
Abstract
Cardiomyocytes undergo a variety of cell death events during myocardial ischemia‒reperfusion injury (MIRI). Understanding the causes of cardiomyocyte mortality is critical for the prevention and treatment of MIRI. Among the various types of cell death, autosis is a recently identified type of autophagic cell death with distinct morphological and chemical characteristics. Autosis can be attenuated by autophagy inhibitors but not reversed by apoptosis or necrosis inhibitors. In recent years, it has been shown that during the late phase of reperfusion, autosis is activated, which exacerbates myocardial injury. This article describes the characteristics of autosis, autophagic cell death, and the relationship between autophagic cell death and autosis; reviews the mechanism of autosis in MIRI; and discusses its clinical significance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MIRI is a common clinical problem that often occurs during reperfusion therapy after acute myocardial infarction (AMI)[1]. Despite extensive research on MIRI, medical interventions have had minimal effects on MIRI[2]. Ischemia causes a sharp decrease in tissue oxygen levels, which inhibits aerobic energy metabolism. The cessation of mitochondrial oxidative phosphorylation and decrease in tissue ATP levels stimulate glycolysis, which rapidly depletes fermentable substrates[3]. Subsequent reperfusion and reoxygenation cause an excessive inflammatory response that exacerbates ischemic tissue damage, which is termed I/R[4]. I/R injury is closely associated with oxidative damage, including impaired endothelial barrier function, mitochondrial dysfunction, the activation of cell death, calcium overload, aseptic inflammation, and autoimmune responses[4]. Cardiomyocyte structure and function are especially dependent on calcium homeostasis. Ca2+ plays a crucial role in excitation–contraction coupling (ECC) in cardiac tissue[5]. Intracellular H+ accumulation following cardiac ischemia causes a decrease in intracellular pH and an increase in the intracellular Na+/H+ exchanger (HNX). Excess intracellular Na+ promotes Na+ excretion and Ca2+ uptake via Na+/Ca2+ exchanger (NCX), significantly increasing intracellular calcium levels and leading to calcium overload. Extracellular pH levels increase significantly during reperfusion, and HNX and NCX activity are increased, exacerbating intracellular calcium overload[5, 6]. These alterations typically lead to aberrant changes in the membrane potential, a higher likelihood of diastolic Ca2+ release, increased susceptibility to arrhythmias (both early and delayed after depolarization), and injury to cells[5, 6]. KATP channels are closely associated with the pathophysiological sequelae of myocardial ischemia, and their opening protects the heart from the effects of ischemia. Sarcolemmal KATP channels induce action potential shortening and intracardiac K+ loss, thereby maintaining energy expenditure and reducing Ca2+ accumulation during ischemia[7].
Cell death is a stable pathological indicator of I/R injury, and increasing evidence suggests that targeting cell death to counteract I/R injury is a new therapeutic strategy. Different forms of cell death have been identified in I/R injury, including apoptosis, necroptosis, mitochondrial-mediated cell death, pyroptosis, ferroptosis, and autophagic cell death[8]. An increase in cardiac-specific caspase-3 during MIRI increases infarct size and the risk of death after MIRI[9]. In addition, apoptosis was also induced by elevated Bax, TNF-α, and TRAIL at the onset of reperfusion in an I/R model[10]. In contrast, cardiac-specific overexpression of Bcl-2 significantly attenuated cardiomyocyte apoptosis and infarct size after I/R injury[11]. Necroptosis is a form of regulated cell necrosis characterized by both necrosis and apoptosis. Necroptosis is triggered by death receptors (e.g. tumor necrosis factor receptor 1, TNFR1) and requires the kinase activity of RIPK1 and RIPK3. Necroptosis is often accompanied by the release of damage-associated molecular patterns (DAMPs) and cytokines, which induce an immune-inflammatory response and exacerbate tissue damage[12, 13]. In contrast to the necroptosis pathway, the defining event in the mitochondrial-mediated cell death pathway is the calcium (Ca2+)-dependent opening of the mitochondrial permeability transition pore (mPTP), which is located in the inner mitochondrial membrane (IMM)[14]. Griffiths and Halestrap confirmed that mitochondrial permeability transition (MPT) occurs during reperfusion of the ischemic heart[15]. During reperfusion, mitochondria with reduced electron transfer capacity due to ischemia are at high risk of PTP opening due to elevated Ca and Pi levels, ROS bursts, and acidosis clearance. The fate of the cell is determined by the degree of PTP opening. If it is minimal, the cell may recover and survive; if it is moderate, the cell may undergo programmed cell death even if energy production is adequate; and if it is severe, the cell will die of necrosis due to insufficient energy production[16]. Necroptosis is a type of receptor-mediated cell death that occurs following the recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). DAMPs cause MIRI through the activation of the transcription factor NF-κB[17]. Ferroptosis is a process in which iron-dependent ROS accumulation exceeds the ability of the cell to maintain REDOX homeostasis, leading to lipid peroxidation and ultimately cell death[18]. Notably, there is evidence that iron and lipid peroxidation are required for ferroptosis but not for cell rupture[19]. During MIRI, increased intracellular iron concentrations and lipid peroxidation may be important for the increase in the infarct size during reperfusion[20]. Ferroptosis occurs during the reperfusion phase but not the ischemic phase, the levels of the key ferroptosis enzymes GPX4 and long-chain fatty acid coenzyme a ligase 4 (ACSL4) are significantly regulated only during reperfusion and are accompanied by elevated iron concentrations and malondialdehyde (MDA) levels[21]. During the reperfusion phase, mitochondrial respiration is enhanced, which triggers ROS release and ferroptosi22s[22]. Although ROS may cause other forms of cell death, they do not cause autosis[23, 24].
