research techniques are insufficient to support this inference, given the important role played by YAP/TAZ in cardiac fibroblasts [96] and recent-related studies on cardiac fibroblasts reprogrammed into cardiomyocytes [113, 114], it is speculated that Irisin’s regulatory effects on YAP/TAZ and its downstream-specific transcription factors may play an important role in inducing cardiomyocyte proliferation.
Piezo1 and FNDC5/Irisin
Irisin is a skeletal muscle-derived factor and expressed in the heart, liver, and kidney. During exercise, peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) induces increased expression of the membrane-bound protein precursor fibronectin type III domain-containing protein 5 (FNDC5) in skeletal muscle. FNDC5 cleaves two projects to form a new polypeptide, Irisin. In cardiac hypertrophy induced by TAC, injection of recombinant Irisin protein activated AMPK and inhibited mTOR, resulting in significant alleviation of cardiac hypertrophy and myocardial fibrosis [115]. Previous studies have shown that resistance training induces skeletal muscle Irisin secretion and activates the AMPK-PINK1/Parkin-LC3/P62 pathway, which inhibits mitochondrial oxidative stress in cardiomyocytes [116]. In oxidized low-density lipoprotein (oxLDL)-induced vascular injury, Irisin treatment inhibited endothelial cell apoptosis and improved endothelial function via the Akt/mTOR/Nrf2 signaling pathway [117]. Exercise increases the secretion of Irisin from the skeletal muscle, and then Irisin enters the target tissue through the circulation and interacts with its receptor, Integrin αV/β5, which activates signaling pathways such as AMPK, ERK1/2, MAPK, etc. and performs its biological functions [118, 119].
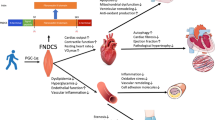
Activation of Piezo1 by different blood flow conditions produces different biological effects that can lead to atherosclerosis and myocardial fibrosis or atherosclerosis- and myocardial fibrosis-protective effects [120]. Our integration of Piezo1 downstream signaling pathways revealed a possible potential link between Piezo1 and FNDC5/Irisin. Integrins interact with Piezo1 as described previously. In the ECM, Irisin interacts with the integrin family and has great potential in participating in cytoskeleton regulation and cell proliferation [112, 121]. Mechanical signaling and changes in the ECM environment alter YAP/TAZ expression and nuclear translocation levels, and Piezo1 and FNDC5/Irisin may regulate the nuclear translocation of YAP/TAZ through a common molecular mechanism. In that both Piezo1 and FNDC5/Irisin regulation of organismal blood pressure is accomplished through the AKT/eNOS signaling pathway, Piezo1 mediates ATP generation to regulate AKT/eNOS to regulate blood pressure [11]; and FNDC5/Irisin activates AMPK to participate in the regulation of arterial blood pressure [122], the two may synergize to regulate blood pressure changes. Turbulence-activated Piezo1 stimulates the NF-κB signaling pathway to promote atherosclerosis development [32], whereas Irisin targets the ROS/p38MAPK signaling pathway to inhibit the activation of NF-κB and produce atherosclerosis protective effects [123]. Exercise upregulation of Irisin expression may antagonize the inflammatory response and atherogenic effects generated by aberrant activation of Piezo1 in pathological states (Fig. 4). In conclusion, the link between Piezo1 and FNDC5/Irisin is extensive, and there may be a regulatory relationship between the two, which needs more studies to further confirm.

Exercise modulates the signaling pathway mechanisms by which mechanosensitive ion channels and FNDC5/Irisin exert cardiovascular protective functions. ECM, extracellular matrix; EC, endothelial cell
Exercise and downstream signaling pathway mechanisms of TRP channels
TRPC protein is a non-selective cation channel that can be activated by mechanical stress and phospholipase C-coupled surface receptor stimulation. Among them, TRPC3 and TRPC6 play a key role in cardiovascular remodeling. Hypoxic stimulation in mice promotes the interaction of TRPC3 and NADPH oxidase 2 (Nox2), leading to Nox2-dependent ROS production that mediates adverse cardiac remodeling. Physical exercise down-regulates the expression of TRPC3 and Nox2 in the mice heart, thereby enhancing myocardial compliance and flexibility and improving cardiac function [124]. Stretch-sensitive TRPV2 channels have a role in regulating Ca2+ concentration and cardiac function. Cardiac TRPV2 expression was increased after exercise, and TRPV2 deficiency led to decreased cardiac contractile function and a significant reduction in exercise capacity, TRPV2-KO mice were unable to complete compulsory exercise, and cardiac function was further impaired after voluntary wheel running exercise [78]. TRPV4 protein is one of the important calcium channels in vascular endothelial cells. It was found that TRPV4 mediated Irisin-induced Ca2+ influx and endothelium-dependent vasodilation, and that inhibition of TRPV4 prevented Irisin-induced increases in Ca2+ concentration and vasodilatory dysfunction [80] (Fig. 4).
