
Abstract
Cell death is a fundamental physiological process in all living organisms. Processes such as embryonic development, organ formation, tissue growth, organismal immunity, and drug response are accompanied by cell death. In recent years with the development of electron microscopy as well as biological techniques, especially the discovery of novel death modes such as ferroptosis, cuprotosis, alkaliptosis, oxeiptosis, and disulfidptosis, researchers have been promoted to have a deeper understanding of cell death modes. In this systematic review, we examined the current understanding of modes of cell death, including the recently discovered novel death modes. Our analysis highlights the common and unique pathways of these death modes, as well as their impact on surrounding cells and the organism as a whole. Our aim was to provide a comprehensive overview of the current state of research on cell death, with a focus on identifying gaps in our knowledge and opportunities for future investigation. We also presented a new insight for macroscopic intracellular survival patterns, namely that intracellular molecular homeostasis is central to the balance of different cell death modes, and this viewpoint can be well justified by the signaling crosstalk of different death modes. These concepts can facilitate the future research about cell death in clinical diagnosis, drug development, and therapeutic modalities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The lifespan of human cells can vary depending on their type, ranging from a few days to several years, as all cells have their own unique life cycle [1]. For example, epithelial cells renew themselves faster [2], while neuronal cells can work extra-long because of their unique DNA repair mechanisms, maintaining a healthy mitochondrial population, effective removal of aggregated proteins and aged or defective organelles [3, 4]. Cell death is a fundamental process in all living organisms, serving both physiological and pathological functions [5]. It plays an important role in physiological processes such as growth and development, organ formation, body aging, tissue renewal, immune modulation, and drug response [6]. Hundreds of millions of cells die and renew themselves every day in the human body, which is essential to maintain the health of the organism [7]. And humans have been generating new knowledge about the mechanisms of cell death and clearance [8].
There is a long-standing consensus in the research field that cell death is an inevitable consequence of cell life and is significant for understanding of the pathogenesis of various disorders and attempts to combat them [9]; therefore, research on cell death continues to evolve [10]. Cell death has been defined as the irreversible degeneration of important cellular functions (especially ATP production and retention of redox homeostasis), culminating in the loss of cellular integrity (permanent cell membrane permeability or cell fragmentation) [11]. One of the more authoritative definitions and explanations of cell death from a biochemical, morphological, and functional perspective is that of the Nomenclature Committee on Cell Death (NCCD) [8].
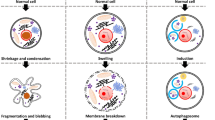
Cell death patterns are generally distinguished into accidental cell death (ACD) and regulated cell death (RCD) [8]. ACD refers to the physical disintegration of the plasma membrane in response to severe physical (e.g., mechanical stress, temperature, or osmotic forces), chemical (e.g., extreme pH changes), or mechanical (e.g., shear forces) stimuli, and the resulting almost instantaneous and uncontrollable cell death [12]. In contrast, RCD is a distinct form of cell death that results from the activation of one or more signal transduction modules, exhibiting unique cellular and morphological characteristics. As such, RCD is potentially modifiable through drug or gene intervention [13]. RCD was observed in dying cells of the toad as early as 1842 [14], a concept that was extensively studied with the discovery of apoptosis in 1972 [15]. In contrast, in strict physiological conditions, the organism deliberately eliminates redundant or irreversibly damaged cells through a process known as programmed cell death (PCD). This specific form of RCD provides programmed renewal for organismal development and tissue renewal [16]. Unlike other forms of cell death, PCD is not associated with disruptions to homeostasis and does not occur in response to the failure of adaptation to stressors. Recently, novel modes of cell death, such as ferroptosis and cuproptosis, have been discovered [12]. These novel cell death modes can also contribute to clinical diagnosis, drug development, and therapeutic modalities. In this review, we provided a systematic review of the currently discovered cell death modes, as shown in Fig. 1, and we also discussed the differences and associations among different cell deaths modes.

A landscape of the currently discovered cell death modes
Types of cell death
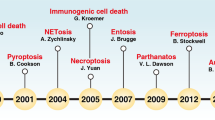
At the early research stage, limited by the inadequacy of microscopic techniques and studies on the regulation of gene coding, researchers’ understanding of the concept about cell death relied more on its macroscopic morphological changes (including the disposal mechanism of dead cells and their fragments) [17], and thus, classified cell death into three main different forms: (1) Type I cell death, represented by apoptosis, morphologically manifested mainly by cytoplasmic contraction, chromatin condensation, nuclear fragmentation, and membrane blistering, culminating in the formation of apparently intact apoptotic vesicles [18, 19], which are efficiently taken up by phagocytically active neighboring cells and degraded within lysosomes [20]; (2) type II cell death, represented by autophagy, manifests as extensive cytoplasmic vacuolization [21], characterized by phagocytic uptake and subsequent lysosomal degradation; (3) type III cell death, represented by necrosis, does not exhibit the distinctive features of type I or type II cell death and terminates with disposal of cell carcasses in the absence of significant phagocytosis and lysosomal involvement [22, 23]. However, this above classification has multiple limitations and does not accurately distinguish between specific cell death characteristics. Therefore, since 2005, NCCD has precisely defined the main cell death modes on the basis of genetics, biochemistry, pharmacology, function (rather than morphology), and identified criteria to identify dead cells as those with irreversible plasma membrane permeabilization or complete fragmentation [13, 24,25,26]. In order to make the research process of cell death clearer, here we listed the timeline of the discovery of various cell death modes (Fig. 2). And we reviewed them in detail below.

Timeline of the discovery of various cell death modes
Apoptosis
For a long time, apoptosis was considered as the only regulated cell death model [27]. Apoptosis currently refers to active, ordered cell death under physiological or pathological conditions in order to maintain the homeostasis of its own internal environment through genetic regulation and is a form of programmed cell death [18]. Apoptosis involves a series of gene regulation and its main morphological features cell rounding, nuclear contraction, membrane blistering, apoptotic bodies formation [28] and is often accompanied by caspase-3 activation, DNA fragmentation and phosphatidylserine exposure [29].
Apoptosis can be triggered by two different pathways: the mitochondrial pathway as well as the death receptor pathway. The mitochondrial pathway is regulated by members of the BCL-2 protein family [30], and cysteine proteases regulate the mitochondrial pathway when they act after intracellular sensors detect severe damage to the cell. The receptor death pathway is activated when members of the tumor necrosis factor (TNF) family bind to the “death receptor” (TNFR). The linkage of these receptors initiates the formation of a multi-lipoprotein death-inducing signaling complex. Cysteine proteases are central mediators of apoptosis. Activated CASP8 (receptor death pathway) and CASP9 (mitochondrial pathway) in turn activate CASP3, 6 and 7, and the proteases damage cells by cleaving large amounts of proteins, leading to apoptosis [31]. Apoptosis is now known to consist of two major isoforms: extrinsic and intrinsic apoptosis [32].
Extrinsic apoptosis
An RCD activated by perturbations of the extracellular microenvironment sensed by receptors on the cell membrane [33], mainly mediated by membrane receptors (especially cell surface death receptors (CD95) and TNF receptor superfamily member 1A (TNFR1), etc.) and driven by CASP8 and CASP10 [34]. In addition, the dependent receptor UNC5B and DCC may also activate Extrinsic apoptosis by activating CASP9 or death-associated protein kinase 1 (DAPK1), while CASP3 is considered to be a major player [35].
Intrinsic apoptosis
A form of RCD triggered by multiple microenvironmental perturbations such as mitochondrial stress, DNA damage, endoplasmic reticulum stress, mitotic defects, oxidative stress, and metabolic stress [36, 37]. Mitochondrial outer membrane permeabilization (MOMP), tightly controlled by the BCL-2 family, plays an important regulatory role by affecting the release of mitochondrial proteins (cytochrome C, somatic (CYCS), diablo IAP-binding mitochondrial protein (DIABLO), and HtrA serine peptidase 2 (HTRA2)), and subsequent activation of initiator caspases [38,39,40]. CASP3, CASP6, and CASP7 are considered to be co-effectors [41]. Intrinsic apoptosis includes two unique death modes, they are anoikis and mitotic death.
Anoikis
This is a specific form of Intrinsic apoptosis triggered by integrin-dependent anchorage deficiency [42]. This mode of death caused by the interaction of normal epithelial cells and extracellular matrix (ECM) was first reported in 1994 and was known as anoikis [43]. Anoikis prevents tumor-anchored proliferation and attachment to inappropriate stroma and is, therefore, usually considered as a tumor suppressor process [42]. Due to the unique mechanism of interaction between anoikis and ECM [44], this apoptotic modality is of great value in the study of tumor metastasis [45], immune infiltration [46], drug resistance [47], and disease prognosis [48]. Recent interesting studies have reported that cells adopt rounded morphologies and formed small hemispherical plasma membrane protrusions, which could promote the formation of plasma membrane-proximal signaling hubs, thereby conferring anoikis resistance [49].