It has now been shown that autosis occurs in MIRI, and autosis is a new form of autophagic cell death; therefore, we will first examine autophagy[23, 25, 26]. Autophagy is involved in a variety of cellular functions, including the maintenance of homeostasis, metabolism, and defense mechanisms, and has traditionally been classified as a beneficial (adaptive) process associated with cellular function and survival[27]. However, many morphological features of autophagy are often observed in dead cells; therefore, whether autophagy promotes or inhibits cell death has been a topic of debate[24, 26, 27]. Many researchers have shown that autophagy has dual roles: an increase in autophagy promotes cell survival under many stress conditions, but excessive autophagy leads to cell death under certain conditions. For instance, during MIRI, autophagy has dual roles and can support survival or death[28, 29]. During myocardial reperfusion, an inadequate exogenous energy supply induces cardiomyocytes through adenosine phosphate-activated protein (adenosine monophosphate-activated protein kinase, AMPK), which mediates mammalian target of rapamycin (mTOR)-dependent autophagy to supply amino acids and fatty acids to maintain cell survival and thus has a cytoprotective effect[30, 31]. During myocardial reperfusion, the restoration of the energy supply inhibits the AMPK/mTOR signaling pathway, and processes such as the inflammatory response and oxidative stress induce excessive autophagy in cardiomyocytes by upregulating the expression of the autophagy-associated protein Beclin 1, which leads to autophagic cell death[32, 33]. In 2013, Yang Liu et al. introduced the term "autosis," which is a new form of autophagic cell death, and provided new insights into the direct link between dysregulated autophagy and cell death[23].
Autophagic cell death and autosis
Autophagic cell death
In yeast and mammals, there are three types of autophagy (macroautophagy, microautophagy, and chaperone-mediated autophagy), and autophagy is typically referred to as macroautophagy[27]. Macroautophagy begins with the formation of autophagic vesicles by a series of autophagy-related (ATG) proteins at the assembly sites, and substrate molecules are recruited to form an autophagosome with a double-membrane structure through vesicle expansion and closure. Subsequently, the outer membrane of the autophagosome fuses with the lysosomal or vesicular membrane, releasing the contents into the lysosomal lumen or vesicle as single-membrane autophagic vesicles and degrading them into small molecules for recycling by acidic hydrolases[34]. Autophagy is regulated at multiple steps from autophagosome formation to lysosomal degradation, and under certain conditions, autophagy may become dysregulated and lead to autophagic cell death[35]. Historically, three types of programmed cell death have been defined based on morphological criteria: Type I apoptotic cell death, Type II autophagic cell death, and Type III, which includes necrosis and cytoplasmic cell death[36]. Autophagic cell death was initially defined as a type of cell death that occurred in the absence of chromatin condensation and was accompanied by massive autophagic vacuolization of the cytoplasm[37].
Autosis
Autosis was first identified in HeLa cells and later reported in mouse kidneys during ischemia‒reperfusion and newborn rat brains during hypoxia–ischemia[23, 38]. Recently, autosis has been observed in cardiomyocytes during late reperfusion[25]. To date, the best method for determining autosis is electron microscopy, and immunofluorescence assays can also help to differentiate cells undergoing autosis by detecting fragmented endoplasmic reticulum or mitochondria with nuclear depressions[26]. The development of autosis is associated with dysregulated autophagy and cannot be prevented by inhibiting apoptosis or necrosis[35]; it can be inhibited by knocking down ATG13 or ATG14 and by treatment with the autophagy inhibitor 3-methyladenine (3MA)[23, 35]. Chemical inhibitors of autophagosome-lysosome fusion, such as bafilomycin A1, also do not inhibit autosis[23, 25]. These features of autosis may support the hypothesis that excessive autophagosome accumulation rather than excessive lysosomal degradation can induce autosis[35]. Autosis differs from other forms of cell death, such as necrosis and apoptosis, and is characterized by the presence of unique morphological and biochemical features[39]. According to electron microscopic observations, autosis involves a unique morphology (Fig. 1); during the early stage (phase 1a), the formation of large numbers of autophagosomes and autophagic lysosomes, electron-dense mitochondria, convoluted nuclear membranes, and a dilated and fragmented endoplasmic reticulum. In the middle stage (stage 1b), the inner and outer nuclear membranes are separated, and a dense membrane-bound region is formed. In the late stage (stage 2), focal balloon-like changes in the perinuclear space, mitochondrial swelling, and the beginning of the disappearance of the endoplasmic reticulum, autophagosomes, and autophagic lysosomes occur[23]. Thus, autosis fulfills the two basic criteria proposed by the Nomenclature Committee on Cell Death: it is inhibited by inhibition of the autophagy pathway and is characterized by a lack of apoptosis and necrosis[40].

During the early phase of autosis, the numbers of autophagosomes (APs), autolysosomes (ALs), and empty vacuoles (EVs) are drastically increased, and separation of the inner and outer nuclear membranes is observed. The later phase is characterized by focal nuclear concavity, focal ballooning of the perinuclear space (PNS), and the disappearance of subcellular organelles. ER endoplasmic reticulum, Nu nucleus
The relationship between autophagic cell death and autosis
Although autosis has certain characteristics, it shares several biochemical and morphological traits with other types of autophagic cell death[35]. Whether autosis is a type of autophagic cell death is one of the main questions surrounding it[35, 39,40,41]. Autosis acts independently of other modes of cell death, whereas autophagic cell death occurs primarily during the developmental phase and otherwise delays the onset of apoptosis or necrosis; hence, the hypothesis is that autosis is a more invasive form of autophagic cell death[35]. Because autosis is independent and specific, it is a desirable therapeutic target.