Conclusions and prospects
The mechanosensitive cation channel Piezo1 plays an important role in the regulation of cardiovascular function, and as one of the mediators regulating the cardiovascular mechanical transduction process, research on Piezo1 has certain basic and clinical application value. In the normal heart, Piezo1 responds to appropriate mechanical stimulation to improve cardiac contractility and participates in the regulation of blood pressure, while Piezo1 deficiency is detrimental to vascular development and embryonic lethality; in the pathological heart, Piezo1 can improve ischemic damage to the heart and produce an atherosclerosis-protective effect. FNDC5/Irisin responds to the cardiovascular protective effects of exercise by increasing myocardial contractility and improving mitochondrial function. Piezo1 has a role in regulating the localization of the YAP nucleus, and aberrant activation of Piezo1 promotes the translocation of the YAP nucleus to generate an inflammatory response and is atherogenic. Mechanistic studies have found that regulation of YAP by Piezo1 and Irisin involves the intersection of multiple signaling pathways and a high degree of involvement of the integrin family, but direct evidence of exercise-stimulated myocardial Piezo1 regulation of YAP activity has not been clarified. In addition, exercise stimulates Irisin secretion, and in addition to targeting integrin αV/β5, whether other specific receptors are involved in exerting oxidative stress and apoptosis inhibition, autophagy modulation, and YAP function is not known. These are also hot issues for future research. This paper reviews the relationship between exercise, mechanosensitive ion channels, FNDC5/Irisin, YAP and cardiac protection from a new perspective, and proposes that exercise improves cardiovascular function by stimulating the mechanosensitive channels and that Irisin may play a role in this process, providing theoretical basis for cardiovascular disease prevention and cardiac rehabilitation of clinical patients.
Data availability
Enquiries about data availability should be directed to the authors.
References
Kefauver JM, Ward AB, Patapoutian A (2020) Discoveries in structure and physiology of mechanically activated ion channels. Nature 587:567–576. https://doi.org/10.1038/s41586-020-2933-1
Brohawn SG (2015) How ion channels sense mechanical force: insights from mechanosensitive K2P channels TRAAK, TREK1, and TREK2. Ann N Y Acad Sci 1352:20–32. https://doi.org/10.1111/nyas.12874
Brohawn SG, Su Z, MacKinnon R (2014) Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci USA 111:3614–3619. https://doi.org/10.1073/pnas.1320768111
Startek JB, Boonen B, Talavera K, Meseguer V (2019) TRP channels as sensors of chemically-induced changes in cell membrane mechanical properties. Int J Mol Sci. https://doi.org/10.3390/ijms20020371
Servin-Vences MR, Moroni M, Lewin GR, Poole K (2017) Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes. Elife. https://doi.org/10.7554/eLife.21074
Ben-Shahar Y (2011) Sensory functions for degenerin/epithelial sodium channels (DEG/ENaC). Adv Genet 76:1–26. https://doi.org/10.1016/B978-0-12-386481-9.00001-8
Lin SH, Cheng YR, Banks RW, Min MY, Bewick GS, Chen CC (2016) Evidence for the involvement of ASIC3 in sensory mechanotransduction in proprioceptors. Nat Commun 7:11460. https://doi.org/10.1038/ncomms11460
Knoepp F, Ashley Z, Barth D, Baldin JP, Jennings M, Kazantseva M, Saw EL, Katare R, Alvarez de la Rosa D, Weissmann N, Fronius M (2020) Shear force sensing of epithelial Na(+) channel (ENaC) relies on N-glycosylated asparagines in the palm and knuckle domains of alphaENaC. Proc Natl Acad Sci USA 117:717–726. https://doi.org/10.1073/pnas.1911243117
Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, Montal M, Patapoutian A (2012) Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483:176–181. https://doi.org/10.1038/nature10812
Ge J, Li W, Zhao Q, Li N, Chen M, Zhi P, Li R, Gao N, Xiao B, Yang M (2015) Architecture of the mammalian mechanosensitive Piezo1 channel. Nature 527:64–69. https://doi.org/10.1038/nature15247
Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S (2016) Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest 126:4527–4536. https://doi.org/10.1172/JCI87343
Wehrwein EA, Joyner MJ (2013) Regulation of blood pressure by the arterial baroreflex and autonomic nervous system. Handb Clin Neurol 117:89–102. https://doi.org/10.1016/B978-0-444-53491-0.00008-0
Zeng WZ, Marshall KL, Min S, Daou I, Chapleau MW, Abboud FM, Liberles SD, Patapoutian A (2018) PIEZOs mediate neuronal sensing of blood pressure and the baroreceptor reflex. Science 362:464–467. https://doi.org/10.1126/science.aau6324
