
Abstract
Retinoblastoma (RB) is an intraocular malignancy that is most common in children and rare in adults. Addressing novel biomarkers and therapeutic targets for RB to modulate tumor progression has become a challenge. The aim of the present study was to investigate the function of long non-coding RNAs (LncRNAs) LOXL1-AS1 in RB cell proliferation and metastasis. It was found that LOXL1-AS1 was overexpressed in RB tissues and cells. In order to evaluate cell viability and colony formation potential, the knockdown of LOXL1-AS1 has been established. Knockdown of LOXL1-AS1 was also inhibited cells migration and invasion. In addition, the proportion of cells in the G2/M phase of the sh-LOXL1-AS1 group increased significantly, and the proportion of cells in the sh-NC group decreased significantly. In the xenograft model of RB, the tumors in the sh-LOXL1-AS1 group grow slowly compared to the sh-NC group. Western blot analysis revealed that LOXL1-AS1 can regulate the progression of RB cells through MAPK signaling pathway in vitro and in vivo. These results indicated that LncRNA LOXL1-AS1 promotes proliferation, invasion and inhibits apoptosis of retinoblastoma by regulating MAPK signaling pathway, and might be expected to be a novel basis for clinical diagnosis and treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Retinoblastoma (RB) is the most common primary intraocular malignant tumor in the pediatric population worldwide [1]. RB remains the most common intraocular malignant tumor in children, with an incidence of 1/15000–1/20000 cases annually and approximately 30–40% of patients exhibiting bilateral onset [2]. The early manifestations of RB mainly include leukocoria and strabismus, which can be severe enough to cause blindness or even fatality [3]. Therefore, early treatment and diagnosis are particularly important due to the high degree of malignancy in RB. Current therapies for RB including intravenous chemotherapy, ophthalmic artery interventional chemotherapy, and local laser irradiation, can increase the survival rate, but tumor invasion through the optic nerve or choroid with the potential to spread regionally and distally remains a serious clinical complication [4]. Resistance to chemotherapeutic drugs and their serious toxic effects contribute to the fatal consequences of metastatic disease in the central nervous system and distant organs [5]. Thus, new therapeutic strategies targeting critical regulatory pathways in the development and progression of RB have been explored. Long noncoding RNA (lncRNA) is a type of noncoding RNA that is longer than 200 nucleotides but does not encode a protein [6]. Recently, lncRNAs have been found to participate in a variety of cellular and physiological functions with advances in high-throughput sequencing technology [7]. LncRNAs are increasingly recognized as associated with cellular homeostasis in cancers, such as cell proliferation, migration, invasion, and differentiation [8]. Moreover, the role of lncRNAs as novel biomarkers for diagnosis, clinical treatment and prognosis in tumor development has received attention from worldwide studies [9]. For example, Rajagopal et al. reported that lncRNA HOTAIR is critical in suppressing drug responses and driving genomic instability in multiple malignant behaviors [10]. Gao has shown that lncRNA MALAT1 and lncRNA NKILA can promote cell apoptosis and inhibit the growth of RB cells [11].
Though there are some limitations to our comprehensive understanding of lncRNAs, it has provided a theoretical basis for elucidating the cancer promotion or inhibition function of lncRNAs for clinical application to RB. LncRNA LOXL1 antisense RNA 1 (LOXL1-AS1) is located on human chromosome 15q24.1 and contains 10,781 nucleotides and five exons. It is encoded on the opposite strand of LOXL1, which has been shown to have an oncogenic effect in diverse human cancers [12, 13]. Previous research investigated whether LOXL1‐AS1 increased medulloblastoma metastasis by regulating the PI3K/AKT signaling pathway [14]. LOXL1‐AS1 is upregulated in cervical squamous cell carcinoma and plays a key role as an oncogene [15]. However, its biological functions and underlying mechanisms in RB tumorigenesis and progression have not been fully elucidated. The current study sought to assess the expression profile and functional properties of LOXL1-AS1 in RB cells in vitro and in vivo. We hypothesized that LOXL1-AS1 was overexpressed in a series of RB cell lines, which suggested that LOXL1-AS1 might be involved in the regulation of RB. However, the role of MAPK signaling pathway modulated by LOXL1-AS1 remains unexplored in RB, which might be an innovative regulatory mechanism contributing to LOXL1-AS1-mediated RB progression.