Mitotic death
It is thought to be a specific mode of cell death (most commonly intrinsic apoptosis) driven by mitotic catastrophe. Because mitotic catastrophe does not always lead to RCD (but can also drive cellular senescence), it is not defined as a form of cell death. In 2018, the cell death nomenclature committee recommended using the term mitotic death to denote RCD driven by mitotic catastrophe [8]. Mitotic death was reported as early as 1968, when researchers found that X-irradiation in HeLa cells resulted in chromosomal aberrations and mitotic death [50]. Mitotic catastrophe is a regulated tumor suppressor mechanism that prevents the proliferation and survival of cells that are unable to complete mitosis due to extensive DNA damage, problems with the mitotic machinery, or failure of the mitotic checkpoint [51]. The failure of mitotic catastrophe is a critical event in tumor transformation and progression, as it leads to the generation or survival of polyploid and aneuploid cells [51]. On the other hand, the failure of mitotic catastrophe has emerged as an important mechanism of resistance to anticancer chemotherapeutic agents. This resistance mainly reflects the increased resistance of tumor cells to organism-induced intrinsic apoptosis [52].
Necroptosis
A chemical inhibitor of non-apoptotic cell death with therapeutic potential for ischemic brain injury was first reported in 2005 [53], and the identification of the pharmacological necrosis inhibitor-1 (RIPK1 inhibitor) drove the study of this mode of death and eventually defined this mode of RCD death distinct from apoptosis [54]. Necroptosis is currently considered to be an RCD mode triggered by extracellular or intracellular homeostatic perturbations that are heavily dependent on the activity of MLKL, RIPK3, and RIPK1 [55]. It is usually triggered by perturbations of the extracellular or intracellular microenvironment detected by specific death receptors (CD95 and TNFR1, etc.) or pathogen recognition receptors (TLR3 and TLR4, etc.) [56]. The main morphological features of necroptosis are cell swelling, rupture of plasma membrane, and moderate chromatin condensation [12, 53].
As mentioned previously, the biological consequences of TNFR1 signaling from cell survival and death can be accomplished through a variety of RCD processes, particularly apoptosis and necroptosis [57]. The differences between necroptosis and apoptosis are the period of loss of cell membrane integrity, changes in cellular volume and chromatine, and the signaling pathways involved [58]. In necroptosis, the integrity of the plasma membrane is lost in the early stage, allowing the influx of extracellular ions and fluids, which leads to the swelling of the cell and its organelles and mild chromatin condensation [59]. In apoptosis, the integrity of the plasma membrane persists until late stage in the process. Its morphological changes included decreased cell size and chromatin fragmentation [59]. Necroptosis is a cellular regulatory process which was initiated by the auto-transphosphorylation of RIPK1 and RIPK3, and the recruitment of mixed lineage kinase domain-like (MLKL) [60,61,62]. RIPK3-dependent necroptosis is usually activated in the inhibited state of the CASP apoptotic pathway, since it is considered as a complementary pathway to apoptosis. Meanwhile, necroptosis is also commonly considered as an intracellular defense mechanism in infection condition [63, 64].
In recent years, there have also been an increasing number of studies on necroptosis, a large part of which has focused on tumor-related studies [65,66,67]. Early studies reported that necroptosis acts as a tumor suppressor in most cases, but more cutting-edge studies have seen its bidirectional role in pathology studies [68]. Several chemo modulators of RIPK1 have been developed including Necrostatin-1 (Nec-1) and its derivatives (Nec-1 s), who have shown inhibition of TNFR1-driven death in vitro and in vivo [69, 70]. In addition, the regulation of the body’s immune microenvironment by necroptosis, such as the maintenance of adult T-cell homeostasis [71] and antitumor immunity [72] are of interest to clinicians. Its roles in inflammation [73], neurodevelopment [74], and death signaling crosstalk [75] are also current areas of high interest for research.
Necrosis
The term necrosis is very old, dating back more than 200 years [76], but it is often confused with the concept of tissue necrosis because they both described by the same word “necrosis” [77]. Necrosis is usually considered to be non-programmed and unregulated and is commonly seen in infectious or non-infectious diseases and cancer. Morphological features of necrosis include cell swelling, spillage of cell contents, loss of biofilm integrity, and dissipation of ion gradients [10]. What is more, it is often accompanied by loss of inter-nucleosomal DNA fragments and ATP depletion [78]. Therefore, necrosis is considered the only type of ADC and often refers to a mode of death that is not specifically defined.
The early phenomena of necrosis include increased intracellular calcium ion and reactive oxygen species concentrations, with the end result being irreversible cellular damage [79]. There is no clear cellular signaling mechanism for necrosis, and although there are recent reports of Ninj1 on cell membrane rupture [80], its unique function in cellular necrosis needs further confirmation. On the other hand, cell necrosis is often found in conjunction with diseases, such as stroke, neurodegenerative diseases, and cancer [81]. Notably some tissue damage in neurodegenerative diseases is actually caused by uncontrolled necrosis rather than any of the programmed cell death pathways.
Mitochondrial permeability transition (MPT)-driven necrosis
MPT was observed in the 1950s [82]. It is often observed during apoptosis, necroptosis, etc. [83] and was once considered a common mechanism of apoptosis and necroptosis [84]. In 2018, the Cell Death Nomenclature Committee defined MPT-driven necrosis as a specific form of RCD triggered by perturbations in the intracellular microenvironment and dependent on Cyclophilin D (CYPD) [8]. MPT-driven necrosis is profoundly involved in the etiology of several pathological conditions characterized by irrational loss of cells after mitosis, which may be closer to mitochondrial outer membrane permeabilization (MOMP)-dependent apoptosis [85]. MPT-driven necrosis is initiated when some injury leads to a sudden loss of osmotic homeostasis of the inner mitochondrial membrane. The manifest morphology is mainly that of necroptosis [85], characterized by mitochondrial membrane potential collapse, respiratory chain uncoupling, ATP depletion, and mitochondrial swelling [62].
At the biochemical level, MPT-driven necrosis is believed to occur after the permeability transition pore complex (PTPC), a supramolecular complex assembled at the junction between the inner and outer mitochondrial membranes, is opened [86]. Several molecules that can interact with PTPC have been shown to regulate MPT-driven necrosis, including members of the BCL-2 family (BAX, BAK, and BID) [87], DRP1 [88], P53 [89], and others.
Autophagy-dependent cell death
Autophagy is a pathway of catabolism, a central molecular pathway for maintaining cellular and organismal homeostasis [90], which is centered on the degradation of cytoplasmic proteins and damaged organelles via lysosomes [91]. Cellular autophagy is a cellular response in the face of stressful conditions and includes basal autophagy under physiological conditions and induced autophagy under stressful conditions, also referred to as lethal and protective autophagy. Autophagy-dependent cell death (ADCD) is a type of RCD that requires the autophagic machinery or its constituents for its mechanism and is distinguished by the presence of autophagic vacuoles. While both ADCD and adaptive autophagy share molecular mechanisms, there are differences between them. Several genes, including PI3K/Akt, Bcl-2, and mTOR, impede autophagy, whereas tumor suppressors such as PTEN, TSC2, and HIF1α usually promote autophagy [92,93,94]. Autophagy has an important role in inhibiting tumor growth, deleting toxic misfolded proteins, and eliminating intracellular microbes and antigen presentation [95].
Autosis
This is a new form of autophagy-dependent cell death identified in 2013, which is highly reliant on plasma membrane Na( +), K( +)-ATPase [96]. Autosis is triggered by starvation, high doses of autophagy-inducing peptides, or permanent cerebral ischemia. It has unique morphological and biochemical features, including early observable expansion of autophagosomes, autolysosomes, and empty vacuoles. Dilated and fragmented endoplasmic reticulum can also be observed in the early stage of autosis. What is more, a swollen perinuclear space (PNS) containing cytoplasmic materials and electron-dense mitochondria was observed by transmission electron microscopy. During the advanced stage of autosis, a significant reduction in cytoplasmic organelles was observed, and focal nuclear concavity, as well as focal ballooning of the PNS, was apparent [97]. Another important feature of autosis is the increased cellular substrate adherence [98].
