
Abstract
Adriamycin (ADR) causes irreversible damage to the heart, leading to ADR-induced cardiomyopathy (ACM). Angiotensin-(1–9) [Ang-(1–9)] is a peptide from the counter-regulatory renin-angiotensin system, but the effects on ACM is unclear. Our study was aimed to explore the effects and underlying molecular mechanisms of Ang-(1–9) against ACM in Wistar rats. Rats were injected intraperitoneally with ADR via six equal doses (each containing 2.5 mg/kg) within a period of 2 weeks to induce ACM. After 2 weeks of ADR treatment, the rats were treated with Ang-(1–9) (200 ng/kg/min) or angiotensin type 2 receptor (AT2R) antagonist PD123319 (100 ng/kg/min) for 4 weeks. Although Ang-(1–9) treatment did not influence blood pressure, it significantly improved left ventricular function and remodeling in ADR-treated rats, by inhibiting collagen deposition, the expression of TGF-β1, inflammatory response, cardiomyocyte apoptosis and oxidative stress. Moreover, Ang-(1–9) reduced ERK1/2 and P38 MAPK phosphorylation. The therapeutic effects of Ang-(1–9) were blocked by the AT2R antagonist PD123319, which also offset the down-regulation protein expression of pERK1/2 and pP38 MAPK induced by Ang-(1–9). These data suggest that Ang-(1–9) improved left ventricular function and remodeling in ADR-treated rats by an AT2R/ ERK1/2 and P38 MAPK-dependent mechanism. Thus, the Ang-(1–9)/AT2R axis may provide a novel and promising target to the prevention and treatment of ACM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adriamycin (ADR), under the brand name doxorubicin, is one of the most effective antitumor drugs with broad anti-tumor spectrum and strong anti-tumor activity, and is widely used in the treatment of hematogenous and solid cancers in clinic. However, this drug can cause dosage-dependent cardiotoxicity, which limits the clinic administration of ADR in pediatric, adult, and recurrent cancer patients. Patients receiving long-term ADR therapies develop cardiac dysfunction even at a lower dosage [1]; the incidence of congestive heart failure was approximately 26% when patients received ADR at a cumulative dose over 550 mg/m2. Adriamycin-induced cardiomyopathy (ACM) caused by ADR is characterized by cardiomyocyte loss, progressive cardiac enlargement, and ultimately congestive heart failure. Previous publications indicated that multiple mechanisms have been shown to be involved in ACM, including oxidative stress, myocardial fibrosis, inflammation, apoptosis and mitochondrial disruption [2], however the exact mechanism is indistinct. In addition, there is no specific evidence-based treatment of ACM. Therefore, exploring novel promising targets for ACM treatment and uncovering the underlying mechanism is urgently needed.
Angiotensin-(1–9) [Ang-(1–9)] is a nine-amino acid peptide of the non-canonical renin-angiotensin system (RAS) produced from Ang I by the monopeptidase angiotensin-converting enzyme type 2 (ACE2) [3], which also produces Ang-(1–7) from Ang II hydrolysis. Moreover, Ang-(1–7) can be synthesized from Ang-(1–9) through the action of ACE [4]. Ang-(1–7) is proposed to function via the G-protein-coupled receptor Mas [5]. ACE2, Ang-(1–7), Mas receptor and Ang-(1–9) are considered the counter-regulatory arm of RAS and has been shown to counteract the effects of the classical RAS [6]. The classic RAS pathway converts the cleavage of the peptide angiotensinogen into angiotensin I under the protease renin [7]. Then, ACE hydrolyze Ang I to form Ang II, which contributes to the pathophysiology of cardiovascular disease and induces aldosterone release through AT1R. Studies demonstrated that Ang-(1–9) could inhibit myocardial hypertrophy induced by norepinephrine or Ang II [8, 9]. In the stroke-prone spontaneously hypertensive rat, Ang-(1–9) improve endothelial function and reduce oxidative stress, collagen deposition and cardiac fibrosis [10]. Furthermore, gene therapy with Ang-(1–9) prevented sudden cardiac death and greatly preserved left ventricular systolic function after myocardial infarction [11]. Similar favorable effects for Ang-(1–9) were reported in streptozotocin-induced diabetic rats, and Ang 1–9 suppressed the oxidative stress and the generation of pro-inflammatory cytokines in the myocardium [12]. Ang-(1–9) has significant cardiovascular bioactivity in vivo and in vitro by activating angiotensin receptor type 2 (AT2R) [4, 9, 10]. The protective effects of Ang-(1–9) on cardiovascular disease were not dependent on Ang-(1–9) to Ang-(1–7) conversion, because co-treatment of the Mas receptor antagonist A779 did not impact on the effects of Ang-(1–9) [13]. Moreover, the Ang-(1–9)/AT2R axis should be investigated as a new alternative for treatment of cardiovascular disease.
In the current study, we hypothesized that Ang-(1–9) may improve cardiac function of rats with ACM via through AT2R-dependent mechanisms. Based on this hypothesis, we determined investigate the therapeutic effect of Ang-(1–9) on ACM, including myocardial fibrosis, inflammation, apoptosis and oxidative stress.