Materials and methods
Human tissues and ethical approval
Human RB tissues from 16 patients cases were obtained from The Third Affiliated Hospital of Nanchang University and Affiliated Eye Hospital of Nanchang University. All patients had signed informed consent. No patients underwent chemotherapy or radiotherapy before the surgery. This study has been approved by the Ethics Committee of The Third Affiliated Hospital of Nanchang University. The clinical information of the patients is mentioned in Table 1.
Cell culture and reagents
Three cell lines, the human retinoblastoma cell Lines Y79 and WERI-Rb1 and the human retinal pigment epithelium cell line ARPE-19, were purchased from the Institute of Biochemistry and Cell Biology (Shanghai, China). The WERI-Rb1 cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS) (Gibco, Invitrogen, Waltham, MA, USA) and 1% penicillin/streptomycin (P/S) (Solarbio, Beijing, China). Y79 and ARPE-19 cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS and 1% P/S. All cells were cultured at 37 °C in a humidified incubator with 5% CO2.
Lentivirus transfection and construction
Lentiviruses encoding shRNAs targeting the knockdown of lncRNA (Lv-LOXL1-AS1), with the sequences 5′-GGGTGCCACGGCTTACCAATG-3′ and 5′GCTTTGGCATCCAAGAAATCA-3′, were constructed by GenePharma (Shanghai, China) and contained a GFP fluorescent tag. The negative control (NC) shRNA sequence was 5′-TTCTCCGAACGTGTCACGT-3′ (Lv-NC). The lentivirus (5 × 108 TU/mL) and polybrene (5 μg/ml, GenePharma) were added to Y79 and WERI-Rb1 cells, respectively, for 24 h. Cells were then selected with 0.5 μg/ml puromycin for the next week and received stable knockdown cell lines (Table 2).
Quantitative real-time PCR
Total RNA was extracted from tissues and cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and complementary DNA was obtained using a reverse transcriptase kit (TaKaRa, Kusatsu, Japan) according to the manufacturer’s protocol. All real-time PCRs were performed with SYBR Green Reaction Mix (TaKaRa, Dalian, China) in a CFX96 Real-Time System (Bio-Rad, CFX96 optics module). Afterward, the expression levels of each RNA were calculated using the 2−ΔΔCT method relative to the level of the control gene.
Immunofluorescent assay
Stable knockdown cell lines were washed three times with PBS and fixed with 4% paraformaldehyde for 30 min at 37 °C. The cells were rinsed with PBS for 3 × 5 min, permeabilized with 0.5% Triton X-100 in PBS for 20 min, and then blocked in 10% FBS for 2 h. A GFP tag was inserted into the cells, which were then stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired with a fluorescence inverted microscope (Olympus, IX73).
Cell viability assay
Cell viability was assessed using a cell counting kit-8 (CCK-8) assay (Solarbio, Beijing, China) according to the manufacturer’s protocol. Stable knockdown cell lines of Y79 (5 × 103 cells/well) and WERI-Rb1 (8 × 103 cells/well) were seeded into 96-well plates with 100 μl medium containing 10% CCK-8 reagent. The cells were incubated for 2 h at 37 °C with 5% CO2. The absorbance value of each well was measured by a microplate reader at 450 nm. All experiments were performed in triplicate.
Soft agar colony formation assay
Soft agarose (A9045-5G, Sigma‒Aldrich, St. Louis, MO) was prepared at concentrations of 1.2% and 0.6% in a high-pressure and steam-sterilized environment. The medium was supplemented with 20% FBS and 2% P/S. Then, 1.2% soft agarose was mixed at a 1:1 ratio, and 2 ml was added to each well of a six-well plate and cooled at room temperature for 30 min. Cells were added to the same volume of medium with 0.6% soft agarose for the upper agar layer. After cells were cultured for at least 2 weeks, the morphology of the colonies was analyzed using cell staining, and the number of colonies formed per well was quantified using ImageJ (version 1.52; National Institutes of Health) software.