Pyroptosis
The first studies on pyroptosis date back to 1986 [99]. Pyroptosis was observed in macrophages infected with the Gram-negative bacterial pathogen Shigella flexneri in 1992, although researchers were unaware of this newly discovered type of cell death at the time [100]. It was not until 2001 that the term pyroptosis was introduced by D’Souza et al. [101]. For a long time, pyroptosis was considered as CASP1-induced monocyte death [102]. Contemporary research recognizes pyroptosis as a type of RCD stimulated by disruptions to intracellular or extracellular homeostasis associated with innate immunity, including exposure to bacteria, viruses, toxins, and chemotherapeutic drugs. Its activation is heavily dependent on members of the GSDMD protein family, inducing the pore formation in the plasma membrane [103].
Although there are similarities between apoptosis and pyroptosis, such as DNA damage and chromatin condensation, pyroptosis differs in that swollen cells with numerous bubble-like protrusions are visible on the cell membrane surface before rupture [104]. In addition, in the early stages of pyroptosis, dUTP cut end labeling (TUNEL) staining was positive, which is a method for detection of DNA damage [105]. On the other hand, pyroptosis cells have lower DNA damage and the nucleus remains intact [106].
Pyroptosis has gained attention for its important role in tumors as a mode of inflammation and programmed cell death [107]. Pyroptosis has shown to inhibit tumor cell proliferation studies while its role in promoting tumor growth has also been reported that pyroptosis can help form suitable tumor microenvironment for tumor cell growth [108]. In addition, it has a unique response to some small molecule compounds. For example, FL118, a camptothecin analog, activates caspase-1 dependent pyroptosis by upregulating the expression of IL-1 and IL-18β, as well as upstream markers NLRP1 and ASC [109]. Disulfiram covalently modifies Cys191/Cys192 in human/mouse GSDMD to prevent pore formation and, thus, inhibits pyroptosis [110]. And the role of some pyroptosis inhibitors and agonists in the tumor microenvironment is under further clinical investigation due to its dual mechanism of promoting and inhibiting tumorigenesis and progression [108].
Ferroptosis
Ferroptosis is one of the most reported modes of cell death in the last decade. Ferroptosis is a type of RCD that occurs due to oxidative imbalances within the intracellular microenvironment, primarily caused by the buildup of intracellular iron accumulation and lipid peroxidation [111]. This non-apoptotic death process drawn the attention of researchers as early as 2003 when they screened tumor cells for compounds and found that erastin, a cell-permeable compound from a high-inclusion screen, preferentially killed cells that had been genetically modified to carry oncogenic RAS mutations [112]. In 2012, the term “Ferroptosis” was formally used to describe this iron-dependent non-apoptotic mode of death [113]. The morphological features include mitochondrial aberrations, reduced cristae, increased membrane density, and increased mitochondrial membrane rupture, and are accompanied by increased lipid peroxidation, elevated ROS, and regulation of several ferroptosis-related genes [114].
Ferroptosis does not show any apoptotic features and occurs through an Fe2+-catalyzed lipid peroxidation process. This process is initiated by non-enzymatic (Fenton reaction) and enzymatic mechanisms (lipoxygenase) [115]. LPCAT3, ALOXs, and ACSL4-mediated polyunsaturated fatty acid oxidation pathways are required for lipid peroxidation in Ferroptosis and relatively, some antioxidant systems including SLC7A11, GPX4, and NFE2L2 can inhibit the lipid peroxidation process in ferroptosis.
In fact, the new understanding of ferroptosis is that its occurrence depends on the balance between the production of ROS induced by iron accumulation and the antioxidant system that avoids lipid peroxidation [116]. The intracellular environment is often disturbed by various stresses, but cells often have the ability to self-balance to maintain their healthy survival [117]. However, various factors, such as physical stimuli, chemical molecules, metallic elements, and temperature, can disrupt this balance. The ionic elements including Fe, Cu, Zn, Na, and Mg are all reported to have important effects on intracellular ion homeostasis, so the final outcome of the abnormal intracellular homeostasis caused by them often depends on a combination of conditions [118]. Based on this, therapeutic strategies to induce ferroptosis have become a hot topic of research, agonists and inhibitors of ferroptosis have entered the clinic. For neurodegenerative diseases, iron overload diseases and current drug resistance issues in cancer therapy are potential research directions [119].
Cuproptosis
Researchers in 2022 reported a copper ion-dependent cell death pattern, similar to the previously reported ferroptosis, a novel death pattern triggered by metal ions called cuproptosis [120]. Cuproptosis is mainly triggered by intracellular copper accumulation and is dependent on mitochondrial respiration. First, copper binds directly to the lipoylated components of the tricarboxylic acid (TCA) cycle, resulting in lipoylated protein aggregation and subsequent iron-sulfur cluster protein loss leading to proteotoxic stress and ultimately cell death.
Key regulators of copper ion carrier-induced cell death include FDX1 and proteolipid acylation [120], but there is no consensus report on the specific morphological features of death. Compared to the conventional view that oxidative stress is the underlying molecular mechanism of metal-induced toxicity [121], cuproptosis does not depend on oxidative stress but is induced by mitochondrial stress, has further advanced the study of mitochondria in cellular life processes. However, there is no doubt that with the discovery of ferroptosis and cuproptosis, metal ion homeostasis will receive more and more attention in cell physiology, disease pathology, drug pharmacology, organ development, and many other fields [122, 123].
Disulfidptosis
Disulfidptosis is the most recent report on the current mode of cell death, a previously unexplained type of cell death. Disulfidptosis is usually triggered when cells with high expression of the solute carrier family member protein SLC7A11 are subjected to glucose starvation [124]. As mentioned previously, SLC7A11 is also one of the most critical regulators of ferroptosis, and is, therefore, considered a potent target of ferroptosis. SLC7A11-mediated cystine uptake inhibits ferroptosis but in turn promotes disulfidptosis under glucose starvation [125]. The discovery of disulfidptosis could help explain the phenomenon that the process of which involves that SLC7A11-highly expressing cells subjected to glucose starvation induce aberrant disulfide bonding between actin cytoskeletal proteins in an SLC7A11-dependent manner, disrupting their organization and ultimately leading to actin network collapse and cell death. Therefore, therapeutic strategies targeting disulfide may become a new strategy in cancer treatment. In current preclinical trials, treatment with glucose inhibitors induces disulfuration in cancer cells with high SLC7A11 expression and effectively inhibits tumor growth.
Parthanatos
PARP (poly ADP-ribose polymerase) is a DNA repair enzyme and a cleavage substrate for cysteine aspartase. Therefore, it plays an important role in DNA damage repair and apoptosis [126]. Parthanatos is triggered by PARP1 overactivation and accompanied by oxidative stress, DNA damage and chromatin condensation [127], which was first reported in 2008 [128]. Parthanatos occurs without produce apoptotic vesicles and small-sized DNA fragments, and it is possible to observe plasma membrane rupture in the absence of cell swelling [129].
The apoptosis-inducing factor mitochondria-associated protein 1 (AIFM1) is required for the execution of parthanatos, and overactive PARP1 causes its release from mitochondria into the nucleus by binding AIFM1 to produce chromatinolysis [130]. Macrophage migration inhibitory factor (MIF) has also been identified as a protein that binds to AIFM1 and has nuclease activity, which produces large DNA fragments when parthanatos is induced [131]. The NCCD proposes to define parthanatos as a form of RCD initiated by PARP1 hyperactivation and precipitated by the consequent bioenergetic catastrophe coupled to AIF-dependent and MIF-dependent DNA degradation [8].
Based on its mechanism in DNA damage repair, parthanatos should also contribute to the oxidative DNA damage-related diseases, including myocardial infarction, diabetes, and trans-degenerative diseases. Inhibition of PARP1 by pharmacological or genetic intervention mediates powerful cytoprotective effects in animal models of multiple diseases [132]. Interestingly, PARP inhibitor, a novel drug has achieved good clinical benefit in recent years [133]. PARP inhibitor can target PARP and achieve antitumor effects by inhibiting DNA damage repair and promoting apoptosis [126]. It can be seen that the same action mechanism may lead to different death modes in different genomic cellular environments.