Autosis in ischemia‒reperfusion injury
Autophagosomes and autosis
A growing number of studies have demonstrated that an imbalance between autophagosome synthesis and degradation causes the excessive build-up of autophagosomes, which triggers autosis[26]. Jihoon Nah et al. examined autophagic flux-related parameters in mouse hearts following 30 min of ischemia and subsequent reperfusion to determine whether autosis could occur from an imbalance between the synthesis and destruction of cardiac autophagosomes during I/R. The findings showed that autophagic flux increased during ischemia and early reperfusion (up to 6 h); however, it deteriorated in the later stages of reperfusion, leading to further accumulation of autophagosomes in the border zone during myocardial infarction[25]. The area of myocardial infarction after 24 h of reperfusion was significantly larger than that after 6 h of reperfusion. Electron microscopy analysis revealed a time-dependent increase in the number of both autophagosomes and autolysosomes during MIRI. These results suggest that autophagy may be induced by increasing autophagosome accumulation, thereby promoting myocardial injury in the late phase of reperfusion. However, it has also been suggested that the death of cardiomyocytes in the late phase of reperfusion may be caused by a decrease in autophagic flux and can be exacerbated by the injection of Tat-Beclin 1[25]. Tat-Beclin 1 is a potent autophagy-inducing cell-permeable peptide that leads to the accumulation of autophagosomes rather than activating the lysosomal degradation pathway for the induction of autosis[23, 25], suggesting that cardiomyocyte autosis is caused by excessive autophagosome accumulation rather than a decrease in autophagic flux[25]. Therefore, suppressing the excessive accumulation of autophagosomes by inhibiting autophagy in the late phase of reperfusion may be effective in normalizing autophagic flux and thus preventing autosis[29].
To create autophagosomes, cells enlarge the membranes of the plasma membrane and organelles such as mitochondria and the endoplasmic reticulum[42]. The occurrence of autosis in cardiomyocytes increases the depletion of endoplasmic reticulum membranes for autophagosome biogenesis. The level of intracellular membrane proteins decreases in a dose-dependent manner when Tat-Beclin 1 is present. Additionally, high levels of autophagosome production deplete the plasma membrane, which causes intracellular organelle degradation and, eventually, organelle dysfunction. It has been suggested that the excessive consumption of organelle membranes is the cause of autosis[25, 26].
Rubicon and autosis
Rubicon directly interacts with Rab7 is a negative regulator of class III PI3K complex-2, and suppresses autophagy and endosomal transport[43,44,45]. In 2009, it was discovered that Rubicon interacted with Beclin 1 to suppress autophagy[46, 47]. According to recent research, Rubicon plays a role in cardiovascular disease in response to cardiac damage[25, 48].
Two hours after reperfusion, Rubicon protein levels increased, and six hours later, they started increasing dramatically[25]. However, during reperfusion, autophagic flux was inhibited in a time-dependent manner, and Rubicon levels increased. Following this inactivation, autophagosomes continued to form, and significant levels of autophagic vacuoles accumulated, which causes a deficiency in autophagosome cell-derived membranes, resulting in cardiotoxicity[25, 26]. Since autosis is accompanied by a lack of essential cytoplasmic membranes, the overproduction of autophagosomes due to an increase in Rubicon inhibits autophagosome degradation by lysosomes and recirculation and promotes autosis in the mouse heart during the late phase of reperfusion[25, 49]. Reductions in autophagosome formation during the late phase of ischemia/reperfusion, autophagy, and cardiac damage are all benefits of downregulating Rubicon during the late phase of ischemia/reperfusion[25]. Rubicon inhibits autophagic flux, which leads to a substantial increase in autophagosomes; however, autosis cannot be induced by Rubicon upregulation alone, indicating that additional mechanisms are needed to boost autophagosome production to induce autosis[25]. Jihoon Nah et al. demonstrated that the upregulation of Tfeb (transcription factor EB) was one of the mechanisms that promoted autosis during reperfusion to promote cardiac autosis in the later stages of reperfusion[50]. Interestingly, it has also been shown that the interaction of Rubicon with Beclin 1 leads to autophagosome accumulation by inhibiting autophagosome-lysosome fusion[47]. Therefore, eliminating Rubicon upregulation-induced autophagosome formation or inhibiting Rubicon upregulation and targeting of Beclin 1 are important for preventing autosis-mediated cardiac injury during the late phase of reperfusion.
Na+-K+-ATPase and autosis
Na+-K+-ATPase, which is a ubiquitous membrane pump involved in the pathophysiological mechanisms of human disorders, is necessary for autosis and creates a sodium‒potassium gradient across the plasma membrane by depleting ATP[23, 51]. Cardiac glycosides, which are a broad family of naturally occurring steroid chemicals that are antagonists of Na+-K+-ATPase, were first reported to be used in 1785 to treat heart illness[52]. Cardiac glycosides have been shown to prevent autosis in recent studies [3]. Jihoon Nah et al. injected the cardiac glycoside ouabain at 3 and 6 h after reperfusion to determine whether death in cardiomyocytes with advanced I/R was sensitive to cardiac glycosides. They then examined the MI and showed that autosis was reduced by inhibiting Na+-K+-ATPase activity to inhibit autosis could reduce MIRI[25]. However, treatment of cardiomyocytes with the calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N’,’N-tetraacetic acid tetrakis (acetoxymethyl ester) did not affect autosis[25]. This finding indicates that intracellular autosis signaling is not mediated by calcium levels[25, 37]. It has been found that activation of the local calcium-related signaling pathway PKA regulates autophagic flux by increasing autophagic lysosomes and decreasing autophagosomes[53]. Since excessive accumulation of autophagosomes can lead to autosis, we hypothesize that activation of PKA may have a protective effect on the cell by attenuating autosis, but this needs to be investigated extensively.