Transwell cell migration and invasion assays
Stable knockdown cell lines of Y79 and WERI-Rb1 cells were seeded into the upper level of a Transwell chamber (6.5 mm diameter, 8 μm pores, Corning, NY, USA) at a density of 2.0 × 106 cells/chamber, and the lower level was filled with 600 μl medium containing 20% FBS. Cells were cultured at 37 °C with 5% CO2 for 48 h. Subsequently, the cells that had invaded the lower surface of the membrane were fixed for 30 min with 4% paraformaldehyde, stained with 0.25% crystal violet dye for 10 min, and rinsed with PBS several times. In the invasion experiment, Matrigel matrix (Corning, New York, USA) was diluted 1:3 with precooled basal medium at 4 °C and coated on the bottom of the Transwell chamber. The next procedure was the same as the migration experiment. Photographs of three random regions were taken, and the number of migrated cells was counted with a microscope.
Western blot assay
Cells were harvested and lysed with RIPA buffer (Beyotime Institute of Biotechnology, Beijing, China) supplemented with protease inhibitors (CWBIO, Beijing, China). After the protein concentration was determined by a bicinchoninic acid (BCA) Protein Assay Kit (Beyotime, Shanghai, China) to ensure the use of equal amounts of protein in each group, the protein was separated by 10% SDS‒PAGE gel and transferred to 0.45 µm polyvinylidene fluoride (PVDF) membranes (Millipore, Burlington, MA, USA) at a constant current of 350 mA. The PVDF membranes were placed in 5% skim milk for 1 h to block nonspecific binding sites and incubated with primary antibodies at 4 °C for 12 h. The following antibodies were used as primary antibodies: Bcl-2 antibody (1;1000, No. 12789-1-AP), Bax antibody (1;1000, No. 50599-2-lg) (Proteintech, USA), p44/42 MAPK (Erk1/2) antibody (1:1000, No. 4695S), phospho-p44/42 MAPK antibody (P-Erk1/2) (1:1000, No. 4377T), MEK1/2 antibody (1;1000, No. 9122), phospho-MEK1/2 antibody (1;1000, No. 9154S), and β-actin antibody (1:1500, No. 8457). (Cell Signaling Technology, Danvers, MA, USA).The membranes were washed three times in PBST and incubated with the appropriate secondary antibody at room temperature for 1 h. Then, the membranes were visualized via an enhanced chemiluminescence (ECL) imaging system and analyzed with ImageJ software.
Enzyme‐linked immunosorbent assay
The enzyme-linked immunosorbent assay (ELISA) was performed according to the manufacturer’s guidelines (Boster Biological Technology, Wuhan, China). Y79 and WERI-Rb1 cells were collected and diluted five times in PBS and centrifuged at 4 °C for 10 min at 12,000×g. Next, the supernatants were collected for secretion measurement. The detection reagent and each group of cells were pipetted into precoated plates that had been coated with MMP-2 and MMP-9 antibodies for 60 min at 37 °C. The OD value was determined at 450 nm by an instant enzyme-labeled analyzer.
Cell apoptosis assay
An Annexin V fluorescein (FITC)/PI double-staining cell apoptosis detection kit (No. KGA107, KeyGen Biotech, Nanjing, China) was used to identify apoptotic cells according to the manufacturer’s instructions. Briefly, cells were collected and washed twice with PBS, then resuspended in 300 μl binding buffer containing 3 μl Annexin V FITC and 3 μl PI. Cell suspensions were incubated at room temperature in the dark for approximately 15 min. Samples were then analyzed by FlowJo software (BD FACSCalibur). All experiments were performed in triplicate.
Cell cycle assay
Flow cytometry analysis was performed to analyze cell cycle progression. Briefly, cells were fixed with 70% ethanol at – 20 °C overnight. After washing twice with PBS, the cells were incubated with 50 μg/ml RNase A and 50 μg/ml propidium iodide (PI) at room temperature in the dark for 15 min. The results were analyzed using FlowJo software.