Entotic cell death
Entotic cell death is a form of cell death that occurs when one cell inserts itself into the neighboring cell, which results in the ultimate death of the invading cell [134]. Entotic cell death was first proposed in 2007 by Michael Overholtzer et al. [135], who found that breast cancer cells enter neighboring cells to form cell intracellular (CIC) structures, with most of the living internalized cells being degraded by lysosomal enzymes and a small fraction being released. Unlike phagocytosis of apoptotic cells, internalization of suspended cells is not associated with caspase activation and is not driven by phosphatidylserine exposure [136] but is dependent on adherens junctions and driven by Rho and ROCK activity in internalized cells, consistent with a cell invasion process [135]. It has been shown that entotic cell death can inhibit the transformed growth of tumor cells in soft agar and may be a potential mechanism for cancer suppression [135]. However, some researchers have also found that in contrast to tumor suppression, entotic cell death also leads to aneuploidization and polyploidization, which promotes tumor development [137, 138]. This mode of cell death deserves further investigation.
Immunogenic cell death
Immunogenic cell death (ICD) is a term coined in 2005 [139] to describe a type of regulated cell death (RCD) that is capable of triggering an adaptive immune response in a host with active immunological capabilities [8]. It is a form of cancer cell death which can be triggered by certain chemotherapeutic agents, lysing viruses, photodynamic therapy, and radiation therapy [140]. ICD involves the release of damage-associated molecules from dying tumor cells and activates tumor-specific immune responses, while long-term efficacy of anticancer drugs is obtained through a combination of both direct killing of cancer cells and antitumor immune activation. Damage-associated molecular patterns (DAMPs) for immunogenic cell death include calreticulin (CALR), HMGB1 (High-mobility group box 1), ATP, ANXA1 (Annexin A1), and type I IFN [141]. ICD inducers trigger several types of cellular stress responses, including autophagy, endoplasmic reticulum stress, or non-folded protein responses, leading to various forms of cell death, including typical apoptosis and necroptosis, and ultimately the release of DAMPs required to activate the anticancer immune response [142]. Current research on immunogenic cell death has facilitated the development of new therapeutic agents, therapeutic combinations, and personalized treatment strategies.
Lysosome-dependent cell death
The idea of lysosome-dependent death was first proposed in 1955 by Christian de Duve, who discovered lysosomes as a mechanism of cellular degradation and coined the term “lysosome-dependent cell death” in 2000 [143]. Lysosome-dependent cell death is a form of RCD mediated by hydrolytic enzymes that are released into the cytoplasm following lysosomal membrane permeabilization (LMP), which is characterized by lysosomal membrane damage. Massive lysosomal leakage leads to increased cytoplasmic acidity, uncontrolled breakdown of cellular components, and cell death due to necrosis [144]. Many external and internal stimuli can initiate LMP and cause lysosome-dependent death. These include lysosomal pro-osmotic agents that have detergent-like effects or cause osmotic lysis, reactive oxygen species (ROS), and apoptosis inducers [145].
NETotic cell death
NETotic cell death is a form of RCD driven by the release of neutrophil extracellular traps (NET), an extracellular reticulated DNA–protein structure released by cells in response to infection or injury [12]. NET-like structures can be released by cells other than neutrophils, including eosinophils [146], mast cells [147], and basophils [148]. NET production and release, or NETosis, was first observed by Arturo Zychlinsky et al. in 2004 [149]. NETosis is a dynamic process dependent on multiple signals and steps, including NADPH oxidase-mediated ROS production, autophagy, the release and translocation of granular enzymes, and peptides from the cathelecidin family from the cytosol to the nucleus [12].
Alkaliptosis
Alkaliptosis is a form of pH-dependent cell death that was only first reported in 2018 [12]. Researchers performing compound antitumor drug sensitivity experiments identified a compound, JTC801 that specifically induced pH-dependent death in cancer cells and slowed tumor growth in mice [150]. And then, this novel RCD driven by intracellular alkalinization was defined. Alkaliptosis is currently known to be pathway directed by IKBKB-NF-κB-dependent carbonic anhydrase 9 (CA9), but the exact molecular mechanism remains unclear. The role of alkaliptosis has been preliminarily studied in tumors, diabetes and other diseases [151, 152], and further studies are needed to facilitate the development of novel treatment based on alkaliptosis.
Oxeiptosis
Oxeiptosis is also a cell death mode first reported named in 2018, which is a ROS-induced cysteinase-non-dependent apoptosis-like cell death pathway [153]. Oxeiptosis is driven by the KEAP1-PGAM5-AIFM1 pathway, and overactivated KEAP1 can mediate H2O2 to induce oxeiptosis in an NFE2L2 independent manner. The role of oxeiptosis in human disease remains unknown, but its study in tumors has been a step ahead [154].
Oncosis
The term “Oncosis” was introduced more than a century ago to denote cell death due to swelling [155]. Oncosis, which is characterized by swelling, is characterized morphologically by increased size, vacuolization of the cytoplasm, cell swelling, and disruption of intracellular structures such as the nucleus, endoplasmic reticulum, and mitochondria, as well as disruption of cell membrane integrity, and a marked inflammatory response could be found in oncosis cells [156]. Based on these features, oncosis has been considered as a non-apoptotic mode of death [157].
Cellular senescence
In contrast to the various modes of death described above, cellular senescence is actually the most common and healthy life posture of cells. The term “cellular senescence” refers to the irreversible decline in cellular proliferative capacity, accompanied by distinct morphological and biochemical features, including the expression of the senescence-associated secretory phenotype (SASP). The term “cellular senescence” refers to the irreversible loss of cellular proliferative capacity, accompanied by distinct morphological and biochemical features, including the expression of the senescence-associated secretory phenotype (SASP) [8]. It is a pathophysiological process in which cells permanently lose their proliferative capacity while maintaining viability and metabolic activity [158], and exhibit specific morphological features including flattening, intracellular vacuolization, nuclear enlargement, and altered chromatin structure.
Cellular senescence is a beneficial process during embryonic development and may also have important roles in tissue repair and regeneration, immune responses, preservation of stem cell populations, and tumor suppression mechanisms [159]. Senescent cells accumulated during the aging of an organism, and with increased generation and inefficient clearance of senescent cells by the organism [160]. Cellular senescence can likewise lead to poor outcomes in many age-related diseases [161]. It is worth noting that cellular senescence is a natural process, and it is not considered as a form of RCD.
Detection methods and related genes of cell death
There are no very clear criteria for the identification of cell death patterns. In both in vivo and in vitro cell culture environments, cell growth environments of millions of orders of magnitude or more imply multiple life-regulated forms, which makes the identification of cell death types more difficult.
The traditional way to identify cell death is through observation by photoelectric microscopes, which has so far been the gold standard for identifying death patterns [162,163,164]. In particular, with the improvement of observation precision by TEM and scanning electron microscopy, the observation of subcellular morphology and intracellular components at the nanoscale has become possible, which is the reason why novel death modes have been gradually observed [165]. With the development of molecular biology, more biological tools are also used to help study the life course of cells, including HE staining, immunohistochemistry, immunofluorescence, mitochondrial membrane potential, reactive oxygen levels, DNA damage, flow cytometry, intracellular protein, and RNA composition assays can help us to make broad distinctions between cell death types [166]. Further, the emergence of some cell death detection kits, including apoptosis kits, cell cycle assay kits, and TUNNEL assay kits, has further facilitated our investigation of cell physiological processes. In the future, with the development of sequencing technology, especially single-cell sequencing technology, and the popularization of new spatio-temporomics technologies such as spatial transcriptome, there are undoubtedly new challenges and opportunities for the identification of traditional cell death patterns. The subdivision of traditional RCD patterns and the discovery of novel death patterns will drive the basic life disciplines, including cell biology, forward.
As discussed above, a series of genes could be activated in different cell death modes, triggered expression and regulation of downstream genes are also involved. In Table 1 and Table S1, we summarized the genes related to partial cell death modes. It is worth noting that sometimes there is not a single death mode in cells, and multiple death modes coexist due to the complex microenvironment in cells.
It has been reported that chalcone derivative chalcone-24 killed the cancer cells by inducing autophagy and necroptosis [167]. Some of these core genes and proteins are involved in crosstalk between different death modes. For example, although BCL-2 is closely related to mitochondria-mediated apoptosis, it also plays an additional role in autophagy [168]. Thus, we intersected the genes involved in different death modes, as shown in Fig. 3. We hope that the summary of these related genes can provide some clues for researchers to explore the mechanism of cell death.

The intersection of related genes between different cell death modes
Discussion
Human beings develop from a fertilized egg, and new cells are produced and expanded along with cell division and differentiation. Senescent or damaged cells are phagocytosed and metabolized through efferocytosis, and different cells have different functions and work together to maintain the proper functioning of each system of the body. As the smallest unit of the organism’s tissue structure, the life course of the cell may seem insignificant, but it has a profound impact on the organism. The studies focused on the end of cell life, cell death, are of great importance in many fields such as tissue development, disease pathogenesis, and drug pharmacology [169, 170].