Materials and methods
Animal model
All animal experimental procedures were performed according to the recommendations in U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 8523 revised 1996). All male Wistar rats (8–10 weeks of age, 250–300 g body weight) were housed in temperature-controlled and humidity-controlled cages with a 12 h light and 12 h dark cycle and given free access to water and food. The cages were kept clean and dry. The animal protocol was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University.
All rats were randomly divided into 2 groups: the control group (n = 15) and the treatment group (n = 150). As previously described [14], ADR (Sigma, Saint Louis, USA) was injected intraperitoneally (i.p.) via six equal doses (each containing 2.5 mg/kg ADR) to rats in the treatment group within a period of 2 weeks for a total cumulative dose of 15 mg/kg body weight. Control rats received saline injections in the same regimen as ADR. Two weeks later the surviving rats (n = 144) in the treatment group were randomly divided into 3 groups (n = 48 per group): the mock group, the Ang-(1–9) group and the Ang-(1–9) + PD123319 group. All rats in these three groups were anesthetized with 2% isofluorane in 1.5 L/min O2 and implanted Alzet osmatic minipumps (Cupertino, California, USA) to deliver saline vehicle in the mock group, Ang-(1–9) (Phoenix Pharmaceuticals, Burlingame, CA, USA, 200 ng/kg/min) in the Ang-(1–9) group and Ang-(1–9) (200 ng/kg/min) + angiotensin type 2 receptor (AT2R) antagonist PD123319 (Sigma, St Louis, MO, USA, 100 ng/kg/min) in the Ang-(1–9) + PD123319 group for 28 days. At the end of experiment, all rats were sacrificed under anesthesia. The hearts of rats were extracted for further morphological and histological studies.
Body weight, heart weight and blood pressure measurement
The body weights (BW) and heart weights (HW) were measured in rats using an electronic scale (Seca Company, Germany) at the end of the experiment.
Systolic blood pressure (sBP) and diastolic blood pressure (dBP) of the conscious rats in the four groups were obtained by a noninvasive tail-cuff system (Softron BP-98A. Tokyo, Japan). The same operator measured blood pressure between 8:00 and 10:00 am and recorded it as the average of three consecutive measurements.
Echocardiography
At the end of experiment, transthoracic echocardiographic scanning was performed in rats anesthetized with 1.5% isoflurane using Vevo2100 high-resolution imaging system (VisualSonics, Toronto, Canada). Echocardiographers were blinded to the animals’ identity. Left ventricular end-systolic diameter (LVESD), Left ventricular end-diastolic diameter (LVEDD), interventricular septum thickness (IVS), left ventricular posterior wall thickness (LVPW) were measured by M-mode echocardiography of the LV at the level of the papillary muscles. Left ventricular ejection fraction (LVEF) was calculated using the formula: LVEF = (LVEDV − LVESV)/LVEDV × 100. Left ventricular end-diastolic volume (LVEDV) and left ventricular end-systolic volume (LVESV) were also calculated by the formula. Fractional shortening (FS) was calculated using the formula: FS = (LVEDD − LVESD)/ LVEDD × 100. The early (E) and late (A) diastolic mitral flow velocities were measured by pulsed Doppler in the four-chamber view and the ratio of E/A was calculated. All measurements represent the mean of 6 consecutive cardiac cycles.
Histopathology and immunohistochemistry
Freshly isolated hearts were fixed in 10% formalin, paraffin-embedded, and cut into serial sections 4 μm thick. The sections of myocardial tissue were dewaxed, rehydrated and stained with hematoxylin–eosin to display cardiomyocyte morphology. Masson’s trichrome staining was performed to detect myocardial fibrosis. The collagen components were expressed as the proportion of area positively stained with Masson’s trichrome to the total left ventricular area in the section. The collagen I and III, VCAM-1 and IL-6 protein expressions in the myocardium were detected by immunohistochemical staining. The sections were incubated with corresponding primary antibodies against collagen I (1:100; Abcam), collagen III (1:50; Abcam), VCAM-1 (1:100; Abcam) and IL-6 (1:100; Abcam) overnight at 4 °C, and subsequently incubated with secondary antibody for 30 min at 37 °C. Then sections were stained with diaminobenzidine and hematoxylin. The images from all histopathological sections were obtained with a microscope (BX41, Olympus). Positive areas were measured with Image Pro Plus software (Media Cybernetics Inc.).
Western blot analysis
Total proteins from myocardial tissues were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. After blocking nonspecific proteins with 5% skim milk for 1.5 h at room temperature, the PVDF membranes were incubated with different primary antibodies overnight at 4 °C. The specific primary antibodies included collagen I (1:1000; Abcam, UK), collagen III (1:1000; Abcam, UK), TGF-β1 (1:1000; Abcam, UK), β-actin (1:1000; Cell Signaling Technology (CST), USA), CD68 (1:1000; CST, USA), ICAM-1 (1:1000; Abcam, UK), TNF-α (1:1000; Abcam, UK), pERK1/2 (1:800; CST, USA), ERK1/2 (1:800; CST, USA), pP38 (1:1000; CST, USA), P38 (1:1000; CST, USA), Bcl-2 (1:1000; Abcam, UK), c-caspase3 (1:1000; Abcam, UK), caspase3 (1:1000; Abcam, UK), P47 (1:500; Santa Cruz Biotechnology, USA), NOX4 (1:1000; Abcam, USA), AT2R (1:1000; Abcam, UK). Then, all membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibody (1:5000, Proteintech, Wuhan, China) for 1 h at room temperature. The protein bands were visualized by use of an enhanced chemiluminescence reagent (Millipore Corp., MA, USA). The levels of protein expression were normalized to that of β-actin.