Baratchi S, Khoshmanesh K, Woodman OL, Potocnik S, Peter K, McIntyre P (2017) Molecular sensors of blood flow in endothelial cells. Trends Mol Med 23:850–868. https://doi.org/10.1016/j.molmed.2017.07.007
Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DA, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad RK, Evans PC, Ainscough JF, Beech DJ (2014) Piezo1 integration of vascular architecture with physiological force. Nature 515:279–282. https://doi.org/10.1038/nature13701
Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A (2014) Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA 111:10347–10352. https://doi.org/10.1073/pnas.1409233111
Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, Jodar M, Moro C, Offermanns S, Feng Y, Demolombe S, Patel A, Honore E (2015) Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep 13:1161–1171. https://doi.org/10.1016/j.celrep.2015.09.072
Kang H, Hong Z, Zhong M, Klomp J, Bayless KJ, Mehta D, Karginov AV, Hu G, Malik AB (2019) Piezo1 mediates angiogenesis through activation of MT1-MMP signaling. Am J Physiol Cell Physiol 316:C92–C103. https://doi.org/10.1152/ajpcell.00346.2018
Beech DJ (2018) Endothelial Piezo1 channels as sensors of exercise. J Physiol 596:979–984. https://doi.org/10.1113/JP274396
Douguet D, Patel A, Xu A, Vanhoutte PM, Honore E (2019) Piezo ion channels in cardiovascular mechanobiology. Trends Pharmacol Sci 40:956–970. https://doi.org/10.1016/j.tips.2019.10.002
Gimbrone MA Jr, Garcia-Cardena G (2016) Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 118:620–636. https://doi.org/10.1161/CIRCRESAHA.115.306301
Carmeliet E (1999) Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 79:917–1017. https://doi.org/10.1152/physrev.1999.79.3.917
Dutta S, Minchole A, Quinn TA, Rodriguez B (2017) Electrophysiological properties of computational human ventricular cell action potential models under acute ischemic conditions. Prog Biophys Mol Biol 129:40–52. https://doi.org/10.1016/j.pbiomolbio.2017.02.007
Tran K, Smith NP, Loiselle DS, Crampin EJ (2009) A thermodynamic model of the cardiac sarcoplasmic/endoplasmic Ca(2+) (SERCA) pump. Biophys J 96:2029–2042. https://doi.org/10.1016/j.bpj.2008.11.045
Kormos A, Nagy N, Acsai K, Vaczi K, Agoston S, Pollesello P, Levijoki J, Szentandrassy N, Papp JG, Varro A, Toth A (2014) Efficacy of selective NCX inhibition by ORM-10103 during simulated ischemia/reperfusion. Eur J Pharmacol 740:539–551. https://doi.org/10.1016/j.ejphar.2014.06.033
Clasen L, Angendohr S, Becher S, Bartsch B, Enkel S, Meyer C, Kelm M, Makimoto H, Klocker N (2023) Cardiac ischemia and reperfusion in mice: a comprehensive hemodynamic, electrocardiographic and electrophysiological characterization. Sci Rep 13:5693. https://doi.org/10.1038/s41598-023-32346-5
Jiang F, Yin K, Wu K, Zhang M, Wang S, Cheng H, Zhou Z, Xiao B (2021) The mechanosensitive Piezo1 channel mediates heart mechano-chemo transduction. Nat Commun 12:869. https://doi.org/10.1038/s41467-021-21178-4
Murthy SE, Dubin AE, Patapoutian A (2017) Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat Rev Mol Cell Biol 18:771–783. https://doi.org/10.1038/nrm.2017.92
Liang J, Huang B, Yuan G, Chen Y, Liang F, Zeng H, Zheng S, Cao L, Geng D, Zhou S (2017) Stretch-activated channel Piezo1 is up-regulated in failure heart and cardiomyocyte stimulated by AngII. Am J Transl Res 9:2945–2955
Niu L, Cheng B, Huang G, Nan K, Han S, Ren H, Liu N, Li Y, Genin GM, Xu F (2022) A positive mechanobiological feedback loop controls bistable switching of cardiac fibroblast phenotype. Cell Discov 8:84. https://doi.org/10.1038/s41421-022-00427-w
Blythe NM, Muraki K, Ludlow MJ, Stylianidis V, Gilbert HTJ, Evans EL, Cuthbertson K, Foster R, Swift J, Li J, Drinkhill MJ, van Nieuwenhoven FA, Porter KE, Beech DJ, Turner NA (2019) Mechanically activated Piezo1 channels of cardiac fibroblasts stimulate p38 mitogen-activated protein kinase activity and interleukin-6 secretion. J Biol Chem 294:17395–17408. https://doi.org/10.1074/jbc.RA119.009167
Albarran-Juarez J, Iring A, Wang S, Joseph S, Grimm M, Strilic B, Wettschureck N, Althoff TF, Offermanns S (2018) Piezo1 and G(q)/G(11) promote endothelial inflammation depending on flow pattern and integrin activation. J Exp Med 215:2655–2672. https://doi.org/10.1084/jem.20180483
Zhang Y, Su SA, Li W, Ma Y, Shen J, Wang Y, Shen Y, Chen J, Ji Y, Xie Y, Ma H, Xiang M (2021) Piezo1-mediated mechanotransduction promotes cardiac hypertrophy by impairing calcium homeostasis to activate calpain/calcineurin signaling. Hypertension 78:647–660. https://doi.org/10.1161/HYPERTENSIONAHA.121.17177