When the earliest concept of necrosis was introduced, humans did not have a precise knowledge of the concept of cell death [76]. It was not until the discovery of apoptosis in 1972 [15] that humans began to focus on the microscopic differences in cellular life processes. Along with the development of molecular biology techniques and in-depth investigation of sequencing and genetic inheritance, humans gradually began to resolve the microscopic processes and regulatory mechanisms of cell death. The discovery of necroptosis, pyroptosis, and autophagy-dependent apoptosis has greatly advanced the current research on tumor development and drug therapy [171]. In particular, the recent discoveries of metal ion homeostasis-induced cell death, including ferroptosis and cuproptosis, have undoubtedly opened up new areas of research for researchers [172]. Immediately following from 2018 when death modes such as Alkaliptosis, Oxeiptosis were investigated [150, 153], 2022 when cuproptosis was established [120] to 2023 when disulfidptosis was discovered [124], the fervor of inquiring cell death modes is unabated continuously.
Interestingly, this broad pattern of cell death is not exclusive, and there are often complex crosstalk of death signals between hundreds of millions of cell populations, such as a program-dependent mode of cell death, apoptosis often does not exist alone, and the complex intracellular microenvironment and epigenetic regulation often make this mode of death intricately crosstalk with necroptosis, pyroptosis, and other forms of cell death [10]. Another study found that the post-ubiquitinated state of RIPK1, a downstream signaling molecule of tumor necrosis factor (TNF), has implications for apoptosis and, thus, changes cell fate [173]; phosphorylation levels of RIPK1 also appear to affect the function of FAD and CASP8, thereby inhibiting RIPK1-dependent RCD and favoring RIPK1-independent apoptosis [174, 175]. Of course, it is noteworthy that this would also imply that a form of RCD similar to TNFR1 for initiation point gene regulatory activation may not be a single-cell death pathway. The latest research based on this view is the study of PANoptosis, which is considered to be a new form of PCD. PANoptosis is a combination of pyroptosis, apoptosis, and necroptosis. It is characterized by the selection of the best cell death modes to remove damaged or infected cells, thereby maintaining the homeostasis of tissues and organs [176].
Certainly, researches about cell death models in the molecular level is also an important field. Since the current research mainly focused on cell morphological phenotypes and unique molecular pathways, it is difficult to accurately interpret the fate of cells. Besides, scientists are still committed to exploring the molecular processes of unique cell death modes. For example, in two recent consecutive reports, Hiller’s team explored the whole process of membrane protein NINJ1 regulating plasma membrane rupture by super-resolution imaging technology. When the cells release a PCD signal, NINJ1 is activated and aggregates on the surface of the cell membrane sequentially, forming a “zipper”-like polymer that opens the cell membrane like a zipper and causes cell lysis[177]. Dixit’s team found that blocking the aggregation of NINJ1 with monoclonal antibodies can inhibit the rupture of cell membranes and protect tissues from damage [178]. These two studies confirmed that cell disintegration caused by PCD is not caused by the changed osmotic pressure, but by cutting the cell membranes autonomously with NINJ1. These interesting finds undoubtedly give us subversive understandings of PCD. Therefore, although we have systematically reviewed the history of cell death which recorded the important roles of these cell death modes in this review, fresh chapters on cell death will be opened as the research continues to deepen.
Conclusion
In summary, the discovery of cell death is often accompanied by the discovery of signaling pathways. With the development of technology for basic research and the deepness of researchers’ cognition, it undoubtedly helps us to have a deeper understanding of cell fate and even life fate. Currently, we have to admit that a single death mode is no longer sufficient to explain the phenomena due to the coexistence of tens of billions of cells. Neither electron microscopic observations nor biological experiments can fully clarify the life state of the whole cell population. It is now a consensus that cell death forms do not exist singularly, but often multiple modes of cell deaths exist in parallel (Fig. 4). The microscopic regulatory mechanisms involved are crosstalk, with molecular signals balancing each other to maintain the stability of the cell population. The future exploration of cell death patterns will undoubtedly be the discovery of new death patterns and the interconnections between the original death patterns. With the development of sequencing technologies, especially single-cell sequencing, the combination of multi-omics such as spatial transcriptome, and the increase of observable precision of electron microscopy, researchers are confident that they can explain the microscopic processes of cell population death more comprehensively in the near future.

Multiple modes of cell deaths exist in parallel
References
Bree RT, Stenson-Cox C, Grealy M, Byrnes L, Gorman AM, Samali A (2002) Cellular longevity: role of apoptosis and replicative senescence. Biogerontology 3(4):195–206. https://doi.org/10.1023/a:1016299812327
Semont A, Mouiseddine M, Francois A, Demarquay C, Mathieu N, Chapel A et al (2010) Mesenchymal stem cells improve small intestinal integrity through regulation of endogenous epithelial cell homeostasis. Cell Death Differ 17(6):952–961. https://doi.org/10.1038/cdd.2009.187
Pollina EA, Gilliam DT, Landau AT, Lin C, Pajarillo N, Davis CP et al (2023) A NPAS4-NuA4 complex couples synaptic activity to DNA repair. Nature 614(7949):732–741. https://doi.org/10.1038/s41586-023-05711-7
Misgeld T, Schwarz TL (2017) Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96(3):651–666. https://doi.org/10.1016/j.neuron.2017.09.055
Wyllie AH (1987) Cell death. Cytol Cell Physiol. https://doi.org/10.1016/B978-0-08-091882-2.50024-5
Vaux DL, Korsmeyer SJ (1999) Cell death in development. Cell 96(2):245–254. https://doi.org/10.1016/s0092-8674(00)80564-4
Bertalanffy FD, Lau C (1962) Cell renewal. In: Jeon KW (ed) International review of cytology. Elsevier, Amsterdam, pp 357–366
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P et al (2018) Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 25(3):486–541. https://doi.org/10.1038/s41418-017-0012-4
Evan G, Littlewood T (1998) A matter of life and cell death. Science 281(5381):1317–1322. https://doi.org/10.1126/science.281.5381.1317
D’Arcy MS (2019) Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int 43(6):582–592. https://doi.org/10.1002/cbin.11137
Loo DT, Rillema JR (1998) Measurement of cell death. Methods Cell Biol 57:251–264. https://doi.org/10.1016/s0091-679x(08)61583-6
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G (2019) The molecular machinery of regulated cell death. Cell Res 29(5):347–364. https://doi.org/10.1038/s41422-019-0164-5
Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D et al (2015) Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 22(1):58–73. https://doi.org/10.1038/cdd.2014.137
Huang Y, Xu W, Zhou R (2021) NLRP3 inflammasome activation and cell death. Cell Mol Immunol 18(9):2114–2127. https://doi.org/10.1038/s41423-021-00740-6
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257. https://doi.org/10.1038/bjc.1972.33
Jacobson MD, Weil M, Raff MC (1997) Programmed cell death in animal development. Cell 88(3):347–354. https://doi.org/10.1016/s0092-8674(00)81873-5
Wyllie AH, Kerr JF, Currie AR (1980) Cell death: the significance of apoptosis. Int Rev Cytol 68:251–306. https://doi.org/10.1016/s0074-7696(08)62312-8
Lawen A (2003) Apoptosis-an introduction. BioEssays 25(9):888–896. https://doi.org/10.1002/bies.10329
Ndozangue-Touriguine O, Hamelin J, Breard J (2008) Cytoskeleton and apoptosis. Biochem Pharmacol 76(1):11–18. https://doi.org/10.1016/j.bcp.2008.03.016
Guicciardi ME, Leist M, Gores GJ (2004) Lysosomes in cell death. Oncogene 23(16):2881–2890. https://doi.org/10.1038/sj.onc.1207512
Gozuacik D, Kimchi A (2007) Autophagy and cell death. Curr Top Dev Biol 78:217–245. https://doi.org/10.1016/S0070-2153(06)78006-1
Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L et al (2007) Cell death modalities: classification and pathophysiological implications. Cell Death Differ 14(7):1237–1243. https://doi.org/10.1038/sj.cdd.4402148
Schweichel JU, Merker HJ (1973) The morphology of various types of cell death in prenatal tissues. Teratology 7(3):253–266. https://doi.org/10.1002/tera.1420070306