According to a recent study, autosis-inducing conditions such as ischemia, exercise, hunger, and I/R can cause physical interactions between Na+-K+-ATPase and Beclin 1, which can stimulate autosis[38]. Interestingly, cardiac glycosides decrease autosis and the degree of ischemic injury by decreasing the interaction between Na+-K+-ATPase and Beclin 1[23, 38]. However, how Na+-K+-ATPase interacts with Beclin 1 to control autophagy is unknown. This interaction may alter the ion-pumping activity or ion-exchange-dependent effects of Na+-K+-ATPase, which can cause autosis[26]. Another possibility is that Na+-K+-ATPase alters the function of Beclin 1, which causes autosis by influencing vesicular transport or autophagic activity[26]. Since Beclin 1 and Rubicon physically interact, it has been suggested that Beclin 1 plays a key role in autosis; however, more research is needed to determine how this interaction controls autosis, as well as the motifs and residues that it binds and where that binding occurs within the cell[47]. Downregulating Rubicon and inhibiting Na+-K+-ATPase did not synergistically reduce myocardial infarct size, and so further studies are needed to determine how the interaction between Beclin 1 and Na+-K+-ATPase or Rubicon affects autosis[25].
Tfeb and autosis
Tfeb is a major regulatory molecule associated with lysosomal biogenesis that coordinates autophagosome-lysosome fusion and substrate degradation by driving autophagy and lysosomal gene expression[53]. Tfeb binds a coordinated lysosomal expression and regulation (CLEAR) motif (GTCACGTGAC) and regulates lysosomal gene expression[54, 54].
Tfeb is activated during ischemia‒reperfusion in the mouse heart and remains activated during the later stages of reperfusion. In addition, Tfeb targets such as Beclin 1 and VPS11 are also upregulated in the late reperfusion phase[50]. Autophagosome accumulation and autosis were dysregulated in cardiomyocytes when Tfeb was activated in the presence of autophagic flux blockers. Conversely, after cardiac reperfusion, suppressing endogenous Tfeb decreased cardiomyocyte autosis in mice. Jihoon Nah et al. assessed the extent of myocardial infarction following the injection of 3,4-dimethoxychalone (3,4-DC), a chemical activator of Tfeb, after reperfusion in mice to further explore whether the activation of Tfeb in the late stage of reperfusion could exacerbate MIRI. The findings demonstrated that injecting 3,4-DC worsened myocardial damage. This finding suggests that MIRI is promoted by Tfeb activation during the late phase of reperfusion[50, 55]. Consequently, during the late phase of reperfusion, cardiomyocyte autosis is promoted by Tfeb activation[50]. Tfeb knockdown reduces autophagosome and autophagic lysosome accumulation during reperfusion, which decreases cardiomyocyte autosis but exacerbates myocardial infarction[50]. This finding suggests that Tfeb is beneficial in the early stages of ischemia and reperfusion and that its upregulation in the later stages of ischemia and reperfusion is functionally harmful but that sustained downregulation throughout ischemia and reperfusion may also be detrimental[50]. Tfeb activity appears to be time dependent, similar to the role of autophagy in ischemia and reperfusion[25]. Tfeb should therefore be targeted only in the later phases of reperfusion to prevent autosis but not autophagy[50]. This group also reported that Tfeb overexpression enhanced high-dose Tat-Beclin 1-induced cardiomyocyte autosis, whereas Atg7 knockdown reversed Tfeb overexpression-induced cardiomyocyte death[25, 50].
Sodium–glucose cotransporter 2 inhibitors and autosis
SGLT2 inhibitors have shown unprecedented cardiac and renal benefits in large-scale clinical trials in patients with type 2 diabetes mellitus with preexisting cardiovascular disease or multiple cardiovascular risk factors[56, 57]. It is unknown how SGLT2, which is mostly expressed in the kidney, protects the cardiovascular system[58,59,60,61]. The theory that SGLT2 inhibitors may protect the heart by promoting autophagy was proposed by Avogaro A, Packer M, et al.[62,63,64]. An emerging hypothesis is that SGLT2 inhibitors can directly inhibit the Na+/H+ exchanger (NHE) 1 isoform in the myocardium[58, 61]. NHE1 is predominantly expressed in cardiomyocytes, and its activity is markedly increased in pathological conditions such as diabetes mellitus, heart failure, and acute ischemia‒reperfusion injury[65].
According to a recent study, the primary target by which SGLT2 inhibitors act on cardiomyocytes is NHE1[67]. NHE1 knockdown ameliorates MIRI in mice, and NHE1 activation increases the intracellular sodium load in cardiomyocytes, which causes calcium overload during ischemia‒reperfusion and exacerbates reperfusion injury[67]. Autosis is strongly stimulated by ischemia and nutrient and glucose deprivation, and the SGLT2 inhibitor empagliflozin (EMPA) attenuates excessive autosis by directly inhibiting NHE1 activity in cardiomyocytes. NHE1 knockdown significantly reversed glucose deprivation-induced autosis. Furthermore, a study demonstrated that the cardioprotective effect of EMPA was achieved by reducing the excessive accumulation of autophagosomes[67]. Consequently, NHE1 is a target by which EMPA can reduce autosis in cardiomyocytes and exert cardioprotective effects on a mouse model of MI without diabetes. By creating cardiac-specific NHE1 inhibitors, autosis can be reduced for the benefit of MIRI patients.
In another study, cardiomyocyte-specific NHE1-knockout (NHE1 cKO) mice did not exhibit cardioprotection in a model of nondiabetic myocardial infarction with acute hyperglycemia, and EMPA exerted significant cardioprotective effects by inhibiting autophagy levels by targeting Beclin 1 but not NHE1. Additionally, EMPA decreased autosis and exerted cardioprotective effects by reversing the autophagic cell death caused by Tat-beclin1 or GD in cardiomyocytes[68]. SGLT2 inhibitors can effectively ameliorate myocardial injury during nondiabetic myocardial infarction with acute hyperglycemia by inhibiting Beclin 1-dependent autophagy in cardiomyocytes rather than by targeting NHE1[68]. The reason for this is that diabetes induces a process that prevents EMPA from binding to NHE1 and exerting its effect. Beclin 1 is therefore a therapeutic target for EMPA in the treatment of acute hyperglycemia during nondiabetic myocardial infarction with acute hyperglycemia.