TUNEL staining
Apoptosis of myocardial samples was measured by TUNEL staining using the In Situ Cell Death Detection Kit, TMR red (Roche, Germany) according to the manufacturer’s instructions. The paraffin-embedded sections of myocardial tissue were dewaxed and rehydrated using xylene and ethanol gradings, incubated with proteinase K working solution, washed by PBS for 2 times, and then treated for 1 h at 37 °C with the TUNEL reaction mixture containing TdT and fluorescein labeled dUTP. Sections were washed by PBS for 3 times and stained with DAPI to show cellular nuclei for 10 min at 37 °C. The images were obtained with a fluorescence microscope (Ni-E, Nikon, Japan) using an excitation wavelength. The ratio of apoptosis was expressed as the proportion of apoptotic cells to the total number of cardiomyocytes.
NOX activity assay
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) activity in myocardial homogenate samples was measured according to the method using an assay kit (Solarbio, Beijing, China). NOX oxidized NADPH to NAD, and the oxidation of NADPH was coupled with the reduction of 2,6 dichlorophenol indigo (DCPIP). The blue DCPIP was reduced to colorless DCPIP. The reduction rate of blue DCPIP at 600 nm was measured to calculate the activity of NOX.
SOD activity assay
The activity of superoxide dismutase (SOD) in the myocardium of rats was determined using an assay kit (WST-1; Beijing, Solarbio, China) following the manufacturer’s instructions. SOD catalyzed the dismutation of the superoxide anion (O2–) into hydrogen peroxide and molecular oxygen. O2− was generated through the reaction system of xanthine and xanthine oxidase, and it acted with WST-1 to produce a water-soluble yellow formazan dye which can be detected by a spectrophotometer in absorbance at 450 nm. The SOD activity was expressed as IU/mg protein.
RNA extraction and real-time quantitative PCR
Freshly isolated heart samples from the rats of four groups was homogenized and total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA). according to the manufacturer’s instruction. The concentrations of total RNA were detected by the absorbance at 260 nm using a spectrophotometer. The mRNA expression of myocardial AT2R and β-actin was determined by real-time quantitative PCR, which was performed with the Applied Biosystems cDNA Reverse Transcription and Takara SYBR RT-PCR kits (Applied Biosystems, Life Technologies). The primer sequences for AT2R and β-actin genes were listed as follows: AT2R, forward: 5′-TGGCTTGTCTGTCCTCAT-3′, reverse: 5′-AGACTTGGTCACGG GTAA-3′; β-actin, forward 5′-TGTTGCCCTAGACTTCGAGCA-3′, reverse 5′-GGACCCAGGAAGGAAGGCT-3′. Quantitative values were obtained using the threshold cycle value (Ct) and the levels of relative mRNA expression were determined by the 2−∆∆CT method.
Statistical analysis
Continuous data were reported as mean ± standard error (SEM). One-way ANOVA with Post hoc Tukey tests was used to compare parameters among more than two groups. Statistical analyses were determined using SPSS 21.0 (SPSS Inc., Chicago, IL). P value < 0.05 was considered statistically significant.
Results
Animal basic characteristics
Body weight (BW), heart weight (HW), systolic blood pressure (sBP) and diastolic blood pressure (dBP) were remarkably lower in Mock group rats relative to those in the Control group. Compared with the Mock group, treatment with Ang-(1–9) only significantly increased HW, but had no effect on the other three measures. Co-administration of the AT2R blocker PD123319 abolished the effect of Ang-(1–9) on HW (Table 1).
Cardiac function
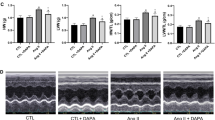
Echocardiographic measurements of LVEF, FS and E/A ratio were remarkably decreased whereas LVESD and LVEDD were increased in the Mock group compared with the Control group (Fig. 1A–F). However, these echocardiographic values were all reversed in the Ang-(1–9) group as compared with the Mock group. In addition, PD123319 co-treatment with Ang-(1–9) increased values of LVESD and LVEDD and decreased that of LVEF, FS and E/A ratio relative to the Ang-(1–9) group. Thus, Ang-(1–9) ameliorated left ventricular remodeling and rescued systolic and diastolic function in rats with ADR treatment, while PD123319 abolished the effects of Ang-(1–9) on ACM in rats.