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al (2012) Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ 19(1):107–120. https://doi.org/10.1038/cdd.2011.96
Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P et al (2005) Classification of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ 12(Suppl 2):1463–1467. https://doi.org/10.1038/sj.cdd.4401724
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al (2009) Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ 16(1):3–11. https://doi.org/10.1038/cdd.2008.150
Lockshin RA, Zakeri Z (2001) Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol 2(7):545–550. https://doi.org/10.1038/35080097
Hacker G (2000) The morphology of apoptosis. Cell Tissue Res 301(1):5–17. https://doi.org/10.1007/s004410000193
Cohen GM (1997) Caspases: the executioners of apoptosis. Biochem J 326(1):1–16. https://doi.org/10.1042/bj3260001
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21(1):92–101. https://doi.org/10.1016/j.devcel.2011.06.017
Nunez G, Benedict MA, Hu Y, Inohara N (1998) Caspases: the proteases of the apoptotic pathway. Oncogene 17(25):3237–3245. https://doi.org/10.1038/sj.onc.1202581
Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP (2000) Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol 59(1):65–85. https://doi.org/10.1016/s0006-2952(99)00310-x
Rossi D, Gaidano G (2003) Messengers of cell death: apoptotic signaling in health and disease. Haematologica 88(2):212–218
Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE et al (2016) Proteome-wide covalent ligand discovery in native biological systems. Nature 534(7608):570–574. https://doi.org/10.1038/nature18002
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES (2019) Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10(1):1689. https://doi.org/10.1038/s41467-019-09397-2
Pihan P, Carreras-Sureda A, Hetz C (2017) BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ 24(9):1478–1487. https://doi.org/10.1038/cdd.2017.82
Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L (2017) DNA damage in stem cells. Mol Cell 66(3):306–319. https://doi.org/10.1016/j.molcel.2017.04.006
Allavena G, Cuomo F, Baumgartner G, Bele T, Sellgren AY, Oo KS et al (2018) Suppressed translation as a mechanism of initiation of CASP8 (caspase 8)-dependent apoptosis in autophagy-deficient NSCLC cells under nutrient limitation. Autophagy 14(2):252–268. https://doi.org/10.1080/15548627.2017.1405192
Liu HR, Gao E, Hu A, Tao L, Qu Y, Most P et al (2005) Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation 111(1):90–96. https://doi.org/10.1161/01.cir.0000151613.90994.17
Chipuk JE, Bouchier-Hayes L, Green DR (2006) Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ 13(8):1396–1402. https://doi.org/10.1038/sj.cdd.4401963
Galluzzi L, Lopez-Soto A, Kumar S, Kroemer G (2016) Caspases connect cell-death signaling to organismal homeostasis. Immunity 44(2):221–231. https://doi.org/10.1016/j.immuni.2016.01.020
Taddei ML, Giannoni E, Fiaschi T, Chiarugi P (2012) Anoikis: an emerging hallmark in health and diseases. J Pathol 226(2):380–393. https://doi.org/10.1002/path.3000
Frisch SM, Francis H (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 124(4):619–626. https://doi.org/10.1083/jcb.124.4.619
Zhang D, Zhou X, Zhang K, Yu Y, Cui SW, Nie S (2023) Glucomannan from Aloe vera gel maintains intestinal barrier integrity via mitigating anoikis mediated by Nrf2-mitochondria axis. Int J Biol Macromol 235:123803. https://doi.org/10.1016/j.ijbiomac.2023.123803
Li D, Park Y, Hemati H, Liu X (2023) Cell aggregation prevents anoikis and induces CD44 cleavage by maintaining lipid raft integrity to promote triple negative breast cancer metastasis. Res Sq. https://doi.org/10.21203/rs.3.rs-2535728/v1
Zhao Z, Li C, Peng Y, Liu R, Li Q (2022) Construction of an original anoikis-related prognostic model closely related to immune infiltration in gastric cancer. Front Genet 13:1087201. https://doi.org/10.3389/fgene.2022.1087201
Bose M, Sanders A, De C, Zhou R, Lala P, Shwartz S et al (2023) Targeting tumor-associated MUC1 overcomes anoikis-resistance in pancreatic cancer. Transl Res 253:41–56. https://doi.org/10.1016/j.trsl.2022.08.010
Cai C, Peng Y, Shen E, Wan R, Gao L, Gao Y et al (2022) Identification of tumour immune infiltration-associated snoRNAs (TIIsno) for predicting prognosis and immune landscape in patients with colon cancer via a TIIsno score model. EBioMedicine 76:103866. https://doi.org/10.1016/j.ebiom.2022.103866
Weems AD, Welf ES, Driscoll MK, Zhou FY, Mazloom-Farsibaf H, Chang BJ et al (2023) Blebs promote cell survival by assembling oncogenic signalling hubs. Nature 615(7952):517–525. https://doi.org/10.1038/s41586-023-05758-6
Terasima T, Ohara H (1968) Chromosome aberration and mitotic death in x-irradiated HeLa cells. Mutat Res 5(1):195–197. https://doi.org/10.1016/0027-5107(68)90094-8
Vitale I, Galluzzi L, Castedo M, Kroemer G (2011) Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 12(6):385–392. https://doi.org/10.1038/nrm3115
Huun J, Lonning PE, Knappskog S (2017) Effects of concomitant inactivation of p53 and pRb on response to doxorubicin treatment in breast cancer cell lines. Cell Death Discov 3:17026. https://doi.org/10.1038/cddiscovery.2017.26
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1(2):112–119. https://doi.org/10.1038/nchembio711
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X et al (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4(5):313–321. https://doi.org/10.1038/nchembio.83
Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K et al (2019) The role of necroptosis in cancer biology and therapy. Mol Cancer 18(1):100. https://doi.org/10.1186/s12943-019-1029-8
Moriwaki K, Chan FKM, Miyoshi E (2021) Sweet modification and regulation of death receptor signalling pathway. J Biochem 169(6):643–652. https://doi.org/10.1093/jb/mvab034
Borghi A, Verstrepen L, Beyaert R (2016) TRAF2 multitasking in TNF receptor-induced signaling to NF-kappaB, MAP kinases and cell death. Biochem Pharmacol 116:1–10. https://doi.org/10.1016/j.bcp.2016.03.009
Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X et al (2021) Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol 9:789948. https://doi.org/10.3389/fcell.2021.789948
Zhang Y, Chen X, Gueydan C, Han J (2018) Plasma membrane changes during programmed cell deaths. Cell Res 28(1):9–21. https://doi.org/10.1038/cr.2017.133
Round-table conference (1969) The surgical therapy of the metastases of the central nervous system. Minerva Neurochir 13(3):265–287
Nadaud B, Depaepe L, Zaharia D, Balme B (2014) Chondrodermatitis nodularis helicis. Ann Dermatol Venereol 141(4):306–307. https://doi.org/10.1016/j.annder.2014.02.001
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147. https://doi.org/10.1038/nrm3737
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C et al (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471(7338):363–367. https://doi.org/10.1038/nature09852
Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17(3):151–164. https://doi.org/10.1038/nri.2016.147
Baik JY, Liu Z, Jiao D, Kwon HJ, Yan J, Kadigamuwa C et al (2021) ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat Commun 12(1):2666. https://doi.org/10.1038/s41467-021-23004-3
Park HH, Kim HR, Park SY, Hwang SM, Hong SM, Park S et al (2021) RIPK3 activation induces TRIM28 derepression in cancer cells and enhances the anti-tumor microenvironment. Mol Cancer 20(1):107. https://doi.org/10.1186/s12943-021-01399-3
Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N et al (2022) ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 606(7914):594–602. https://doi.org/10.1038/s41586-022-04753-7
Wu NY, Huang HS, Chao TH, Chou HM, Fang C, Qin CZ et al (2017) Progesterone prevents high-grade serous ovarian cancer by inducing necroptosis of p53-defective fallopian tube epithelial cells. Cell Rep 18(11):2557–2565. https://doi.org/10.1016/j.celrep.2017.02.049
Chen S, Lv X, Hu B, Shao Z, Wang B, Ma K et al (2017) RIPK1/RIPK3/MLKL-mediated necroptosis contributes to compression-induced rat nucleus pulposus cells death. Apoptosis 22(5):626–638. https://doi.org/10.1007/s10495-017-1358-2
Deng XX, Li SS, Sun FY (2019) Necrostatin-1 prevents necroptosis in brains after ischemic stroke via inhibition of RIPK1-mediated RIPK3/MLKL signaling. Aging Dis 10(4):807–817. https://doi.org/10.14336/AD.2018.0728
Kamiya M, Mizoguchi F, Kawahata K, Wang D, Nishibori M, Day J et al (2022) Targeting necroptosis in muscle fibers ameliorates inflammatory myopathies. Nat Commun 13(1):166. https://doi.org/10.1038/s41467-021-27875-4
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J et al (2020) Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol 13(1):110. https://doi.org/10.1186/s13045-020-00946-7
Yuan J, Amin P, Ofengeim D (2019) Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci 20(1):19–33. https://doi.org/10.1038/s41583-018-0093-1
Klempner LB, Preobrazhenskaia TM, Sotnikov VM (1975) Combined lung cancer and systemic scleroderma. Arkh Patol 37(6):55–61