Cariporide is an NHE1 inhibitor that is used to prevent or reduce ventricular fibrillation when used in acute therapy, and it has also been shown to be cardioprotective during IR in vivo and in isolated hearts[69,70,71,72]. The cardioprotective mechanism involves attenuating myocardial injury by regulating cellular pH, reducing excess sodium entry into cardiomyocytes, reducing calcium overload via the Na+/Ca+ exchanger (NCX), and reducing myocardial infarct size but does not involve attenuating autosis[73, 74]. Its protective effect on the heart occurs by regulating cellular pH, reducing excessive sodium entry into cardiomyocytes, and reducing calcium overload through NCX to reduce myocardial injury and myocardial infarct size but does not involve reducing autosis [73, 74]. Cariporide reduces the incidence of myocardial infarction, but there is an adverse effect of cerebrovascular occlusion leading to ischemic stroke[73].
Conclusion
In summary, autosis is a new form of cell death that occurs in the late stages of myocardial reperfusion and has therapeutic potential in MIRI. Further studies on the mechanism of autosis occurrence will not only contribute to a clearer understanding of the relationship between autophagy and autosis but also provide new strategies for treating MIRI in the clinic.
Clinical significance
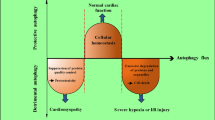
Studies have demonstrated that the activation of autophagy in early ischemia protects the myocardium, and overactivation of autophagy can occur in the reperfusion stage; overactivation of autophagy can exacerbate MIRI by inducing autophagic cell death (Fig. 2). Thus, understanding the point at which autophagy shifts from protecting the heart to harming the heart is essential for the medical treatment of MIRI.

The occurrence of autophagy during MIRI. During myocardial ischemia, ATP is deficient, and ADP levels are increased, which decreases the ATP/ADP ratio, leading to the activation of AMPK, which activates ULK1, thereby initiating autophagy. Moreover, AMPK can induce autophagy by inhibiting the activity of mTOR, and the induction of autophagy during the ischemic phase of myocardial ischemia exerts protective effects on the myocardium. During myocardial reperfusion, the energy supply is restored to the myocardium during the reperfusion phase; therefore, autophagy is initiated by the large amounts of reactive oxygen species generated during reperfusion, and Bcl-2 protein expression is decreased and attenuates the inhibition of the Beclin 1 protein, which activates the Beclin 1-dependent autophagy pathway, leading to excessive organelle and protein depletion and ultimately causing autophagic death
Autosis is a novel form of autophagic cell death induced by high doses of autophagy-inducing peptides, starvation, and ischemia-hypoxia (Fig. 3). Because autosis can be inhibited by autophagy inhibitors but cannot be reversed by apoptosis or necrosis inhibitors, the discovery that autosis is involved in MIRI is highly clinically relevant. Therefore, the effects of using autophagy inhibitors in combination with other medications that prevent cell death during the late phase of reperfusion may be additive. The upregulation of Tfeb and Rubicon during the late phase of reperfusion results in autosis by inactivating autophagic flux and autophagosome accumulation. Rubicon upregulation induces autosis mainly by promoting autophagosome accumulation. As a result, interventions that block the upregulation of Rubicon and prevent the excessive build-up of autophagosomes during the late phase of reperfusion may be cardioprotective. Tfeb upregulation, however, protects the myocardium during the early stages of ischemia, but it can cause myocardial damage during the late stages of reperfusion. For this reason, the exact time at which Tfeb changes from having a beneficial to a harmful effect on the heart is crucial when treating MIRI patients, and altering the upregulation of Tfeb should be used to inhibit autosis only in the late phase of reperfusion. Beclin 1 may play a key role in autosis, and it has been demonstrated that Rubicon and Na+-K+-ATPase can interact with this factor to induce autosis. However, but the exact mechanism remains to be investigated, and further studies will be conducted to determine the regulatory relationship between Rubicon and Na+-K+-ATPase and Beclin 1 and identify a more effective target for the treatment of MIRI in the clinic. Since the onset of autosis during MIRI is accompanied by the activation of NHE1, NHE1 could also be a novel target for the development of cardiac-specific NHE1 inhibitors to attenuate autosis. It has been demonstrated that cardiac glycosides inhibit NHE1 to reduce autosis during MIRI; however, since arrhythmias are a potential side effect of cardiac glycosides and the infarcted heart is prone to them, cardiac glycosides may not be the best treatment for MIRI[25]. Autosis is also observed in various cardiovascular disorders; the coadministration of copper and homocysteine causes autosis and cardiomyocyte apoptosis in a p62-dependent manner[76]. The mechanisms underlying autosis are not entirely understood, even though many factors have been linked to this process [76]. There are no other effective treatments available to prevent cell death in ischemic regions. Therefore, the development of new drugs targeting the late phase of reperfusion has important clinical implications for the treatment of autosis.

Mechanisms of autosis in MIRI. During MIRI, the upregulation of Tfeb and Rubicon inhibits the fusion of autophagosomes with lysosomes, leading to excessive autophagosome accumulation and causing autosis. The occurrence of autosis depends on Na+-K+-ATPase. The interaction of Na+-K+-ATPase with Beclin 1 and Rubicon can promote autosis, while cardiac glycosides, the Na+-K+-ATPase antagonist, can inhibit the interaction and thus inhibit autosis. EMPA inhibits autosis by targeting NHE1. The Na+-K+-ATPase antagonists cardiac glycosides inhibit this interaction and autosis, and EMPA inhibits autosis by targeting NHE1
Data availability
No datasets were generated or analysed during the current study.