Xenotransplantation experiments in vivo
All animal experiments were carried out in compliance with a protocol specifically approved by the Experimental Animal Center of Nanchang University Animal Care and Use Committee (approval No. NCUFLLL-20220722). Twelve male athymic BALB/c nude mice (18–20 g, 4–5 weeks old) were maintained in a special pathogen-free (SPF) condition and handled as described previously [16]. Twelve mice were randomly divided into two groups, namely, scramble groups and the sh-LOXL1-AS1 group (n = 6 per group). A total of 1 × 107 stable knockdown cell lines of Y79 cells were injected subcutaneously into the right side in each mouse in 200 μl PBS and 30% (v/v) Matrigel matrix. The tumor volume (length, L, and width, W) and body weights of the mice were monitored every 7 days. The tumor size was calculated as follows: tumor volume (mm3) = L × W2/2. After 28 days of feeding, the mice were euthanized by i.v. injection of 100 mg/kg pentobarbital and tumor tissues were dissected for subsequent analyses.
Hematoxylin and eosin staining and immunohistochemistry
Tumor tissues from the mouse model were embedded in paraffin in 5-µm serial sections. Hematoxylin and eosin staining (H&E) was used to confirm the pathology results. The slides were examined under a light microscope with a photomicrographic attachment. After H&E staining, the slides were washed, blocked, and incubated with primary antibody against Ki-67 (1:1000, No. ab15580, Abcam) at 4 °C overnight. The sections were incubated with the corresponding secondary antibody (No. ab205718, Abcam) for 30 min. The immunostaining reactivity was captured by diaminobenzidine (DAB) and images were taken for each slide.
Statistical analysis
All data are expressed as the mean ± SD and each experiment was repeated more than three times. The statistical significance of differences was determined using Student’s t test for the study. P < 0.05 was considered to indicate a statistically significant difference.
Results
LncRNA LOXL1-AS1 was overexpressed in RB tissues and cells
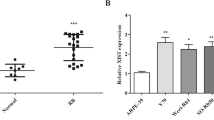
To further evaluate the effects of LOXL1-AS1 in RB, the expression level of LOXL1-AS1 was examined using qRT‒PCR in tumor tissues collected from 16 RB patients and adjacent tissues, as shown in Fig. 1a. Fourteen of the 16 patients demonstrated obviously higher expression of LOXL1-AS1 in RB tissues than in the paired adjacent tissues (Fig. 1b). Furthermore, its expression in RB cell lines (Y79 and WERI-Rb1) and the human normal retinal pigment epithelium cell line ARPE-19 was compared. As shown in Fig. 1c, the LOXL1-AS1 expression level was significantly higher in RB cell lines than in normal cell lines. Together, these data suggested that LOXL1-AS1 was overexpressed in RB tissues and cells.

Expression of LncRNA LOXL1-AS1 in RB tissues and cells. a qRT-PCR analysis was used to detect LOXL1-AS1 expression in 16 cases of clinical RB tissues and adjacent normal tissues. b The paired expression of LOXL1-AS1 was shown in the 16 RB patients. c qRT-PCR was used to compare the expression of LOXL1-AS1 in RB cell lines to the human normal cell line (ARPE-19). ***p < 0.001, ****p < 0.0001
Knockdown of LncRNA LOXL1-AS1 expression in RB cells inhibited cell viability and colony formation capacity
To investigate the functionality of LOXL1-AS1, we employed lentivirus transfection methods to knockdown LOXL1-AS1 expression.
Specific shRNAs depleted the expression level of LOXL1-AS1 in Y79 cells (Fig. 2a) and WERI-Rb1 cells (Fig. 2b). Immunofluorescence staining showed the distribution of fluorescence in the two cell lines, suggesting that LOXL1-AS1 expression was stably reduced (Fig. 2c, d). Next, the CCK-8 assay was performed to detect the viability of the above cells. The knockdown of LOXL1-AS1 obviously induced the proliferation of Y79 cells and WERI-Rb1 cells (Fig. 2e, f). Likewise, in the soft agar colony formation assay, we found that the maximum suppression effect of proliferation ability was approximately 60% in Y79 cells; similarly formed colonies were also found in WERI-Rb1 cells (Fig. 2g, h). These data indicated that knockdown of lncRNA LOXL1-AS1 inhibited cell viability and colony formation capacity.