Cardiac function assessed by echocardiographic imaging and in Wistar rats treated with ADR, Ang-(1–9) and/or PD123319. A Representative echocardiographic graphs in two-dimensional parasternal long axis view and M-mode of the LV at the level of the papillary muscles. LV, left ventricular. B Left ventricular ejection fraction (LVEF). C Fractional shortening (FS). D Left ventricular end-systolic diameter (LVESD). E Left ventricular end-diastolic diameter (LVEDD). F Ratio of the early to late diastolic mitral flow velocities (E/A ratio). G Representative images of hematoxylin and eosin staining of the myocardium in 4 groups of rats. Scale bar: 20 μm. N = 10–15 per group. Data were presented as mean ± SEM. *P < 0.05 vs Control. &P < 0.05 vs Mock. #P < 0.05 vs Ang-(1–9)
Pathological changes
Relative to the Control group, rats of the Mock group exhibited significant inflammatory cells invasion, loss of myofibrils and disorganization under H&E staining (Fig. 1G). In contrast, these abnormalities in myocardial morphology were largely reversed by Ang-(1–9) treatment. Co-administration of the PD123319 abolished the effect of Ang-(1–9) on the degree of the pathological changes.
Cardiac fibrosis
In comparison with the Control group, interstitial fibrosis area in the myocardium displayed by Masson’s trichrome staining, and collagen I and collagen III in the myocardium revealed by immunohistochemical staining were substantially increased in the Mock group, but significantly decreased in the Ang-(1–9) rat group, relative to the Mock and Ang-(1–9) + PD123319 group (Fig. 2A–D). Immunohistochemical results of collagen I and collagen III in the myocardium were further verified by western blot analysis, a similar distribution in all 4 groups (Fig. 2E). Thus, the myocardial fibrosis in ACM rats was alleviated following treatment with Ang-(1–9), and the anti-fibrotic effects of Ang-(1–9) were mediated via AT2R.

Levels of myocardial fibrosis in 4 groups of rats. A Representative Masson’s trichrome staining of myocardial fibers, and immunohistochemical staining of collagen I and collagen III in the myocardium of 4 groups of rats. Scale bar: 20 μm. B–D quantitative analysis of collagen, collagen I and collagen III in the myocardium of the 4 groups of rats. E Representative Western blot images of collagen I, collagen III and TGF-β1 protein expressions in the myocardium of the 4 groups of rats. F Quantification of the protein expressions of TGF-β1 in the myocardium of the 4 groups of rats. N = 6 per group. Data were presented as mean ± SEM. *P < 0.05 vs Control. &P < 0.05 vs Mock. #P < 0.05 vs Ang-(1–9)
TGF-β1 protein expression
As depicted in Fig. 2E, F, Western blot analyses of myocardial tissues indicated that the protein expression of transforming growth factor-β1 (TGF-β1) by western blot analysis were significantly higher in the Mock than the Control rats, which were lowered by Ang-(1–9) treatment, whereas such effect was completely offset by co-administration of PD123319 and Ang-(1–9).
Inflammation
Western blot and immunohistochemical staining showed that protein expressions of CD68 (a marker of macrophage, Fig. 3A), VCAM-1 (Fig. 3B, C), IL-6 (Fig. 3B, D), ICAM-1 (Fig. 3E, F) and TNF-α (Fig. 3E, G) in the myocardium of the Mock group rats were substantially increased which were completely attenuated by Ang-(1–9) treatment. The results supported the anti-inflammatory effects of Ang-(1–9), whereas PD123319 blocked these effects of Ang-(1–9), suggesting the anti-inflammatory effects of Ang-(1–9) were mediated via AT2R.

Levels of cardiac inflammation in 4 groups of rats. A Representative Western blot images and quantification of CD68 protein expression in the myocardium of the 4 groups of rats. B Representative immunohistochemical staining of VCAM-1 and IL-6 of the myocardium in 4 groups of rats. Scale bar: 20 μm. C, D quantitative analysis of VCAM-1 and IL-6 in the myocardium of the 4 groups of rats. E Representative Western blot images and quantification of ICAM-1 and TNF-α protein expressions in the myocardium of the 4 groups of rats. F, G Quantification of ICAM-1 and TNF-α protein expressions in the myocardium of the 4 groups of rats. N = 6 per group. Data were expressed by mean ± SEM. *P < 0.05 vs Control. &P < 0.05 vs Mock. #P < 0.05 vs Ang-(1–9)
Apoptosis
We also detected the apoptosis in the myocardium by TUNEL staining, which revealed an upsurge of TUNEL-positive signals in the myocardium of Mock group rats compared with the Control group, and this abnormality was effectively normalized by Ang-(1–9) treatment (Fig. 4A, B). However, the proportion of TUNEL-positive apoptotic cardiomyocytes in the myocardium was markedly increased in the Ang-(1–9) + PD123319 group versus the Ang-(1–9) group rats.