Faucherre A, Moha Ou Maati H, Nasr N, Pinard A, Theron A, Odelin G, Desvignes JP, Salgado D, Collod-Beroud G, Avierinos JF, Lebon G, Zaffran S, Jopling C (2020) Piezo1 is required for outflow tract and aortic valve development. J Mol Cell Cardiol 143:51–62. https://doi.org/10.1016/j.yjmcc.2020.03.013
Emig R, Knodt W, Krussig MJ, Zgierski-Johnston CM, Gorka O, Gross O, Kohl P, Ravens U, Peyronnet R (2021) Piezo1 channels contribute to the regulation of human atrial fibroblast mechanical properties and matrix stiffness sensing. Cells. https://doi.org/10.3390/cells10030663
Braidotti N, Chen SN, Long CS, Cojoc D, Sbaizero O (2022) Piezo1 channel as a potential target for hindering cardiac fibrotic remodeling. Int J Mol Sci. https://doi.org/10.3390/ijms23158065
Lim GB (2022) Piezo1 senses pressure overload and initiates cardiac hypertrophy. Nat Rev Cardiol 19:503. https://doi.org/10.1038/s41569-022-00746-1
Bartoli F, Evans EL, Blythe NM, Stewart L, Chuntharpursat-Bon E, Debant M, Musialowski KE, Lichtenstein L, Parsonage G, Futers TS, Turner NA, Beech DJ (2022) Global PIEZO1 gain-of-function mutation causes cardiac hypertrophy and fibrosis in mice. Cells. https://doi.org/10.3390/cells11071199
MacKay CE, Floen M, Leo MD, Hasan R, Garrud TAC, Fernandez-Pena C, Singh P, Malik KU, Jaggar JH (2022) A plasma membrane-localized polycystin-1/polycystin-2 complex in endothelial cells elicits vasodilation. Elife. https://doi.org/10.7554/eLife.74765
MacKay CE, Leo MD, Fernandez-Pena C, Hasan R, Yin W, Mata-Daboin A, Bulley S, Gammons J, Mancarella S, Jaggar JH (2020) Intravascular flow stimulates PKD2 (polycystin-2) channels in endothelial cells to reduce blood pressure. Elife. https://doi.org/10.7554/eLife.56655
Volk T, Schwoerer AP, Thiessen S, Schultz JH, Ehmke H (2003) A polycystin-2-like large conductance cation channel in rat left ventricular myocytes. Cardiovasc Res 58:76–88. https://doi.org/10.1016/s0008-6363(02)00858-1
Wu G, Markowitz GS, Li L, D’Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH, van Adelsberg J, Hou H Jr, Kucherlapati R, Edelmann W, Somlo S (2000) Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet 24:75–78. https://doi.org/10.1038/71724
Hof T, Chaigne S, Recalde A, Salle L, Brette F, Guinamard R (2019) Transient receptor potential channels in cardiac health and disease. Nat Rev Cardiol 16:344–360. https://doi.org/10.1038/s41569-018-0145-2
Numaga-Tomita T, Nishida M (2020) TRPC channels in cardiac plasticity. Cells. https://doi.org/10.3390/cells9020454
Camacho Londono JE, Tian Q, Hammer K, Schroder L, Camacho Londono J, Reil JC, He T, Oberhofer M, Mannebach S, Mathar I, Philipp SE, Tabellion W, Schweda F, Dietrich A, Kaestner L, Laufs U, Birnbaumer L, Flockerzi V, Freichel M, Lipp P (2015) A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J 36:2257–2266. https://doi.org/10.1093/eurheartj/ehv250
He X, Li S, Liu B, Susperreguy S, Formoso K, Yao J, Kang J, Shi A, Birnbaumer L, Liao Y (2017) Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc Natl Acad Sci USA 114:E4582–E4591. https://doi.org/10.1073/pnas.1621384114
Numaga-Tomita T, Oda S, Nishiyama K, Tanaka T, Nishimura A, Nishida M (2019) TRPC channels in exercise-mimetic therapy. Pflugers Arch 471:507–517. https://doi.org/10.1007/s00424-018-2211-3
Sabourin J, Bartoli F, Antigny F, Gomez AM, Benitah JP (2016) Transient receptor potential canonical (TRPC)/Orai1-dependent store-operated Ca2+ channels: new targets of aldosterone in cardiomyocytes. J Biol Chem 291:13394–13409. https://doi.org/10.1074/jbc.M115.693911
Kitajima N, Watanabe K, Morimoto S, Sato Y, Kiyonaka S, Hoshijima M, Ikeda Y, Nakaya M, Ide T, Mori Y, Kurose H, Nishida M (2011) TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochem Biophys Res Commun 409:108–113. https://doi.org/10.1016/j.bbrc.2011.04.124
Bon RS, Wright DJ, Beech DJ, Sukumar P (2022) Pharmacology of TRPC channels and its potential in cardiovascular and metabolic medicine. Annu Rev Pharmacol Toxicol 62:427–446. https://doi.org/10.1146/annurev-pharmtox-030121-122314
Kumar B, Dreja K, Shah SS, Cheong A, Xu SZ, Sukumar P, Naylor J, Forte A, Cipollaro M, McHugh D, Kingston PA, Heagerty AM, Munsch CM, Bergdahl A, Hultgardh-Nilsson A, Gomez MF, Porter KE, Hellstrand P, Beech DJ (2006) Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ Res 98:557–563. https://doi.org/10.1161/01.RES.0000204724.29685.db
