
Abstract
In the present study, we screened multiple melanoma cell lines for treatment of Apigenin and miRNA expression, also studied the role of miR-512-3p in melanoma. RT-PCR analysis was done for screening miRNA in melanoma cell lines (WM1361B, WM983A, WM1341D, SK-MEL-3, SH-4, SK-MEL-24 and RPMI-7951) compared to normal human epidermal melanocytes. Colony formation assay for cell viability studies, cell cycle by flowcytometry and protein expression by immunoblot analysis. For in vivo analysis tumour xenograft mouse model was created. Immunohistochemistry was done for PCNA positive cells. For expression of miR-512-3p in tumour tissues fluorescence in situ hybridization was done. In silico studies were done by molecular docking studies. The WM1361B and WM983A cell lines showed overexpression of miR-512-3p and increased cell proliferation compared to normal human epidermal melanocytes. Treatment of anti-miR-512-3p to WM1361B and WM983A cells halted cell proliferation and also caused G1-phase arrest. We studied the effect of Apigenin on the expression levels of miR-512-3p and associated molecular targets. Apigenin treatment in WM1361B and WM983A cells showed inhibition in expression of miR-512-3p, arrest of G1 phase of cell cycle, cytotoxicity and revival of p27 Kip1. Apigenin treatment significantly suppressed the growth of WM1361B in tumour induced mice, the activity was associated with decreased levels of miR-512-3p, tumour cell proliferation and increased levels of p27 Kip1 protein. Docking studies confirm potential affinity of Apigenin for p27 Kip1. Apigenin acts as an inhibitor of miR-512-3p by suppressing growth of melanoma both in vitro and in vivo targeting the p27 Kip1 axis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Melanoma is a skin malignancy responsible for major skin cancer-related mortalities [1, 2]. It is a matter of concern that cases of melanoma are on high in age groups ranging 1 to 10 [3]. Number of studies have emerged in an effort to understand the aetiology of melanoma for developing effective therapies; they have achieved only limited success. Melanoma have been characterized to be an aggressive cancer with high malignancy, approaches which would inhibit its growth and progression may lead to a potential strategy for treating them.
MicroRNAs (miRs) are small non-coding RNAs responsible for regulating the expression of various genes by interacting with the 3′ untranslated regions (3′UTRs) of mRNAs [4]. miRs are responsible for regulating various cellular process which include differentiation, development, apoptosis along with proliferation, in addition to all these processes these miRs can act as oncogenes or show tumour suppressing activity in various cancers [4, 5]. However, little is known about the role of miRs in melanoma. miR-512-3p is reported to be associated with multiple malignancies, miR-512-3p acts as a tumour suppressor in hepatic cancer [6] and lung cancer [7]. miR-512-3p is also reported to regulate the HER2 pathway in breast cancer [8], however role of this important miRNA in melanoma remains unclear. The p27Kip1 is one of the important proteins which regulates the cell cycle in mammalian cell [9]. Overexpression of p27Kip1 leads to exit of the proliferating cells from the cell cycle, whereas the down-regulation results in resuming the process of cell division [10]. The low levels of p27Kip1 are often linked with increased proliferation in pathological conditions like cancer [11]. The overexpressed p27Kip1 levels are observed in condition of atherosclerosis which encounters late wound healing [12]. The progression of cell cycle is dependent on the activation of cyclins and cyclin dependent kinases (CDKs), both of these proteins in G1 phase are responsible for inducing the S-phase and G2 phase which leads to mitosis [13].
Recently, bioactive compounds have emerged as an approach for treating melanomas [14]. Flavonoids are one of the phytochemicals which are present in almost all the plant tissues. Apigenin is a flavonoid present abundantly in vegetables, fruits and in many Chinese medicinal plants. It is reported to show number of activities such as anti-oxidant, anti-inflammatory, anti-infective and importantly the anti-cancer property [15, 16], hence Apigenin has been used from centuries as important tradition medicine. Apigenin has shown cytotoxic activity in cancer cells without affecting the normal cells [17]. In the present study, we first evaluated the involvement of miR-512-3p in melanoma cancer cells, then we studied the chemotherapeutic activity of Apigenin on the proliferation of melanoma cells and also whether it was mediated via miR-512-3p. For in vitro study, we selected at least three different melanoma cell lines and checked whether Apigenin inhibited the growth of melanoma cells through the inhibitory effect of miR-512-3p. In the present work, we provide an evidence that Apigenin inhibited the proliferation of melanoma cells in vitro and also suppressed the tumour growth in vivo in melanoma xenograft mouse model. We demonstrated that Apigenin suppressed the levels of miR-512-3p and also blocked the process of cell division in the G1 phase via reactivating the p27Kip1 protein.