Levels of apoptosis in 4 groups of rats. A Representative TUNEL-positive cardiomyocyte staining in 4 groups of rats. Scale bar: 20 μm. B Quantification of TUNEL-positive cardiomyocytes in 4 groups of rats. C Representative Western blot images of Bcl-2, c-caspase-3 and caspase-3 protein expressions in the myocardium of the 4 groups of rats. D, E Quantification of Bcl-2 protein expression and c-caspase-3/caspase-3 in the myocardium of the 4 groups of rats. N = 6 per group. Data were shown as mean ± SEM. *P < 0.05 vs Control. &P < 0.05 vs Mock. #P < 0.05 vs Ang-(1–9)
To further evaluate the effect of Ang-(1–9) on apoptosis in the myocardium, the levels of Bcl-2 and cleaved caspase3 (c-caspase3) protein expressions in the myocardium tissue were measured by western blot, which showed a significant downregulation of Bcl-2 protein expression and upregulation of c-caspase3 in the Mock group compared with Control group, whereas Ang-(1–9) treatment counteracted these adverse effects of Adriamycin (Fig. 4C–E). In addition, these cardioprotective effects of Ang-(1–9) were largely offset by the addition of PD123319 to Ang-(1–9) treatment. These results suggested that Ang-(1–9) treatment attenuated cardiac apoptosis in ACM rats, and the anti-apoptotic effects of Ang-(1–9) were mediated via AT2R.
Oxidative stress
Compared to controls, the protein expressions of P47 and nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) (Fig. 5A–C) by western blot analysis, and the levels of NOX activity (Fig. 5D) in heart tissues were higher in ADR-treated rats. Treatment of with Ang-(1–9) reduced the protein expressions of P47 and NOX4, and suppressed NOX activity in hearts. The antioxidative effect of Ang-(1–9) was abolished by co-administration of the PD123319. Moreover, ADR treatment inhibited the activity of SOD (an antioxidant enzyme) in the myocardial homogenate. The adverse effect induced by ADR was ameliorated in rats treated with Ang-(1–9) (Fig. 5E). Nevertheless, co-administration of PD123319 and Ang-(1–9) abolished the effect of Ang-(1–9).

Levels of oxidative stress in 4 groups of rats. A Representative Western blot images of p47 and NOX4 protein expressions in the myocardium of the 4 groups of rats. B, C Quantification of p47 and NOX4 protein expressions in the myocardium of the 4 groups of rats. D The levels of NADPH oxidase activity in the myocardium of the 4 groups of rats. E The levels of SOD activity in the myocardium of the 4 groups of rats. N = 6 per group. Data were expressed as mean ± SEM. *P < 0.05 vs Control. &P < 0.05 vs Mock. #P < 0.05 vs Ang-(1–9)
ERK and P38 protein expression
Phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2) and phosphorylated P38 MAPK (pP38) protein expression quantitated by western blot were markedly higher in the Mock group than the Control group, whereas pERK1/2 and pP38 protein expression were markedly lower in the Ang-(1–9) group than the Mock group (Fig. 6A, B). At the same time, co-administration of PD123319 and Ang-(1–9) abolished such effects of Ang-(1–9).

ERK, P38 and AT2R expressions in 4 groups of rats. A Representative Western blot images of pERK and ERK protein expressions and quantification of pERK/ERK in the myocardium of the 4 groups of rats. B Representative Western blot images of pP38 and P38 protein expressions and quantification of pP38/P38 in the myocardium of the 4 groups of rats. C Representative Western blot images and quantification of AT2R protein expression in the myocardium of the 4 groups of rats. D Quantitative analysis of AT2R messenger ribonucleic acid (mRNA) expression by real-time PCR in the myocardium of the 4 groups. N = 6 per group. Data were shown as mean ± SEM. *P < 0.05 vs Control. &P < 0.05 vs Mock. #P < 0.05 vs Ang-(1–9)
AT2R expression
The levels of AT2R protein and mRNA expression measured by western blot and real-time quantitative PCR were remarkably decreased in ACM rats relative to the control rats whereas Ang-(1–9) treatment significantly increased AT2R protein and mRNA expression levels in ACM rats (Fig. 6C, D). The effects of Ang-(1–9) on AT2R protein and mRNA expression was reversed by co-administration of the PD123319.
Discussion
Although the beneficial effects of Ang-(1–9) on hypertension [15], myocardial infarction [11], cardiomyocyte hypertrophy [8] and diabetic cardiomyopathy [12] have been shown in experimental studies, the exact role of Ang-(1–9) in ACM remains unclear. The major finding of the current study was that Ang-(1–9) treatment improved left ventricular remodeling and function in rats with adriamycin-induced cardiomyopathy by attenuating cardiac fibrosis, inflammation, apoptosis and oxidative stress. As far as we know, this study is the first to report the beneficial effects and the relevant mechanisms of Ang-(1–9) in a rat model of ACM.
ACM is characterized by cardiomyocyte loss, progressive cardiac enlargement, leading to congestive heart failure. Unfortunately, there is, so far, no effective approach to prevent or treat ACM except for Dexrazoxane [16], which still has some limitations in clinical application. Previous experimental study has shown that Ang-(1–9) inhibited oxidative stress and inflammatory response induced by streptozotocin, an animal model of diabetic cardiomyopathy [12]. However, whether Ang-(1–9) alleviated ADR-induced cardiomyopathy and, if it did, what mechanisms were responsible, remained to be explored. In the present study, Ang-(1–9) improved cardiac function and heart wight of rats with ACM. In addition, Ang-(1–9) had no affect blood pressure in ADR-treated rats, indicating that such beneficial effects of Ang-(1–9) cannot contribute to haemodynamic changes. This lack of effect on blood pressure may be due to the Ang-(1–9) dose used. The doses of Ang-(1–9) 600 ng/kg/min or above reduced blood pressure [17], while not at lower doses.