Liu DY, Thilo F, Scholze A, Wittstock A, Zhao ZG, Harteneck C, Zidek W, Zhu ZM, Tepel M (2007) Increased store-operated and 1-oleoyl-2-acetyl-sn-glycerol-induced calcium influx in monocytes is mediated by transient receptor potential canonical channels in human essential hypertension. J Hypertens 25:799–808. https://doi.org/10.1097/HJH.0b013e32803cae2b
Thilo F, Loddenkemper C, Berg E, Zidek W, Tepel M (2009) Increased TRPC3 expression in vascular endothelium of patients with malignant hypertension. Mod Pathol 22:426–430. https://doi.org/10.1038/modpathol.2008.200
Zhang S, Patel HH, Murray F, Remillard CV, Schach C, Thistlethwaite PA, Insel PA, Yuan JX (2007) Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 292:L1202–L1210. https://doi.org/10.1152/ajplung.00214.2006
Guinamard R, Demion M, Magaud C, Potreau D, Bois P (2006) Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension 48:587–594. https://doi.org/10.1161/01.HYP.0000237864.65019.a5
Zhao R, Liu X, Qi Z, Yao X, Tsang SY (2021) TRPV1 channels regulate the automaticity of embryonic stem cell-derived cardiomyocytes through stimulating the Na(+) /Ca(2+) exchanger current. J Cell Physiol 236:6806–6823. https://doi.org/10.1002/jcp.30369
Hong J, Lisco AM, Rudebush TL, Yu L, Gao L, Kitzerow O, Zucker IH, Wang HJ (2020) Identification of cardiac expression pattern of transient receptor potential vanilloid type 1 (TRPV1) receptor using a transgenic reporter mouse model. Neurosci Lett 737:135320. https://doi.org/10.1016/j.neulet.2020.135320
Zhong B, Rubinstein J, Ma S, Wang DH (2018) Genetic ablation of TRPV1 exacerbates pressure overload-induced cardiac hypertrophy. Biomed Pharmacother 99:261–270. https://doi.org/10.1016/j.biopha.2018.01.065
Aguettaz E, Bois P, Cognard C, Sebille S (2017) Stretch-activated TRPV2 channels: role in mediating cardiopathies. Prog Biophys Mol Biol 130:273–280. https://doi.org/10.1016/j.pbiomolbio.2017.05.007
Zhang Q, Qi H, Cao Y, Shi P, Song C, Ba L, Chen Y, Gao J, Li S, Li B, Sun H (2018) Activation of transient receptor potential vanilloid 3 channel (TRPV3) aggravated pathological cardiac hypertrophy via calcineurin/NFATc3 pathway in rats. J Cell Mol Med 22:6055–6067. https://doi.org/10.1111/jcmm.13880
Qi H, Ren J, Zhang Q, Cao Y, Ba L, Song C, Shi P, Fu B, Sun H (2019) MiR-103 inhibiting cardiac hypertrophy through inactivation of myocardial cell autophagy via targeting TRPV3 channel in rat hearts. J Cell Mol Med 23:1926–1939. https://doi.org/10.1111/jcmm.14095
Liu Y, Qi H, Shi P, Zhang Q, Li S, Wang Y, Cao Y, Chen Y, Ba L, Gao J, Huang W, Sun H (2018) Transient receptor potential vanilloid-3 (TRPV3) activation plays a central role in cardiac fibrosis induced by pressure overload in rats via TGF-beta(1) pathway. Naunyn Schmiedebergs Arch Pharmacol 391:131–143. https://doi.org/10.1007/s00210-017-1443-7
Peixoto-Neves D, Wang Q, Leal-Cardoso JH, Rossoni LV, Jaggar JH (2015) Eugenol dilates mesenteric arteries and reduces systemic BP by activating endothelial cell TRPV4 channels. Br J Pharmacol 172:3484–3494. https://doi.org/10.1111/bph.13156
Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M, Gutterman DD (2009) Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53:532–538. https://doi.org/10.1161/HYPERTENSIONAHA.108.127100
Guo S, Huang Y, Zhang Y, Huang H, Hong S, Liu T (2020) Impacts of exercise interventions on different diseases and organ functions in mice. J Sport Health Sci 9:53–73. https://doi.org/10.1016/j.jshs.2019.07.004
Polyak A, Topal L, Zombori-Toth N, Toth N, Prorok J, Kohajda Z, Deri S, Demeter-Haludka V, Hegyi P, Venglovecz V, Agoston G, Husti Z, Gazdag P, Szlovak J, Arpadffy-Lovas T, Naveed M, Sarusi A, Jost N, Virag L, Nagy N, Baczko I, Farkas AS, Varro A (2023) Cardiac electrophysiological remodeling associated with enhanced arrhythmia susceptibility in a canine model of elite exercise. Elife. https://doi.org/10.7554/eLife.80710
Maron BJ, Pelliccia A (2006) The heart of trained athletes: cardiac remodelling and the risks of sports, including sudden death. Circulation 114:1633–1644. https://doi.org/10.1161/CIRCULATIONAHA.106.613562
Gazdag P, Oravecz K, Acsai K, Demeter-Haludka V, Ordog B, Szlovak J, Kohajda Z, Polyak A, Barta BA, Olah A, Radovits T, Merkely B, Papp JG, Baczko I, Varro A, Nagy N, Prorok J (2020) Increased Ca(2+) content of the sarcoplasmic reticulum provides arrhythmogenic trigger source in swimming-induced rat athlete’s heart model. Sci Rep 10:19596. https://doi.org/10.1038/s41598-020-76496-2
Pelliccia A, Day S, Olivotto I (2023) Leisure-time and competitive sport participation: a changing paradigm for HCM patients. Eur J Prev Cardiol. https://doi.org/10.1093/eurjpc/zwad011