The cell viability and colony formation ability of LOXL1-AS1 knockdown in RB cells. a and b qRT-PCR were done to determine the transfection efficacy of Y79 and WERI-Rb1 cells. c and d Immunofluorescence staining revealed that knockdown of LOXL1-AS1 was stable distribution in Y79 and WERB-Rb1 cells. e and f CCK-8 assay was carried out to find the proliferation capacity of knockdown of LOXL1-AS1. g and h Colony formation assay was conducted to discover the cloning ability of cells. **p < 0.01, ***p < 0.001, ****p < 0.0001
Knockdown of LOXL1-AS inhibited cell migration and invasion in RB cells
Cell migration and invasion capacity are closely related to cancer metastasis. The transwell assay revealed that LOXL1-AS1 knockdown cells migrated and invaded significantly less than the sh-NC group in Y79 and WERI-Rb1 cells (Fig. 3a). The percentages of the migration and invasion areas are shown in Fig. 3b–e. MMP-2 and MMP-9 have been consistently associated with increased invasiveness and poor prognosis; their expression levels were detected with ELISA. MMP-2 and MMP-9 expression was significantly reduced in Y79 cell lines with LOXL1-AS1 knockdown compared to the sh-NC group, as shown in Fig. 3f, g. Similarly, the MMP-2 and MMP-9 expression levels were also decreased in WERI-Rb1 cells (Fig. 3h, i).

The cell migration and invasion ability of LOXL1-AS1 knockdown in RB cells. a–e Transwell assay was applied to perform cell migration and invasion. f–i Elisa detected the secretion of MMP-2 and MMP-9 in Y79 and WERB-Rb1 cells. **p < 0.01, ***p < 0.001, ****p < 0.0001
Knockdown of LOXL1-AS1 promoted apoptosis in RB cells and arrested the cell cycle at the G2/M phase
The apoptosis of RB cells was analyzed using flow cytometry. FITC events represent apoptotic cells, and the apoptosis ratio was calculated by summing the percentage of events in the Q2 and Q3 regions. Knockdown of LOXL1-AS1 significantly increased the cell apoptosis ratio compared with the sh-NC group (Fig. 4a, b). The Western blot assay also suggested apoptosis was induced via increased levels of Bcl-2 and Bax (Fig. 4c, d). In addition, knockdown of LOXL1-AS1 arrested cell cycle progression at the G2/M phase in the cell cycle analysis, which was obviously decreased compared to the sh-NC group. Accordingly, the proportion of cells in the S phase increased from approximately 2% in the sh-NC group to nearly 10% in the sh-LOXL1-AS1 WERI-Rb1 cell group (Fig. 4e, f). In conclusion, these data show that knockdown of LOXL1-AS1 promoted cell apoptosis and arrested the cell cycle at the G2/M phase.

Knockdown of LOXL1-AS1 on cell cycle and cell apoptosis of RB cells. a and b Flow cytometry was used to clarify the cell apoptosis in RB cells. c and d Western blot assay was used to detect the protein expression of Bcl-2 and Bax in Y79 and WERI-Rb1 cells with the knockdown of LOXL1-AS1. e and f Flow cytometry revealed that cell cycle progression was halted in both cell lines at the G2/M phase. *p < 0.05, **p < 0.01, ***p < 0.001
LOXL1-AS1 modulates the progression of RB cells through the MAPK pathway in vitro
The molecular mechanisms of the LOXL1-AS1-mediated phenotype were explored. It was observed that the phosphorylated levels of MAPK, including p-Erk and p-MEK, were markedly decreased in LOXL1-AS1 knockdown cells. However, the total protein levels of Erk and MEK had no influence on knockdown cells, implying that LOXL1-AS1 modulates the MAPK pathway in RB (Fig. 5a, b). These data suggested that LOXL1-AS1 can modulate the progression of RB cells through the MAPK pathway.

LOXL1-AS1 can regulate the MAPK pathway in RB cell lines. a and b Western blot analysis revealed that the MAPK pathway’s protein expression changed in both cell lines. *p < 0.05, **p < 0.01, ***p < 0.001
Knockdown of LOXL1-AS1 inhibited tumor growth in vivo
To determine whether LOXL1-AS1 affects tumor growth in vivo, a xenograft model of RB was established by inoculating Y79 cells into nude mice. Four weeks later, the mice were sacrificed and the tumors were monitored. Compared with the scramble group, the tumors in the sh-LOXL1-AS1 group grew slowly under the skin (Fig. 6a, b). The volume and weight of tumors from the sh-LOXL1-AS1 group were significantly lower than those from the scramble group (Fig. 6c, d). On this basis, qRT‒PCR was also conducted to detect the expression of LOXL1-AS1 in a subcutaneous tumorigenesis model (Fig. 6e). Immunohistochemical staining showed that the positivity of Ki-67 was clearly different between the sh-LOXL1-AS1 group and the scramble group (Fig. 6f, g). We also assessed the MAPK signaling pathway in vivo; the protein levels were similar to the results in vitro (Fig. 6h).