Adriamycin administration resulted in collagen deposition within the myocardium in rats [18]. Fibrosis has been suggested to be involved in ventricular remodeling and cardiac dysfunction in a rat model of ACM [19]. In the stroke-prone spontaneously hypertensive rat, Ang-(1–9) treatment resulted in a 50% reduction in cardiac fibrosis compared with control animals [10]. In the present study, Ang-(1–9) treatment attenuated myocardial collagen deposition and improved LV remodeling, which were consistent with the previous report [12]. An important factor regulating collagen production is TGF-β. The increased protein levels of TGF-β1 were induced by ADR stimulation [20], which aggravated myocardial fibrosis. Ang-(1–9) treatment prevented the upregulation of TGF-β1 in hypertension rats [21]. Our present study showed that TGF-β1 protein expression was significantly higher in the Ang-(1–9) group than that in the mock group, indicating that Ang-(1–9) treatment decreased collagen production partially through inhibition of TGF-β1 protein expression.
The infiltration of inflammatory cells was involved in ADR-induced cardiac damage [22]. While CD68+ macrophage were the predominant inflammatory cells in the hearts in rat model of ACM [23]. ADR treatment provoked a host of inflammatory reactions through activating the subsequent production of pro-inflammatory cytokines [24, 25], such as TNF-α and IL-6. Over-activation of the inflammatory response induced by ADR exacerbated collagen deposition, which lead to ventricular pathologic remodeling and cardiac dysfunction [26]. Ang-(1–9) protects against volume overload-induced hypertensive cardiovascular damage by decreasing inflammation in the heart and aortic wall [27]. Consistent with these findings, we found that the protein expressions of CD68 and pro-inflammatory cytokines (VACM-1, IL-6, ICAM-1 and TNF-α) were markedly increased in ADR-induced rats, and were significantly inhibited suppressed by Ang-(1–9) treatment.
Apoptosis has been put forth as being involved in cardiac dysfunction under some experimental and clinical conditions [28]. ADR-induced apoptosis in cardiomyocytes was involved in cardiac dysfunction and was associated with multiple signaling pathways [24, 29]. The expression of cleave-caspase3 was up-regulated and that of Bcl-2 was down-regulated in the ADR-induced cardiomyocyte apoptosis [30]. Ang-(1–9) lowered the expression of apoptosis-related proteins such as Bax, Bcl-2 and caspase-3 in a rat model of pulmonary hypertension [13]. In this study, our results demonstrated that Ang-(1–9) treatment reduced TUNEL-positive cells, the activity of caspase3 and increased bcl-2 protein expression in ADR-induced rats.
The most commonly proposed mechanism for ACM is the generation of reactive oxygen species (ROS), leading to oxidative stress [31]. The experimental evidence also has been manifested that there is overwhelming connection between oxidative stress and ACM [32]. ADR-induced ROS generation can trigger DNA damage and inflammatory cytokines expression [33], causing ultimately lipid peroxidation-dependent ferroptosis [34] and cardiomyocyte apoptosis by activating apoptotic signaling [35]. Activated NOX can generate ROS, which is regulated through transcriptional repression of the p47phox gene [36], a cytoplasmic subunit of NOX2. Previous study also showed that ADR promotes NOX expression and leads to phosphorylation of p47phox [31]. In addition, the enhancement of SOD activity protected mice from doxorubicin-induced injury [37]. Ang-(1–9) reduced ROS formation and suppressed NOX activity in the myocardium of streptozotocin-induced diabetic rats [12]. In the current study, we also found that Ang-(1–9) decreased the protein expression of NOX4 and p47phox, suppressed NOX activity and enhanced the activity of SOD in the myocardium of ADR-treated rats.
Previous studies found that ERK1/2, a member of the mitogen-activated protein kinases (MAPKs) family, and p38 MAPK was associated with inflammatory signaling, apoptotic signaling and ROS generation [38,39,40,41], and involved in the pathogenesis of ADR-induced cardiotoxicity. ADR exposure induced the overexpression of phosphorylated p38 MAPK, which was significantly suppressed by SB203580, a specific p38 MAPK inhibitor [25]. In our study, the expression levels of pERK1/2 and pP38 MAPK were remarkedly increased by ADR injection, which was attenuated by Ang-(1–9) in the myocardium of rats.
AT2R is an inhibitory G-protein-coupled receptor with a high affinity for Ang-(1–9) [9] and previous studies have manifested that in all probability, most protective effects of Ang-(1–9) on cardiovascular disease are mediated by AT2R. Ang-(1–9) acts via an AT2R-dependent mechanism to achieve anti-hypertrophic and anti-hypertensive effects in the cardiomyocytes and heart [9, 21]. Therefore, we used PD123319, a known AT2R antagonist, to examine the action of this receptor in Ang-(1–9)-mediated protective effects on ACM. We found in this study that ADR treatment markedly decreased AT2R protein and mRNA expression, whereas co-administration of Ang-(1–9) upregulated the expression of AT2R. These results indicated that AT2R might play a crucial part in the protective effects of Ang-(1–9) on ACM. After antagonizing AT2R with PD123319, the anti-fibrotic, anti-inflammatory, anti-apoptotic and anti-oxidative effects of Ang-(1–9) on ACM were blocked; moreover, the protein expression of pERK1/2 and pP38 MAPK was up-regulated. Thus, down-regulation of ERK1/2 and P38 MAPK via AT2R may be volved in the molecular mechanism of the salutary effect of Ang-(1–9) on ACM. Our current findings further support the beneficial role of the Ang-(1–9)/AT2R axis in the cardiovascular system.