Rode B, Shi J, Endesh N, Drinkhill MJ, Webster PJ, Lotteau SJ, Bailey MA, Yuldasheva NY, Ludlow MJ, Cubbon RM, Li J, Futers TS, Morley L, Gaunt HJ, Marszalek K, Viswambharan H, Cuthbertson K, Baxter PD, Foster R, Sukumar P, Weightman A, Calaghan SC, Wheatcroft SB, Kearney MT, Beech DJ (2017) Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat Commun 8:350. https://doi.org/10.1038/s41467-017-00429-3
Bartoli F, Debant M, Chuntharpursat-Bon E, Evans EL, Musialowski KE, Parsonage G, Morley LC, Futers TS, Sukumar P, Bowen TS, Kearney MT, Lichtenstein L, Roberts LD, Beech DJ (2022) Endothelial Piezo1 sustains muscle capillary density and contributes to physical activity. J Clin Invest. https://doi.org/10.1172/JCI141775
Wong TY, Juang WC, Tsai CT, Tseng CJ, Lee WH, Chang SN, Cheng PW (2018) Mechanical stretching simulates cardiac physiology and pathology through mechanosensor Piezo1. J Clin Med. https://doi.org/10.3390/jcm7110410
Eisner DA, Caldwell JL, Kistamas K, Trafford AW (2017) Calcium and excitation-contraction coupling in the heart. Circ Res 121:181–195. https://doi.org/10.1161/CIRCRESAHA.117.310230
Jiang Y, Yang X, Jiang J, Xiao B (2021) Structural designs and mechanogating mechanisms of the mechanosensitive Piezo channels. Trends Biochem Sci 46:472–488. https://doi.org/10.1016/j.tibs.2021.01.008
Sciancalepore M, Massaria G, Tramer F, Zacchi P, Lorenzon P, Bernareggi A (2022) A preliminary study on the role of Piezo1 channels in myokine release from cultured mouse myotubes. Biochem Biophys Res Commun 623:148–153. https://doi.org/10.1016/j.bbrc.2022.07.059
Smith JR, Hart CR, Ramos PA, Akinsanya JG, Lanza IR, Joyner MJ, Curry TB, Olson TP (2020) Metabo- and mechanoreceptor expression in human heart failure: relationships with the locomotor muscle afferent influence on exercise responses. Exp Physiol 105:809–818. https://doi.org/10.1113/EP088353
Rubinstein J, Lasko VM, Koch SE, Singh VP, Carreira V, Robbins N, Patel AR, Jiang M, Bidwell P, Kranias EG, Jones WK, Lorenz JN (2014) Novel role of transient receptor potential vanilloid 2 in the regulation of cardiac performance. Am J Physiol Heart Circ Physiol 306:H574–H584. https://doi.org/10.1152/ajpheart.00854.2013
Naticchioni M, Karani R, Smith MA, Onusko E, Robbins N, Jiang M, Radzyukevich T, Fulford L, Gao X, Apel R, Heiny J, Rubinstein J, Koch SE (2015) Transient receptor potential vanilloid 2 regulates myocardial response to exercise. PLoS ONE 10:e0136901. https://doi.org/10.1371/journal.pone.0136901
Cooper MA, Ryals JM, Wu PY, Wright KD, Walter KR, Wright DE (2017) Modulation of diet-induced mechanical allodynia by metabolic parameters and inflammation. J Peripher Nerv Syst 22:39–46. https://doi.org/10.1111/jns.12199
Ye L, Xu M, Hu M, Zhang H, Tan X, Li Q, Shen B, Huang J (2018) TRPV4 is involved in irisin-induced endothelium-dependent vasodilation. Biochem Biophys Res Commun 495:41–45. https://doi.org/10.1016/j.bbrc.2017.10.160
Li X, Han L, Nookaew I, Mannen E, Silva MJ, Almeida M, Xiong J (2019) Stimulation of Piezo1 by mechanical signals promotes bone anabolism. Elife. https://doi.org/10.7554/eLife.49631
Bosutti A, Giniatullin A, Odnoshivkina Y, Giudice L, Malm T, Sciancalepore M, Giniatullin R, D’Andrea P, Lorenzon P, Bernareggi A (2021) “Time window” effect of Yoda1-evoked Piezo1 channel activity during mouse skeletal muscle differentiation. Acta Physiol (Oxf) 233:e13702. https://doi.org/10.1111/apha.13702
Obi S, Nakajima T, Hasegawa T, Kikuchi H, Oguri G, Takahashi M, Nakamura F, Yamasoba T, Sakuma M, Toyoda S, Tei C, Inoue T (1985) Heat induces interleukin-6 in skeletal muscle cells via TRPV1/PKC/CREB pathways. J Appl Physiol 122:683–694. https://doi.org/10.1152/japplphysiol.00139.2016
Fujii N, Kenny GP, McGarr GW, Amano T, Honda Y, Kondo N, Nishiyasu T (2021) TRPV4 channel blockade does not modulate skin vasodilation and sweating during hyperthermia or cutaneous postocclusive reactive and thermal hyperemia. Am J Physiol Regul Integr Comp Physiol 320:R563–R573. https://doi.org/10.1152/ajpregu.00123.2020
Krout D, Schaar A, Sun Y, Sukumaran P, Roemmich JN, Singh BB, Claycombe-Larson KJ (2017) The TRPC1 Ca(2+)-permeable channel inhibits exercise-induced protection against high-fat diet-induced obesity and type II diabetes. J Biol Chem 292:20799–20807. https://doi.org/10.1074/jbc.M117.809954
Craighead DH, Shank SW, Gottschall JS, Passe DH, Murray B, Alexander LM, Kenney WL (2017) Ingestion of transient receptor potential channel agonists attenuates exercise-induced muscle cramps. Muscle Nerve 56:379–385. https://doi.org/10.1002/mus.25611