Knockdown of LOXL1-AS1 regulates tumorigenicity in vivo. a and b The effects of LOXL1-AS1 knockdown in subcutaneous tumorigenesis from the two groups. c and d The tumor volume and weights in xenografts from the two groups. e qRT-PCR analysis found that the expression level of LOXL1-AS1 was decreased in the tumor of the mice from sh-LOXL1-AS1 group. f and g Hematoxylin and eosin staining images and immunohistochemistry images of KI-67 in resected tumor tissues. h Western blot analysis was used to detect the expression of the MAPK pathway in tumor tissues. **p < 0.01, ***p < 0.001, ****p < 0.0001
Discussion
Although multimodal treatment regimens for retinoblastoma have advanced over the decades, systemic chemotherapy supplemented by local ophthalmic artery interventional therapy is recommended as the most vital therapeutic modality [17]. After a period of chemotherapy, RB patients frequently develop drug resistance, and the molecular mechanism of RB chemotherapy resistance remains unclear [18]. In summary, various complications follow in the later stages of treatment, including tumor recurrence, drug resistance, and metastasis; new therapeutic options are urgently needed.
In the human genome, approximately 70–90% of genes are transcribed into RNAs, but only 2% of the total RNAs are translated into proteins [19]. Noncoding RNAs have been confirmed to play critical regulatory roles in a variety of physiological, pathological, and carcinogenesis processes, as gene sequences in the remaining RNAs that are not directly involved in coding for proteins are applied [20]. The transcribed content of lncRNAs in noncoding RNA is up to 80%, and previous reports have shown that lncRNAs are differentially expressed in several cancers, such as MALAT1, CCAT1, and MEG3, which can be potential therapeutic targets associated with clinical outcomes [21,22,23].
LncRNA LOXL1-AS1 was identified based on the impact of its abnormal expression in serious studies linked with the cellular stress response [24]. LOXL1-AS1 has been proven to affect cell proliferation, cell cycle, migration, and invasion as an oncogenic lncRNA in osteosarcoma cells [25]. The present study confirmed that LOXL1-AS1 was upregulated in vitro and in vivo, which manifested potent prooncogenic functions in RB. To date, there are no expression data for LOXL1-AS1 in RB tumor tissues. The expression level of LOXL1-AS1 was compared with RB tissues and adjacent tissues in clinical samples. LOXL1-AS1 participates in cancer regulation. Furthermore, we delivered evidence that knockdown of LOXL1-AS1 can effectively suppress cell viability, colony formation, migration, and invasion. According to the CCK-8 assay results, cell viability clearly declined after 36 h of culture, and knockdown of LOXL1-AS1 impaired cell migration and cell invasion capacities. The maximum suppression efficiency of cell migration reached nearly 36% in Y79 cells and 30% in WERI-Rb1 cells. Flow cytometry detection revealed that LOXL1-AS1 also facilitated cell apoptosis and arrested the tumor cell cycle at the G2/M phase in vitro. The disturbance of cell cycle progression is a hallmark of cancer growth. We constructed a xenograft model with knockdown of LOXL1-AS1 in human RB Y79 cell lines, and tumor validity was inhibited in vivo. MMP-2 and MMP-9 are important members of the MMP family, which are the main enzymes that degrade proteins in the extracellular matrix via their endopeptidase activities [26]. MMP-2 and MMP-9 can digest gelatin by binding type II fibronectin repeat sequences to gelatin/collagen. Early reports suggested that MMPs play an essential role in biological systems, acting as cell adhesion molecules, cytoskeletal proteins, and growth factors [27]. ELISA results showed that knockdown of LOXL1-AS1 decreased the secretion of MMP-2 and MMP-9 in Y79 and WERI-Rb1 cells. The mitogen-activated protein kinase (MAPK) pathway is also an evolutionarily conserved kinase module that is involved in activated cell proliferation, stress response, survival, and apoptosis [28].