Multiple experimental studies have demonstrated that Ang-(1–9) plays its roles by activating AT2R and that its actions are independent of MasR [9, 42]. This confirmation was studied using the AT2R antagonist PD123319, which was not specific to AT2R and also blocked MasR [43]. Thus, our study limitations included two aspects. Firstly, we did not perform further experiments with myocardium-specific AT2R knockout rats, which may help to confirm the actions for AT2R in Ang-(1–9) beneficial effects. Secondly, we also did not use MasR antagonist A779 to verify the effects of Ang-(1–9) independent of MasR in the present study.
In conclusion, our study has shown that in a rat model of ACM induced by ADR, Ang-(1–9) treatment improved left ventricular function and remodeling though the inhibition of the collagen deposition, expression of TGF-β1, inflammatory response, cardiomyocyte apoptosis and oxidative stress. The underlying molecular mechanisms may involve inhibited ERK1/2 and P38 MAPK pathways via Ang-(1–9) stimulation of AT2R. Thus, the Ang-(1–9)/AT2R axis may provide a novel and promising target to the prevention and treatment of ACM.
Data availability
All relevant data are presented in the present paper.
References
Wallace KB, Sardão VA, Oliveira PJ (2020) Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res 126:926–941
Kalyanaraman B (2020) Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree? Redox Biol 29:101394
Donoghue M, Hsieh F, Baronas E et al (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87:E1-9
Ocaranza MP, Michea L, Chiong M et al (2014) Recent insights and therapeutic perspectives of angiotensin-(1–9) in the cardiovascular system. Clin Sci 127:549–557
Santos RA, e Silva ACS, Maric C et al (2003) Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA 100:8258–63
Paz Ocaranza M, Riquelme JA, García L et al (2020) Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat Rev Cardiol 17:116–129
Fountain JH, Lappin SL (2022) Physiology, Renin angiotensin system. StatPearls Treasure Island (FL): StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC
Ocaranza MP, Lavandero S, Jalil JE et al (2010) Angiotensin-(1–9) regulates cardiac hypertrophy in vivo and in vitro. J Hypertens 28:1054–1064
Flores-Muñoz M, Smith NJ, Haggerty C et al (2011) Angiotensin1-9 antagonises pro-hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J Physiol 589:939–951
Flores-Munoz M, Work LM, Douglas K et al (2012) Angiotensin-(1–9) attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rat via the angiotensin type 2 receptor. Hypertension 59:300–307
Fattah C, Nather K, McCarroll CS et al (2016) Gene therapy with angiotensin-(1–9) preserves left ventricular systolic function after myocardial infarction. J Am Coll Cardiol 68:2652–2666
Zheng H, Pu SY, Fan XF et al (2015) Treatment with angiotensin-(1–9) alleviates the cardiomyopathy in streptozotocin-induced diabetic rats. Biochem Pharmacol 95:38–45
Cha SA, Park BM, Kim SH (2018) Angiotensin-(1–9) ameliorates pulmonary arterial hypertension via angiotensin type II receptor. Korean J Physiol Pharmacol 22:447–456
Siveski-Iliskovic N, Kaul N, Singal PK (1994) Probucol promotes endogenous antioxidants and provides protection against adriamycin-induced cardiomyopathy in rats. Circulation 89:2829–2835
Norambuena-Soto I, Lopez-Crisosto C, Martinez-Bilbao J et al (2022) Angiotensin-(1–9) in hypertension. Biochem Pharmacol 203:115183
Chen Y, Shi S, Dai Y (2022) Research progress of therapeutic drugs for doxorubicin-induced cardiomyopathy. Biomed Pharmacother 156:113903
Norambuena-Soto I, Ocaranza MP, Cancino-Arenas N et al (2020) Angiotensin-(1–9) prevents vascular remodeling by decreasing vascular smooth muscle cell dedifferentiation through a FoxO1-dependent mechanism. Biochem Pharmacol 180:114190
Yu SY, Liu L, Li P et al (2013) Rapamycin inhibits the mTOR/p70S6K pathway and attenuates cardiac fibrosis in adriamycin-induced dilated cardiomyopathy. Thorac Cardiovasc Surg 61:223–228
Hong YM, Lee H, Cho MS et al (2017) Apoptosis and remodeling in adriamycin-induced cardiomyopathy rat model. Korean J Pediatr 60:365–372
Xu Y, Qu X, Zhou J et al (2021) Pilose antler peptide-3.2KD ameliorates adriamycin-induced myocardial injury through TGF-β/SMAD signaling pathway. Front Cardiovasc Med 8:659643
Ocaranza MP, Moya J, Barrientos V et al (2014) Angiotensin-(1–9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J Hypertens 32:771–783
Li L, Takemura G, Li Y et al (2006) Preventive effect of erythropoietin on cardiac dysfunction in doxorubicin-induced cardiomyopathy. Circulation 113:535–543