References
Wang Y, Hou M, Duan S et al (2022) Macrophage-targeting gene silencing orchestrates myocardial microenvironment remodeling toward the anti-inflammatory treatment of ischemia-reperfusion (IR) injury. Bioact Mater 17:320–333. https://doi.org/10.1016/j.bioactmat.2022.01.026
Chen M, Li X, Yang H et al (2020) Hype or hope: Vagus nerve stimulation against acute myocardial ischemia-reperfusion injury. Trends Cardiovasc Med 30:481–488. https://doi.org/10.1016/j.tcm.2019.10.011
Carreira RS, Facundo HTF, Kowaltowski AJ (2005) Mitochondrial K+ transport and cardiac protection during ischemia/reperfusion. Braz J Med Biol Res 38:345–352. https://doi.org/10.1590/S0100-879X2005000300004
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion—from mechanism to translation. Nat Med. https://doi.org/10.1038/nm.2507
Wang R, Wang M, He S et al (2020) Targeting calcium homeostasis in myocardial ischemia/reperfusion injury: an overview of regulatory mechanisms and therapeutic reagents. Front Pharmacol 11:872. https://doi.org/10.3389/fphar.2020.00872
Kormos A, Nagy N, Acsai K et al (2014) Efficacy of selective NCX inhibition by ORM-10103 during simulated ischemia/reperfusion. Eur J Pharmacol 740:539–551. https://doi.org/10.1016/j.ejphar.2014.06.033
Lefer DJ, Nichols CG, Coetzee WA (2009) Sulfonylurea receptor 1 subunits of ATP-sensitive potassium channels and myocardial ischemia/reperfusion injury. Trends Cardiovasc Med 19:61–67. https://doi.org/10.1016/j.tcm.2009.04.008
Mishra PK, Adameova A, Hill JA et al (2019) Guidelines for evaluating myocardial cell death. Am J Physiol Heart Circ Physiol 317:H891–H922. https://doi.org/10.1152/ajpheart.00259.2019
Condorelli G, Roncarati R, Ross J et al (2001) Heart-targeted overexpression of caspase3 in mice increases infarct size and depresses cardiac function. Proc Natl Acad Sci U S A 98:9977–9982. https://doi.org/10.1073/pnas.161120198
Jeremias I, Kupatt C, Martin-Villalba A et al (2000) Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia. Circulation 102:915–920. https://doi.org/10.1161/01.cir.102.8.915
Chen Z, Chua CC, Ho YS et al (2001) Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol 280:H2313-2320. https://doi.org/10.1152/ajpheart.2001.280.5.H2313
Zhang X, Ren Z, Xu W, Jiang Z (2022) Necroptosis in atherosclerosis. Clin Chim Acta 534:22–28. https://doi.org/10.1016/j.cca.2022.07.004
Grootjans S, Vanden Berghe T, Vandenabeele P (2017) Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ 24:1184–1195. https://doi.org/10.1038/cdd.2017.65
Del Re DP, Amgalan D, Linkermann A et al (2019) Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev 99:1765–1817. https://doi.org/10.1152/physrev.00022.2018
Griffiths EJ, Halestrap AP (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307(Pt 1):93–98. https://doi.org/10.1042/bj3070093
Honda HM, Korge P, Weiss JN (2005) Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci 1047:248–258. https://doi.org/10.1196/annals.1341.022
Liu Y, Li L, Wang Z et al (2023) Myocardial ischemia-reperfusion injury Molecular mechanisms and prevention. Microvascular Res 149:104565. https://doi.org/10.1016/j.mvr.2023.104565
Sparvero LJ, Tian H, Amoscato AA et al (2021) Direct mapping of phospholipid ferroptotic death signals in cells and tissues by gas cluster ion Beam Secondary Ion Mass Spectrometry (GCIB-SIMS). Angew Chem Int Ed Engl 60:11784–11788. https://doi.org/10.1002/anie.202102001
Riegman M, Sagie L, Galed C et al (2020) Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol 22:1042–1048. https://doi.org/10.1038/s41556-020-0565-1
Fang X, Wang H, Han D et al (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116:2672–2680. https://doi.org/10.1073/pnas.1821022116
Tang L-J, Luo X-J, Tu H et al (2021) Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol 394:401–410. https://doi.org/10.1007/s00210-020-01932-z
Wang H, Liu C, Zhao Y, Gao G (2020) Mitochondria regulation in ferroptosis. Eur J Cell Biol 99:151058. https://doi.org/10.1016/j.ejcb.2019.151058
Liu Y, Shoji-Kawata S, Sumpter RM et al (2013) Autosis is a Na+, K+-ATPase–regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia–ischemia. Proc Natl Acad Sci U S A 110:20364–20371. https://doi.org/10.1073/pnas.1319661110
Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22:367–376. https://doi.org/10.1038/cdd.2014.143
Nah J, Zhai P, Huang C-Y et al (2020) Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J Clin Invest 130:2978–2991. https://doi.org/10.1172/JCI132366
Nah J, Zablocki D, Sadoshima J (2020) Autosis a new target to prevent cell death. JACC Basic Transl Sci 5:857–869. https://doi.org/10.1016/j.jacbts.2020.04.014
Lin X, Xiao W, Xiao L, Liu M (2018) Molecular mechanisms of autophagy in cardiac ischemia/reperfusion injury (Review). Mol Med Report. https://doi.org/10.3892/mmr.2018.9028
Shi B, Ma M, Zheng Y et al (2019) mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J Cellular Physiol 234:12562–12568. https://doi.org/10.1002/jcp.28125
Matsui Y, Takagi H, Qu X et al (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100:914–922. https://doi.org/10.1161/01.RES.0000261924.76669.36