It has been reported that the MAPK signaling pathway can inhibit the expression of proinflammatory cytokines in human osteoarthritis chondrocytes [29]. Zhuang also reported that lncRNA DRHC restrained the MEK/ERK signaling pathway via c-Myb in hepatocellular carcinoma [30]. In our study, the phosphorylation of p44/42 MAPK (Erk1/2) and MEK1/2 was key to their activation. Erk1/2 receives phosphorylation regulatory signals from the upstream molecule MEK, which phosphorylates transcription factors in the cytoplasm or nucleus to exert biological effects. Interestingly, we showed that the phosphorylated levels of Erk (p-Erk) and MEK (p-MEK) were dramatically reduced after LOXL1-AS1 depletion, proving that LOXL1-AS1 positively regulates the MAPK signaling pathway.
These results suggested that abnormal changes in the MAPK pathway were critical processes in tumor growth, differentiation, and other physiological activities in RB and that they represent promising therapeutic opportunities. The result of the study highlight the importance of tailoring treatment strategies for RB patients with high LOXL1-AS1 expression and this is probably the first study, which shows the effectiveness of reagent targeting LOXL1-AS1 or the MAPK pathway might serve as promising clinical therapeutics against RB.
In conclusion, we investigated a novel lncRNA, LOXL1-AS1, which is substantially overexpressed in RB tissues and concluded that LOXL1-AS1 promotes proliferation and invasion and inhibits cell apoptosis by regulating the MAPK signaling pathway as a critical mediator in RB. It is quite interesting to explore whether LOXL1-AS1 plays its role via other methods to provide a new basis for the diagnosis and treatment of RB in the future.
Data availability
All data generated or analyzed during this study are included in this article.
References
Fabian ID, Onadim Z, Karaa E et al (2018) The management of retinoblastoma. Oncogene 37(12):1551–1560
Ancona-Lezama D, Dalvin LA, Shields CL (2020) Modern treatment of retinoblastoma: a 2020 review. Indian J Ophthalmol 68(11):2356–2365
Dimaras H, Corson TW (2019) Retinoblastoma, the visible CNS tumor: a review. J Neurosci Res 97(1):29–44
Selvarajah A, Flegg K, Sim W et al (2022) Clinical audit of retinoblastoma management: a retrospective single-institution study. Can J Ophthalmol 57(4):257–269
Gudiseva HV, Berry JL, Polski A et al (2019) Next-generation technologies and strategies for the management of retinoblastoma. Genes 10(12):1032
Taniue K, Akimitsu N (2021) The functions and unique features of LncRNAs in cancer development and tumorigenesis. Int J Mol Sci 22(2):632
Ouyang J, Zhong Y, Zhang Y et al (2022) Long non-coding RNAs are involved in alternative splicing and promote cancer progression. Br J Cancer 126(8):1113–1124
Yang M, Lu H, Liu J et al (2022) lncRNAfunc: a knowledgebase of lncRNA function in human cancer. Nucleic Acids Res 50(D1):D1295–D1306
Choudhari R, Sedano MJ, Harrison AL et al (2020) Long noncoding RNAs in cancer: from discovery to therapeutic targets. Adv Clin Chem 95:105–147
Rajagopal T, Talluri S, Akshaya RL et al (2020) HOTAIR LncRNA: a novel oncogenic propellant in human cancer. Clin Chim Acta 503:1–18
Gao YX, Gao HX, Xu XY et al (2020) Effects of lncRNA MALAT1 and lncRNA NKILA on proliferation, invasion and apoptosis of retinoblastoma. Eur Rev Med Pharmacol Sci 24(16):8296–8307
Zhang P, Zhao F, Jia K et al (2022) The LOXL1 antisense RNA 1 (LOXL1-AS1)/microRNA-423-5p (miR-423-5p)/ectodermal-neural cortex 1 (ENC1) axis promotes cervical cancer through the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway. Bioengineered 13(2):2567–2584
Yang X, Xing G, Liu S et al (2020) LncRNA LOXL1-AS1 promotes endometrial cancer progression by sponging miR-28-5p to upregulate RAP1B expression. Biomed Pharmacother 125:109839