Frank D, Vince JE (2019) Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 26(1):99–114. https://doi.org/10.1038/s41418-018-0212-6
Bousselin M (1786) Observations on necrosis. Lond Med J 7(3):263–279
Espitia O, Agard C (2021) Scalp necrosis in giant cell arteritis. Mayo Clin Proc 96(4):987–988. https://doi.org/10.1016/j.mayocp.2020.11.025
Nicotera P, Leist M, Ferrando-May E (1998) Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett 102–103:139–142. https://doi.org/10.1016/s0378-4274(98)00298-7
Kroemer G, Dallaporta B, Resche-Rigon M (1998) The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 60:619–642. https://doi.org/10.1146/annurev.physiol.60.1.619
Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q et al (2021) NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591(7848):131–136. https://doi.org/10.1038/s41586-021-03218-7
Zapletal E, Vasiljevic T, Busson P, Matijevic GT (2023) Dialog beyond the grave: necrosis in the tumor microenvironment and its contribution to tumor growth. Int J Mol Sci. https://doi.org/10.3390/ijms24065278
De DC, Berthet J, Berthet L, Appelmans F (1951) Permeability of mitochondria. Nature 167(4245):389–390. https://doi.org/10.1038/167389a0
Lemasters JJ, Qian T, Elmore SP, Trost LC, Nishimura Y, Herman B et al (1998) Confocal microscopy of the mitochondrial permeability transition in necrotic cell killing, apoptosis and autophagy. BioFactors 8(3–4):283–285. https://doi.org/10.1002/biof.5520080316
Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y et al (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1366(1–2):177–196. https://doi.org/10.1016/s0005-2728(98)00112-1
Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L (2016) Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol 26(9):655–667. https://doi.org/10.1016/j.tcb.2016.04.006
Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L et al (2015) Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34(12):1475–1486. https://doi.org/10.1038/onc.2014.96
Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S et al (2012) Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci USA 109(17):6566–6571. https://doi.org/10.1073/pnas.1201608109
Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W et al (2016) CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat Commun 7:13189. https://doi.org/10.1038/ncomms13189
Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149(7):1536–1548. https://doi.org/10.1016/j.cell.2012.05.014
Miller DR, Thorburn A (2021) Autophagy and organelle homeostasis in cancer. Dev Cell 56(7):906–918. https://doi.org/10.1016/j.devcel.2021.02.010
Buratta S, Tancini B, Sagini K, Delo F, Chiaradia E, Urbanelli L et al (2020) Lysosomal exocytosis, exosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int J Mol Sci. https://doi.org/10.3390/ijms21072576
Liu W, Xu L, Wang X, Zhang D, Sun G, Wang M et al (2021) PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy 17(12):4159–4181. https://doi.org/10.1080/15548627.2021.1905466
Wang S, Livingston MJ, Su Y, Dong Z (2015) Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 11(4):607–616. https://doi.org/10.1080/15548627.2015.1023983
Yang J, Pi C, Wang G (2018) Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed Pharmacother 103:699–707. https://doi.org/10.1016/j.biopha.2018.04.072
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132(1):27–42. https://doi.org/10.1016/j.cell.2007.12.018
Liu Y, Shoji-Kawata S, Sumpter RM Jr, Wei Y, Ginet V, Zhang L et al (2013) Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA 110(51):20364–20371. https://doi.org/10.1073/pnas.1319661110
Nah J, Zablocki D, Sadoshima J (2020) Autosis: a new target to prevent cell death. JACC Basic Transl Sci 5(8):857–869. https://doi.org/10.1016/j.jacbts.2020.04.014
Fernandez AF, Liu Y, Ginet V, Shi M, Nah J, Zou Z et al (2020) Interaction between the autophagy protein beclin 1 and Na+, K+-ATPase during starvation, exercise, and ischemia. JCI Insight. https://doi.org/10.1172/jci.insight.133282
Friedlander AM (1986) Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem 261(16):7123–7126
Zychlinsky A, Prevost MC, Sansonetti PJ (1992) Shigella flexneri induces apoptosis in infected macrophages. Nature 358(6382):167–169. https://doi.org/10.1038/358167a0
D’Souza CA, Heitman J (2001) Dismantling the Cryptococcus coat. Trends Microbiol 9(3):112–113. https://doi.org/10.1016/s0966-842x(00)01945-4
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7(2):99–109. https://doi.org/10.1038/nrmicro2070
Jorgensen I, Miao EA (2015) Pyroptotic cell death defends against intracellular pathogens. Immunol Rev 265(1):130–142. https://doi.org/10.1111/imr.12287
Chen X, He WT, Hu L, Li J, Fang Y, Wang X et al (2016) Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26(9):1007–1020. https://doi.org/10.1038/cr.2016.100
Tajima T, Yoshifuji A, Matsui A, Itoh T, Uchiyama K, Kanda T et al (2019) beta-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int 95(5):1120–1137. https://doi.org/10.1016/j.kint.2018.11.034
Xu YJ, Zheng L, Hu YW, Wang Q (2018) Pyroptosis and its relationship to atherosclerosis. Clin Chim Acta 476:28–37. https://doi.org/10.1016/j.cca.2017.11.005
Wei X, Xie F, Zhou X, Wu Y, Yan H, Liu T et al (2022) Role of pyroptosis in inflammation and cancer. Cell Mol Immunol 19(9):971–992. https://doi.org/10.1038/s41423-022-00905-x
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X (2021) Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther 6(1):128. https://doi.org/10.1038/s41392-021-00507-5
Yang F, Bettadapura SN, Smeltzer MS, Zhu H, Wang S (2022) Pyroptosis and pyroptosis-inducing cancer drugs. Acta Pharmacol Sin 43(10):2462–2473. https://doi.org/10.1038/s41401-022-00887-6
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J et al (2020) FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol 21(7):736–745. https://doi.org/10.1038/s41590-020-0669-6
Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26(3):165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Dolma S, Lessnick SL, Hahn WC, Stockwell BR (2003) Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3(3):285–296. https://doi.org/10.1016/s1535-6108(03)00050-3
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31(2):107–125. https://doi.org/10.1038/s41422-020-00441-1
Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X (2018) Emerging strategies of cancer therapy based on ferroptosis. Adv Mater 30(12):e1704007. https://doi.org/10.1002/adma.201704007
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2021) Ferroptosis: machinery and regulation. Autophagy 17(9):2054–2081. https://doi.org/10.1080/15548627.2020.1810918
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X et al (2016) Ferroptosis: process and function. Cell Death Differ 23(3):369–379. https://doi.org/10.1038/cdd.2015.158
Jomova K, Makova M, Alomar SY, Alwasel SH, Nepovimova E, Kuca K et al (2022) Essential metals in health and disease. Chem Biol Interact 367:110173. https://doi.org/10.1016/j.cbi.2022.110173
Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J et al (2022) Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (Lond) 42(2):88–116. https://doi.org/10.1002/cac2.12250
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M et al (2022) Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375(6586):1254–1261. https://doi.org/10.1126/science.abf0529
Ercal N, Gurer-Orhan H, Aykin-Burns N (2001) Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Curr Top Med Chem 1(6):529–539. https://doi.org/10.2174/1568026013394831
Ba LA, Doering M, Burkholz T, Jacob C (2009) Metal trafficking: from maintaining the metal homeostasis to future drug design. Metallomics 1(4):292–311. https://doi.org/10.1039/b904533c
Krzywoszynska K, Witkowska D, Swiatek-Kozlowska J, Szebesczyk A, Kozlowski H (2020) General aspects of metal ions as signaling agents in health and disease. Biomolecules. https://doi.org/10.3390/biom10101417
Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M et al (2023) Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol 25(3):404–414. https://doi.org/10.1038/s41556-023-01091-2
Koppula P, Zhuang L, Gan B (2021) Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12(8):599–620. https://doi.org/10.1007/s13238-020-00789-5
Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E (2020) PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev Biol 8:564601. https://doi.org/10.3389/fcell.2020.564601
David KK, Andrabi SA, Dawson TM, Dawson VL (2009) Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 14(3):1116–1128. https://doi.org/10.2741/3297
Andrabi SA, Dawson TM, Dawson VL (2008) Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann NY Acad Sci 1147:233–241. https://doi.org/10.1196/annals.1427.014
Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W et al (2004) Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci 24(48):10963–10973. https://doi.org/10.1523/JNEUROSCI.3461-04.2004
Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA et al (2011) Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal 4(167):20. https://doi.org/10.1126/scisignal.2000902
Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM et al (2016) A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. https://doi.org/10.1126/science.aad6872
Curtin NJ, Szabo C (2013) Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med 34(6):1217–1256. https://doi.org/10.1016/j.mam.2013.01.006
Pilie PG, Gay CM, Byers LA, O’Connor MJ, Yap TA (2019) PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res 25(13):3759–3771. https://doi.org/10.1158/1078-0432.CCR-18-0968
Zeng C, Zeng B, Dong C, Liu J, Xing F (2020) Rho-ROCK signaling mediates entotic cell death in tumor. Cell Death Discov 6:4. https://doi.org/10.1038/s41420-020-0238-7
Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW et al (2007) A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131(5):966–979. https://doi.org/10.1016/j.cell.2007.10.040
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 148(7):2207–2216
Krajcovic M, Johnson NB, Sun Q, Normand G, Hoover N, Yao E et al (2011) A non-genetic route to aneuploidy in human cancers. Nat Cell Biol 13(3):324–330. https://doi.org/10.1038/ncb2174
Krajcovic M, Overholtzer M (2012) Mechanisms of ploidy increase in human cancers: a new role for cell cannibalism. Cancer Res 72(7):1596–1601. https://doi.org/10.1158/0008-5472.CAN-11-3127
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N et al (2005) Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 202(12):1691–1701. https://doi.org/10.1084/jem.20050915
Ahmed A, Tait SWG (2020) Targeting immunogenic cell death in cancer. Mol Oncol 14(12):2994–3006. https://doi.org/10.1002/1878-0261.12851
Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P et al (2020) Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 11(11):1013. https://doi.org/10.1038/s41419-020-03221-2
Radogna F, Diederich M (2018) Stress-induced cellular responses in immunogenic cell death: Implications for cancer immunotherapy. Biochem Pharmacol 153:12–23. https://doi.org/10.1016/j.bcp.2018.02.006
Franko J, Pomfy M, Prosbova T (2000) Apoptosis and cell death (mechanisms, pharmacology and promise for the future). Acta Medica (Hradec Kralove) 43(2):63–68
Wang F, Gomez-Sintes R, Boya P (2018) Lysosomal membrane permeabilization and cell death. Traffic 19(12):918–931. https://doi.org/10.1111/tra.12613
Aits S, Jaattela M (2013) Lysosomal cell death at a glance. J Cell Sci 126(Pt 9):1905–1912. https://doi.org/10.1242/jcs.091181
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E et al (2008) Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14(9):949–953. https://doi.org/10.1038/nm.1855
Wartha F, Henriques-Normark B (2008) ETosis: a novel cell death pathway. Sci Signal 1(21):pe25. https://doi.org/10.1126/stke.121pe25
Morshed M, Hlushchuk R, Simon D, Walls AF, Obata-Ninomiya K, Karasuyama H et al (2014) NADPH oxidase-independent formation of extracellular DNA traps by basophils. J Immunol 192(11):5314–5323. https://doi.org/10.4049/jimmunol.1303418
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535. https://doi.org/10.1126/science.1092385
Song X, Zhu S, Xie Y, Liu J, Sun L, Zeng D et al (2018) JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology 154(5):1480–1493. https://doi.org/10.1053/j.gastro.2017.12.004
Chen F, Zhu S, Kang R, Tang D, Liu J (2023) ATP6V0D1 promotes alkaliptosis by blocking STAT3-mediated lysosomal pH homeostasis. Cell Rep 42(1):111911. https://doi.org/10.1016/j.celrep.2022.111911
Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G (2020) Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol 11:42. https://doi.org/10.3389/fphar.2020.00042
Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P et al (2018) Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol 19(2):130–140. https://doi.org/10.1038/s41590-017-0013-y
Kang P, Chen J, Zhang W, Guo N, Yi X, Cui T et al (2022) Oxeiptosis: a novel pathway of melanocytes death in response to oxidative stress in vitiligo. Cell Death Discov 8(1):70. https://doi.org/10.1038/s41420-022-00863-3
Majno G, Joris I (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146(1):3–15
Van Cruchten S, Van Den Broeck W (2002) Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol 31(4):214–223. https://doi.org/10.1046/j.1439-0264.2002.00398.x
Sayk F, Bartels C (2004) Oncosis rather than apoptosis? Ann Thorac Surg 77(1):382. https://doi.org/10.1016/s0003-4975(02)05014-2
Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75:685–705. https://doi.org/10.1146/annurev-physiol-030212-183653
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155(5):1119–1130. https://doi.org/10.1016/j.cell.2013.10.041
van Deursen JM (2014) The role of senescent cells in ageing. Nature 509(7501):439–446. https://doi.org/10.1038/nature13193
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J et al (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530(7589):184–189. https://doi.org/10.1038/nature16932
Koenig U, Robenek H, Barresi C, Brandstetter M, Resch GP, Groger M et al (2020) Cell death induced autophagy contributes to terminal differentiation of skin and skin appendages. Autophagy 16(5):932–945. https://doi.org/10.1080/15548627.2019.1646552
Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z et al (2021) Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy 17(12):4266–4285. https://doi.org/10.1080/15548627.2021.1911016
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J et al (2019) Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis 10(3):193. https://doi.org/10.1038/s41419-019-1441-4
Kari S, Subramanian K, Altomonte IA, Murugesan A, Yli-Harja O, Kandhavelu M (2022) Programmed cell death detection methods: a systematic review and a categorical comparison. Apoptosis 27(7–8):482–508. https://doi.org/10.1007/s10495-022-01735-y
Ketelut-Carneiro N, Fitzgerald KA (2022) Apoptosis, pyroptosis, and necroptosis-oh my! the many ways a cell can die. J Mol Biol 434(4):167378. https://doi.org/10.1016/j.jmb.2021.167378
He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z et al (2014) A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 33(23):3004–3013. https://doi.org/10.1038/onc.2013.256
Liu J, Liu W, Yang H (2019) Balancing apoptosis and autophagy for Parkinson’s disease therapy: targeting BCL-2. ACS Chem Neurosci 10(2):792–802. https://doi.org/10.1021/acschemneuro.8b00356
Mei J, Tian H, Huang HS, Hsu CF, Liou Y, Wu N et al (2021) Cellular models of development of ovarian high-grade serous carcinoma: a review of cell of origin and mechanisms of carcinogenesis. Cell Prolif 54(5):e13029. https://doi.org/10.1111/cpr.13029
Raudenská M, Balvan J, Masařík M (2021) Cell death in head and neck cancer pathogenesis and treatment. Cell Death Dis 12(2):192. https://doi.org/10.1038/s41419-021-03474-5
Gao W, Wang X, Zhou Y, Wang X, Yu Y (2022) Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther 7(1):196. https://doi.org/10.1038/s41392-022-01046-3
Chen X, Kang R, Kroemer G, Tang D (2021) Ferroptosis in infection, inflammation, and immunity. J Exp Med. https://doi.org/10.1084/jem.20210518
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ et al (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135(7):1311–1323. https://doi.org/10.1016/j.cell.2008.10.044
Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT et al (2017) Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun 8(1):359. https://doi.org/10.1038/s41467-017-00406-w
Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L et al (2017) MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell 66(5):698-710.e5. https://doi.org/10.1016/j.molcel.2017.05.003
Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti T-D (2021) AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 597(7876):415–419. https://doi.org/10.1038/s41586-021-03875-8
Degen M, Santos JC, Pluhackova K, Cebrero G, Ramos S, Jankevicius G et al (2023) Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Nature 618(7967):1065–1071. https://doi.org/10.1038/s41586-023-05991-z
Kayagaki N, Stowe IB, Alegre K, Deshpande I, Wu S, Lin Z et al (2023) Inhibiting membrane rupture with NINJ1 antibodies limits tissue injury. Nature 618(7967):1072–1077. https://doi.org/10.1038/s41586-023-06191-5
Author information
Authors and Affiliations
Contributions
S-HZ, D-CR and H-XT designed the review and the structure of this manuscript. L-YW and X-JL wrote the manuscript draft. Q-QL, YZ, H-LR, J-NS, JZ and JM helped to search reference of this review and contributed to the writing. S-HZ contributed to the revision and finalization of the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, LY., Liu, XJ., Li, QQ. et al. The romantic history of signaling pathway discovery in cell death: an updated review. Mol Cell Biochem 479, 2255–2272 (2024). https://doi.org/10.1007/s11010-023-04873-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-023-04873-2