Zhang T, Guo J, Gu J et al (2019) Protective role of mTOR in liver ischemia/reperfusion injury: involvement of inflammation and autophagy. Oxid Med Cell Longev 2019:7861290. https://doi.org/10.1155/2019/7861290
Zhao D, Yang J, Yang L (2017) Insights for oxidative stress and mTOR signaling in myocardial ischemia/reperfusion injury under diabetes. Oxid Med Cell Longev 2017:6437467. https://doi.org/10.1155/2017/6437467
Kong L, Xiong F, Sun N et al (2020) CaMKIIδ inhibition protects against myocardial ischemia/reperfusion injury: Role of Beclin-1-dependent autophagy. Eur J Pharmacol 886:173539. https://doi.org/10.1016/j.ejphar.2020.173539
Maejima Y, Isobe M, Sadoshima J (2016) Regulation of autophagy by Beclin 1 in the heart. J Mol Cell Cardiol 95:19–25. https://doi.org/10.1016/j.yjmcc.2015.10.032
Nakatogawa H (2020) Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol 21:439–458. https://doi.org/10.1038/s41580-020-0241-0
Nah J, Zablocki D, Sadoshima J (2022) The role of autophagic cell death in cardiac disease. J Mol Cell Cardiol 173:16–24. https://doi.org/10.1016/j.yjmcc.2022.08.362
Schwartz LM (2021) Autophagic cell death during development—ancient and mysterious. Front Cell Dev Biol 9:656370. https://doi.org/10.3389/fcell.2021.656370
Denton D, Kumar S (2019) Autophagy-dependent cell death. Cell Death Differ 26:605–616. https://doi.org/10.1038/s41418-018-0252-y
Fernández ÁF, Liu Y, Ginet V et al (2020) Interaction between the autophagy protein Beclin 1 and Na+, K+-ATPase during starvation, exercise, and ischemia. JCI Insight. https://doi.org/10.1172/jci.insight.133282
Nah J, Fernández ÁF, Kitsis RN et al (2016) Does autophagy mediate cardiac myocyte death during stress? Circ Res 119:893–895. https://doi.org/10.1161/CIRCRESAHA.116.309765
Galluzzi L, Vitale I, Abrams JM et al (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19:107–120. https://doi.org/10.1038/cdd.2011.96
Wu X, Liu Z, Yu X et al (2021) Autophagy and cardiac diseases: Therapeutic potential of natural products. Med Res Rev 41:314–341. https://doi.org/10.1002/med.21733
Zhao YG, Liu N, Miao G et al (2018) The ER contact proteins VAPA/B interact with multiple autophagy proteins to modulate autophagosome biogenesis. Curr Biol 28:1234-1245.e4. https://doi.org/10.1016/j.cub.2018.03.002
Nah J, Zablocki D, Sadoshima J (2021) The roles of the inhibitory autophagy regulator Rubicon in the heart: A new therapeutic target to prevent cardiac cell death. Exp Mol Med 53:528–536. https://doi.org/10.1038/s12276-021-00600-3
Sun Q, Westphal W, Wong KN et al (2010) Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A 107:19338–19343. https://doi.org/10.1073/pnas.1010554107
Chang C, Young LN, Morris KL et al (2019) Bidirectional control of autophagy by BECN1 BARA domain dynamics. Mol Cell 73:339-353.e6. https://doi.org/10.1016/j.molcel.2018.10.035
Zhong Y, Wang QJ, Li X et al (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 11:468–476. https://doi.org/10.1038/ncb1854
Kaur S, Changotra H (2020) The beclin 1 interactome: Modification and roles in the pathology of autophagy-related disorders. Biochimie 175:34–49. https://doi.org/10.1016/j.biochi.2020.04.025
Liu X, Zhang S, An L et al (2019) Loss of Rubicon ameliorates doxorubicin-induced cardiotoxicity through enhancement of mitochondrial quality. Int J Cardiol 296:129–135. https://doi.org/10.1016/j.ijcard.2019.07.074
Gatica D, Chiong M, Lavandero S, Klionsky DJ (2015) Molecular mechanisms of autophagy in the cardiovascular system. Circ Res 116:456–467. https://doi.org/10.1161/CIRCRESAHA.114.303788
Nah J, Sung E-A, Zhai P et al (2022) Tfeb-mediated transcriptional regulation of autophagy induces autosis during ischemia/reperfusion in the heart. Cells 11:258. https://doi.org/10.3390/cells11020258
Clausen MV, Hilbers F, Poulsen H (2017) The structure and function of the Na, K-ATPase isoforms in health and disease. Front Physiol 8:371. https://doi.org/10.3389/fphys.2017.00371
Gross NB, Abad N, Lichtstein D et al (2019) Endogenous Na+, K+-ATPase inhibitors and CSF [Na+] contribute to migraine formation. PLoS ONE 14:e0218041. https://doi.org/10.1371/journal.pone.0218041
PKA compartmentalization links cAMP signaling and autophagy | Cell Death & Differentiation. https://www.nature.com/articles/s41418-021-00761-8. Accessed 20 Feb 2024
Nirmala JG, Lopus M (2020) Cell death mechanisms in eukaryotes. Cell Biol Toxicol 36:145–164. https://doi.org/10.1007/s10565-019-09496-2
Di Malta C, Cinque L, Settembre C (2019) Transcriptional regulation of autophagy: mechanisms and diseases. Front Cell Dev Biol 7:114. https://doi.org/10.3389/fcell.2019.00114
Xiao L, Nie X, Cheng Y, Wang N (2021) Sodium-Glucose Cotransporter-2 inhibitors in vascular biology: cellular and molecular mechanisms. Cardiovasc Drugs Ther 35:1253–1267. https://doi.org/10.1007/s10557-021-07216-9
Neal B, Perkovic V, Mahaffey KW et al (2017) Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377:644–657. https://doi.org/10.1056/NEJMoa1611925
Verma S, Mazer CD, Fitchett D et al (2018) Empagliflozin reduces cardiovascular events, mortality and renal events in participants with type 2 diabetes after coronary artery bypass graft surgery: subanalysis of the EMPA-REG OUTCOME® randomised trial. Diabetologia 61:1712–1723. https://doi.org/10.1007/s00125-018-4644-9