Gao R, Zhang R, Zhang C et al (2018) LncRNA LOXL1-AS1 promotes the proliferation and metastasis of medulloblastoma by activating the PI3K/AKT pathway. Anal Cell Pathol 2018:9275685
Bai H, Li X, Wu S (2020) Up-regulation of long non-coding RNA LOXL1-AS1 functions as an oncogene in cervical squamous cell carcinoma by sponging miR-21. Arch Physiol Biochem 1–5
Jiang A, Wu W, Xu C et al (2022) SP2509, a selective inhibitor of LSD1, suppresses retinoblastoma growth by downregulating β-catenin signaling. Invest Ophthalmol Vis Sci 63(3):20
Francis JH, Roosipu N, Levin AM et al (2018) Current treatment of bilateral retinoblastoma: the impact of intraarterial and intravitreous chemotherapy. Neoplasia 20(8):757–763
Yang L, Zhang L, Lu L et al (2020) lncRNA UCA1 increases proliferation and multidrug resistance of retinoblastoma cells through downregulating miR-513a-5p. DNA Cell Biol 39(1):69–77
Yang M, Wei W (2019) Long non-coding RNAs in retinoblastoma. Pathol Res Pract 215(8):152435
Beermann J, Piccoli MT, Viereck J et al (2016) Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev 96(4):1297–1325
Tian X, Xu G (2015) Clinical value of lncRNA MALAT1 as a prognostic marker in human cancer: systematic review and meta-analysis. BMJ Open 5(9):e8653
Zhang H, Zhong J, Bian Z et al (2017) Long non-coding RNA CCAT1 promotes human retinoblastoma SO-RB50 and Y79 cells through negative regulation of miR-218-5p. Biomed Pharmacother 87:683–691
Bi H, Wang G, Li Z et al (2020) Long Noncoding RNA (lncRNA) Maternally Expressed Gene 3 (MEG3) participates in chronic obstructive pulmonary disease through regulating human pulmonary microvascular endothelial cell apoptosis. Med Sci Monit 26:e920793
Hauser MA, Aboobakar IF, Liu Y et al (2015) Genetic variants and cellular stressors associated with exfoliation syndrome modulate promoter activity of a lncRNA within the LOXL1 locus. Hum Mol Genet 24(22):6552–6563
Chen S, Li W, Guo A (2019) LOXL1-AS1 predicts poor prognosis and promotes cell proliferation, migration, and invasion in osteosarcoma. Biosci Rep 39:BSR201904474
Wang D, Wang D, Wang N et al (2016) Long non-coding RNA BANCR promotes endometrial cancer cell proliferation and invasion by regulating MMP2 and MMP1 via ERK/MAPK signaling pathway. Cell Physiol Biochem 40(3–4):644–656
Li K, Tay FR, Yiu C (2020) The past, present and future perspectives of matrix metalloproteinase inhibitors. Pharmacol Ther 207:107465
Peng WX, Huang JG, Yang L et al (2017) Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol Cancer 16(1):161
Sun HY, Hu KZ, Yin ZS (2017) Inhibition of the p38-MAPK signaling pathway suppresses the apoptosis and expression of proinflammatory cytokines in human osteoarthritis chondrocytes. Cytokine 90:135–143
Zhuang R, Zhang X, Lu D et al (2019) lncRNA DRHC inhibits proliferation and invasion in hepatocellular carcinoma via c-Myb-regulated MEK/ERK signaling. Mol Carcinog 58(3):366–375
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Foundation of China (No. 82060501) and Natural Science Foundation of Jiangxi Province and Technology Department Fund (No. 20202ACB206005).
Author information
Authors and Affiliations
Contributions
WW contributed to the conception and design of the study and manuscript preparation. YZ and CX performed to data analysis and interpretation. HY and SL confirmed the authenticity of all the raw data. WW and GH drafted and revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, W., Zhang, Y., Xu, C. et al. LncRNA LOXL1-AS1 promotes proliferation and invasion and inhibits apoptosis in retinoblastoma by regulating the MAPK signaling pathway. Mol Cell Biochem 479, 1011–1022 (2024). https://doi.org/10.1007/s11010-023-04774-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-023-04774-4