Iring A, Jin YJ, Albarran-Juarez J, Siragusa M, Wang S, Dancs PT, Nakayama A, Tonack S, Chen M, Kunne C, Sokol AM, Gunther S, Martinez A, Fleming I, Wettschureck N, Graumann J, Weinstein LS, Offermanns S (2019) Shear stress-induced endothelial adrenomedullin signaling regulates vascular tone and blood pressure. J Clin Invest 129:2775–2791. https://doi.org/10.1172/JCI123825
Cinar E, Zhou S, DeCourcey J, Wang Y, Waugh RE, Wan J (2015) Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc Natl Acad Sci U S A 112:11783–11788. https://doi.org/10.1073/pnas.1507309112
Fleming I (2010) Molecular mechanisms underlying the activation of eNOS. Pflugers Arch 459:793–806. https://doi.org/10.1007/s00424-009-0767-7
Davies PF (2009) Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med 6:16–26. https://doi.org/10.1038/ncpcardio1397
Wang C, Baker BM, Chen CS, Schwartz MA (2013) Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol 33:2130–2136. https://doi.org/10.1161/ATVBAHA.113.301826
Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H (2002) Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol 283:H1819–H1828. https://doi.org/10.1152/ajpheart.00214.2002
Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis J (2017) Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation 136:549–561. https://doi.org/10.1161/CIRCULATIONAHA.116.026238
Lai A, Thurgood P, Cox CD, Chheang C, Peter K, Jaworowski A, Khoshmanesh K, Baratchi S (2022) Piezo1 response to shear stress is controlled by the components of the extracellular matrix. ACS Appl Mater Interfaces 14:40559–40568. https://doi.org/10.1021/acsami.2c09169
Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S (2016) Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA 113:11525–11530. https://doi.org/10.1073/pnas.1613121113
Mia MM, Cibi DM, Ghani S, Singh A, Tee N, Sivakumar V, Bogireddi H, Cook SA, Mao J, Singh MK (2022) Loss of Yap/Taz in cardiac fibroblasts attenuates adverse remodelling and improves cardiac function. Cardiovasc Res 118:1785–1804. https://doi.org/10.1093/cvr/cvab205
Yang Y, Wang D, Zhang C, Yang W, Li C, Gao Z, Pei K, Li Y (2022) Piezo1 mediates endothelial atherogenic inflammatory responses via regulation of YAP/TAZ activation. Hum Cell 35:51–62. https://doi.org/10.1007/s13577-021-00600-5
Xiong Y, Dong L, Bai Y, Tang H, Li S, Luo D, Liu F, Bai J, Yang S, Song X (2022) Piezo1 activation facilitates ovarian cancer metastasis via Hippo/YAP signaling axis. Channels (Austin) 16:159–166. https://doi.org/10.1080/19336950.2022.2099381
Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W (2020) Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat Commun 11:282. https://doi.org/10.1038/s41467-019-14146-6
Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DT, Bernardis E, Flanagan LA, Tombola F (2014) Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc Natl Acad Sci USA 111:16148–16153. https://doi.org/10.1073/pnas.1409802111
Liu S, Xu X, Fang Z, Ning Y, Deng B, Pan X, He Y, Yang Z, Huang K, Li J (2021) Piezo1 impairs hepatocellular tumor growth via deregulation of the MAPK-mediated YAP signaling pathway. Cell Calcium 95:102367. https://doi.org/10.1016/j.ceca.2021.102367
Panciera T, Azzolin L, Cordenonsi M, Piccolo S (2017) Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol 18:758–770. https://doi.org/10.1038/nrm.2017.87
Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y, Huang Y (2016) Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540:579–582. https://doi.org/10.1038/nature20602
He L, Si G, Huang J, Samuel ADT, Perrimon N (2018) Mechanical regulation of stem-cell differentiation by the stretch-activated Piezo channel. Nature 555:103–106. https://doi.org/10.1038/nature25744
Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, Ando K, Miyazaki T, Yokota Y, Schmelzer E, Belting HG, Affolter M, Lecaudey V, Mochizuki N (2017) Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev Cell 40(523–536):e6. https://doi.org/10.1016/j.devcel.2017.02.019
Duchemin AL, Vignes H, Vermot J (2019) Mechanically activated piezo channels modulate outflow tract valve development through the Yap1 and Klf2-Notch signaling axis. Elife. https://doi.org/10.7554/eLife.44706
Mohri Z, Del Rio HA, Krams R (2017) The emerging role of YAP/TAZ in mechanotransduction. J Thorac Dis 9:E507–E509. https://doi.org/10.21037/jtd.2017.03.179
Tang Z, Wei X, Li T, Wu H, Xiao X, Hao Y, Li S, Hou W, Shi L, Li X, Guo Z (2021) Three-dimensionally printed Ti2448 with low stiffness enhanced angiogenesis and osteogenesis by regulating macrophage polarization via Piezo1/YAP signaling axis. Front Cell Dev Biol 9:750948. https://doi.org/10.3389/fcell.2021.750948