Uthman L, Baartscheer A, Bleijlevens B et al (2018) Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 61:722–726. https://doi.org/10.1007/s00125-017-4509-7
Bell RM, Yellon DM (2018) SGLT2 inhibitors: hypotheses on the mechanism of cardiovascular protection. Lancet Diabetes Endocrinol 6:435–437. https://doi.org/10.1016/S2213-8587(17)30314-5
Lytvyn Y, Bjornstad P, Udell JA et al (2017) Sodium glucose cotransporter-2 inhibition in heart failure: potential mechanisms, clinical applications, and summary of clinical trials. Circulation 136:1643–1658. https://doi.org/10.1161/CIRCULATIONAHA.117.030012
Packer M, Anker SD, Butler J et al (2017) Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: proposal of a novel mechanism of action. JAMA Cardiol 2:1025–1029. https://doi.org/10.1001/jamacardio.2017.2275
Avogaro A, Fadini GP, Del Prato S (2020) Reinterpreting cardiorenal protection of renal sodium-glucose cotransporter 2 inhibitors via cellular life history programming. Diabetes Care 43:501–507. https://doi.org/10.2337/dc19-1410
Packer M (2020) Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium-glucose cotransporter 2 inhibitors. Eur J Heart Fail 22:618–628. https://doi.org/10.1002/ejhf.1732
Packer M (2020) SGLT2 inhibitors produce cardiorenal benefits by promoting adaptive cellular reprogramming to induce a state of fasting mimicry: a paradigm shift in understanding their mechanism of action. Diabetes Care 43:508–511. https://doi.org/10.2337/dci19-0074
Packer M (2017) Activation and inhibition of sodium-hydrogen exchanger is a mechanism that links the pathophysiology and treatment of diabetes mellitus with that of heart failure. Circulation 136:1548–1559. https://doi.org/10.1161/CIRCULATIONAHA.117.030418
Jiang K, Xu Y, Wang D et al (2022) Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis. Protein Cell 13:336–359. https://doi.org/10.1007/s13238-020-00809-4
Wang Y, Meyer JW, Ashraf M, Shull GE (2003) Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res 93:776–782. https://doi.org/10.1161/01.RES.0000094746.24774.DC
Deng R, Jiang K, Chen F et al (2022) Novel cardioprotective mechanism for Empagliflozin in nondiabetic myocardial infarction with acute hyperglycemia. Biomed Pharmacother 154:113606. https://doi.org/10.1016/j.biopha.2022.113606
Wajima T, Beguier B, Yaguchi M (2004) Effects of cariporide (HOE642) on myocardial infarct size and ventricular arrhythmias in a rat ischemia/reperfusion model: comparison with other drugs. Pharmacology 70:68–73. https://doi.org/10.1159/000074670
Le Grand B, Pignier C, Létienne R et al (2009) Na+ currents in cardioprotection: better to be late. J Med Chem 52:4149–4160. https://doi.org/10.1021/jm900296e
Rodríguez-Sinovas A, García-Dorado D, Padilla F et al (2003) Pre-treatment with the Na+/H+ exchange inhibitor cariporide delays cell-to-cell electrical uncoupling during myocardial ischemia. Cardiovasc Res 58:109–117. https://doi.org/10.1016/s0008-6363(02)00840-4
Linz WJ, Busch AE (2003) NHE-1 inhibition: from protection during acute ischaemia/reperfusion to prevention/reversal of myocardial remodelling. Naunyn Schmiedebergs Arch Pharmacol 368:239–246. https://doi.org/10.1007/s00210-003-0808-2
Chang HB, Gao X, Nepomuceno R et al (2015) Na+/H+ exchanger in regulation of platelet activation and paradoxical effects of cariporide. Exp Neurol 272:11–16. https://doi.org/10.1016/j.expneurol.2014.12.023
Karmazyn M, Sostaric JV, Gan XT (2001) The myocardial Na+/H+ exchanger: a potential therapeutic target for the prevention of myocardial ischaemic and reperfusion injury and attenuation of postinfarction heart failure. Drugs 61:375–389. https://doi.org/10.2165/00003495-200161030-00006
He L, Chu Y, Yang J et al (2022) Activation of autophagic flux maintains mitochondrial homeostasis during cardiac ischemia/reperfusion injury. Cells 11:2111. https://doi.org/10.3390/cells11132111
Kriel J, Loos B (2019) The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ 26:640–652. https://doi.org/10.1038/s41418-018-0267-4
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (Grant Nos.82170260).
Funding
This article was funded by National Natural Science Foundation of China, 82170260, 82170260, 82170260, 82170260, 82170260, 82170260, 82170260.
Author information
Authors and Affiliations
Contributions
Xiaoting Yang contributed toward conceptualization, and writing-original draft preparation. Gang Zhou and Dong Zhang contributed toward data curation. Qingzhuo Yang and Yanfang Liu contributed toward visualization and investigation. Yi Li contributed toward production of matching images. Wu Hui contributed toward supervision. Xiaoting Yang contributed toward writing- reviewing and editing.
Corresponding author
Ethics declarations
Conflict of interest
No potential conflict of interest was reported by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, X., Wu, H., Zhou, G. et al. Autosis: a new form of cell death in myocardial ischemia–reperfusion injury. Mol Cell Biochem (2024). https://doi.org/10.1007/s11010-024-04988-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11010-024-04988-0