Hadi N, Yousif NG, Al-amran FG et al (2012) Vitamin E and telmisartan attenuates doxorubicin induced cardiac injury in rat through down regulation of inflammatory response. BMC Cardiovasc Disord 12:63
Qi W, Boliang W, Xiaoxi T et al (2020) Cardamonin protects against doxorubicin-induced cardiotoxicity in mice by restraining oxidative stress and inflammation associated with Nrf2 signaling. Biomed Pharmacother 122:109547
Guo RM, Xu WM, Lin JC et al (2013) Activation of the p38 MAPK/NF-κB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol Med Rep 8:603–608
Ma Y, Zhang X, Bao H et al (2012) Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS ONE 7:e40763
Gonzalez L, Novoa U, Moya J et al (2018) Angiotensin-(1–9) reduces cardiovascular and renal inflammation in experimental renin-independent hypertension. Biochem Pharmacol 156:357–370
Saraste A, Pulkki K, Kallajoki M et al (1997) Apoptosis in human acute myocardial infarction. Circulation 95:320–323
Kumar D, Kirshenbaum LA, Li T et al (2001) Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Antioxid Redox Signal 3:135–145
Wu S, Ko YS, Teng MS et al (2002) Adriamycin-induced cardiomyocyte and endothelial cell apoptosis: in vitro and in vivo studies. J Mol Cell Cardiol 34:1595–1607
Kong CY, Guo Z, Song P et al (2022) Underlying the mechanisms of doxorubicin-induced acute cardiotoxicity: oxidative stress and cell death. Int J Biol Sci 18:760–770
Bhagat A, Kleinerman ES (2020) Anthracycline-induced cardiotoxicity: causes, mechanisms, and prevention. Adv Exp Med Biol 1257:181–192
Wang L, Zhang TP, Zhang Y et al (2016) Protection against doxorubicin-induced myocardial dysfunction in mice by cardiac-specific expression of carboxyl terminus of hsp70-interacting protein. Sci Rep 6:28399
Fang X, Wang H, Han D et al (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA 116:2672–2680
Nitobe J, Yamaguchi S, Okuyama M et al (2003) Reactive oxygen species regulate FLICE inhibitory protein (FLIP) and susceptibility to Fas-mediated apoptosis in cardiac myocytes. Cardiovasc Res 57:119–128
Berasi SP, Xiu M, Yee AS et al (2004) HBP1 repression of the p47phox gene: cell cycle regulation via the NADPH oxidase. Mol Cell Biol 24:3011–3024
Chen CT, Wang ZH, Hsu CC et al (2015) In vivo protective effects of diosgenin against doxorubicin-induced cardiotoxicity. Nutrients 7:4938–4954
Wen SY, Tsai CY, Pai PY et al (2018) Diallyl trisulfide suppresses doxorubicin-induced cardiomyocyte apoptosis by inhibiting MAPK/NF-κB signaling through attenuation of ROS generation. Environ Toxicol 33:93–103
Guo R, Lin J, Xu W et al (2013) Hydrogen sulfide attenuates doxorubicin-induced cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells. Int J Mol Med 31:644–650
Wan Y, Wang J, Xu JF et al (2021) Panax ginseng and its ginsenosides: potential candidates for the prevention and treatment of chemotherapy-induced side effects. J Ginseng Res 45:617–630
Hirata Y (2021) trans-fatty acids as an enhancer of inflammation and cell death: molecular basis for their pathological actions. Biol Pharm Bull 44:1349–1356
Mendoza-Torres E, Oyarzún A, Mondaca-Ruff D et al (2015) ACE2 and vasoactive peptides: novel players in cardiovascular/renal remodeling and hypertension. Ther Adv Cardiovasc Dis 9:217–237
Tetzner A, Gebolys K, Meinert C et al (2016) G-protein-coupled receptor MrgD is a receptor for angiotensin-(1–7) involving adenylyl cyclase, cAMP, and phosphokinase A. Hypertension 68:185–194
Acknowledgements
We kindly thank all the support and contributions of the participators.
Funding
This project was supported by the Natural Science Foundation of Shandong Province (ZR2021QH073). This project was also sponsored by the Clinical Medicine + X Scientific Research Program, Affiliated Hospital of Qingdao University (QDFY + X202101045).
Author information
Authors and Affiliations
Contributions
The concept and design of the present study were provided by HM and RG. HM, CM, YH, LW, XG, LL and FW partially performed some experiments and analyzed data; HM wrote the manuscript; All authors reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
All animal experimental procedures were performed according to the recommendations in U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 8523 revised 1996). The animal protocol was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ma, H., Mao, C., Hu, Y. et al. Angiotensin-(1–9) attenuates adriamycin-induced cardiomyopathy in rats via the angiotensin type 2 receptor. Mol Cell Biochem 479, 73–83 (2024). https://doi.org/10.1007/s11010-023-04718-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-023-04718-y