Nabel EG, Braunwald E (2012) A tale of coronary artery disease and myocardial infarction. N Engl J Med 366:54–63. https://doi.org/10.1056/NEJMra1112570
He L, Nguyen NB, Ardehali R, Zhou B (2020) Heart regeneration by endogenous stem cells and cardiomyocyte proliferation: controversy, fallacy, and progress. Circulation 142:275–291. https://doi.org/10.1161/CIRCULATIONAHA.119.045566
Xu L, Chen Z, Geng T, Ru B, Wan Q, Zhang J, Li S, Cai W (2022) Irisin promotes the proliferation and tenogenic differentiation of rat tendon-derived stem/progenitor cells via activating YAP/TAZ. In Vitro Cell Dev Biol Anim 58:658–668. https://doi.org/10.1007/s11626-022-00699-2
Yang Y, Geng T, Samara A, Olstad OK, He J, Agger AE, Skallerud BH, Landin MA, Heyward CA, Pullisaar H, Reseland JE (2023) Recombinant irisin enhances the extracellular matrix formation, remodeling potential, and differentiation of human periodontal ligament cells cultured in 3D. J Periodontal Res 58:336–349. https://doi.org/10.1111/jre.13094
Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN (2012) Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485:599–604. https://doi.org/10.1038/nature11139
Huang C, Tu W, Fu Y, Wang J, Xie X (2018) Chemical-induced cardiac reprogramming in vivo. Cell Res 28:686–689. https://doi.org/10.1038/s41422-018-0036-4
Yu Q, Kou W, Xu X, Zhou S, Luan P, Xu X, Li H, Zhuang J, Wang J, Zhao Y, Xu Y, Peng W (2019) FNDC5/Irisin inhibits pathological cardiac hypertrophy. Clin Sci (Lond) 133:611–627. https://doi.org/10.1042/CS20190016
Li H, Qin S, Liang Q, Xi Y, Bo W, Cai M, Tian Z (2021) Exercise training enhances myocardial mitophagy and improves cardiac function via Irisin/FNDC5-PINK1/Parkin pathway in MI mice. Biomedicines. https://doi.org/10.3390/biomedicines9060701
Zhang M, Xu Y, Jiang L (2019) Irisin attenuates oxidized low-density lipoprotein impaired angiogenesis through AKT/mTOR/S6K1/Nrf2 pathway. J Cell Physiol 234:18951–18962. https://doi.org/10.1002/jcp.28535
Maak S, Norheim F, Drevon CA, Erickson HP (2021) Progress and challenges in the biology of FNDC5 and Irisin. Endocr Rev 42:436–456. https://doi.org/10.1210/endrev/bnab003
Liu S, Cui F, Ning K, Wang Z, Fu P, Wang D, Xu H (2022) Role of irisin in physiology and pathology. Front Endocrinol (Lausanne) 13:962968. https://doi.org/10.3389/fendo.2022.962968
Shinge SAU, Zhang D, Din AU, Yu F, Nie Y (2022) Emerging Piezo1 signaling in inflammation and atherosclerosis; a potential therapeutic target. Int J Biol Sci 18:923–941. https://doi.org/10.7150/ijbs.63819
Chen T, Lin Y, Wu Z, Shi H, Hu W, Li S, Que Y, Qiu J, Li P, Qiu X, Liang T, Wang X, Gao B, Zhou H, Deng Z, Chen Y, Zhu Y, Peng Y, Liang A, Gao W, Huang D (2022) Irisin ameliorates intervertebral disc degeneration by activating LATS/YAP/CTGF signaling. Oxid Med Cell Longev 2022:9684062. https://doi.org/10.1155/2022/9684062
Inoue K, Fujie S, Hasegawa N, Horii N, Uchida M, Iemitsu K, Sanada K, Hamaoka T, Iemitsu M (2020) Aerobic exercise training-induced irisin secretion is associated with the reduction of arterial stiffness via nitric oxide production in adults with obesity. Appl Physiol Nutr Metab 45:715–722. https://doi.org/10.1139/apnm-2019-0602
Zhang Y, Mu Q, Zhou Z, Song H, Zhang Y, Wu F, Jiang M, Wang F, Zhang W, Li L, Shao L, Wang X, Li S, Yang L, Wu Q, Zhang M, Tang D (2016) Protective effect of irisin on atherosclerosis via suppressing oxidized low density lipoprotein induced vascular inflammation and endothelial dysfunction. PLoS ONE 11:e0158038. https://doi.org/10.1371/journal.pone.0158038
Shimauchi T, Numaga-Tomita T, Ito T, Nishimura A, Matsukane R, Oda S, Hoka S, Ide T, Koitabashi N, Uchida K, Sumimoto H, Mori Y, Nishida M (2017) TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy. JCI Insight. https://doi.org/10.1172/jci.insight.93358
Funding
This work was supported by National Natural Science Foundation of China Grants (31900828 to Y.X. and 32171128 to Z.T.) and the Fundamental Research Funds for the Central Universities (GK202301013 to Y.X.).
Author information
Authors and Affiliations
Contributions
XD, YX and ZT established the theme of the review and constructed the framework; XD and YX searched the papers and did analysis; XD, RL and YX drafted manuscript; XD and YX prepared the figures; XD, RL, YX and ZT revised manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Duan, X., Liu, R., Xi, Y. et al. The mechanisms of exercise improving cardiovascular function by stimulating Piezo1 and TRP ion channels: a systemic review. Mol Cell Biochem (2024). https://doi.org/10.1007/s11010-024-05000-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11010-024-05000-5