Materials and methods
Reagents and antibodies
Lipofectamine reagent, miR-512-3p specific primers, anti-miR-512-3p inhibitors were obtained from Invitrogen, USA. The antibodies specific for p27Kip1, E2F1, E2F2, CDK were obtained from Santa Cruz Biotech., USA. The loading control Actin, U6 and other secondary antibodies were bought from Takara Biotech, USA. All the reagents were of analytical grade.
Melanoma cell lines and culture conditions
For the in vitro studies we selected WM1361B, WM983A, WM1341D, SK-MEL-3, SH-4, SK-MEL-24 and RPMI-7951 human melanoma cell lines which were obtained from the microbiology department of Xiangyang Central Hospital. The cell lines WM1361B, WM983A, WM1341D and SK-MEL-3 were cultured in DMEM media whereas SH-4, SK-MEL-24 and RPMI-7951 were maintained in RPMI-1640 media. The medium was added with FBS 10%, Streptomycin and Penicillin (100 μg/ml) in an incubator at room temperature with CO2 5%. The Normal human epidermal melanocytes (NHEMs) were supplied by American Type Culture Collection (Manassas, VA), which were maintained in HMGs media which was added with melanocytes growth media-254. The Apigenin was prepared by solubilizing in 50 μl DMSO and then added with complete culture media for obtaining the required concertation. The cells were treated when the confluent reached about 60 to 70%.
Extraction of RNA and RT-PCR
The total RNAs were isolated from the melanoma cells and the NHEMs with the help of Trizol reagent (ThermoFisher USA) as per supplied instructions. Briefly, the cell after reaching confluent of 70 to 80% were rinsed with PBS (Ice cold), followed by treatment of Trizol reagent (1 ml) and were collected immediately. The resultant lysate (20 ml) was transferred to a bend tube and chloroform (0.2 ml) was added, the mixture was mixed by continuous shaking and was centrifuged at 10,000×g for 15 min under cold conditions. After centrifugation, the upper colour-less layer was removed and the resultant was added with isopropyl alcohol (5 ml) for precipitating the RNAs. The precipitate was centrifuged 10,000×g for 5 min, after incubation of 10 min at room temperature. The pellet of RNA was obtained at the bottom of tube which was rinsed with isopropyl alcohol and then resuspended in water (nuclease free). The RNA concentration was studied by spectrophotometer. From the isolated RNA the cDNA was synthesised with the help of iScript cDNA synthesis kit (BioRad USA) as per the supplied procedure. Platinum Taq DNA polymerase (ThermoFisher USA) was used to perform RT-PCR analysis using human specific primers as follows, miR-512-3p Forward: 5′-AAGUGCUGUCAUAGCUGAGGUC-3′ and Reverse 5′-CCUCAGCUAUGACAGCACUUUU-3 U6 forward: CTCGCTTCGGCAGCACA, U6 Reverse: AACGCTTCACG AATTTGCGT. RT-PCR was carried on agarose gel (2.5%) prepared using EDTA buffer with ethidium bromide. The RT-PCR steps were as, Stage 1: Initial temperature 95 °C carried for 3 min, then 55 °C carried for 1 min, 75 °C carried for 30 s (2 cycles), Stage 2: Initial temperature 95 °C carried for 3 min, then 55 °C carried for 1 min and 75 °C carried for 30 s (50 cycles), Stage 3: 75 °C for 5 min seconds.
Cell viability studies and colony formation assay
The cell viability studies were carried for NHEMs and melanoma cell lines following MTT assay [18]. Briefly, 1 × 105 cells were transferred and maintained in 96 well plates for 12 h and then treated with Apigenin (0, 10, 20 and 40 µM) for 24 and 48 h. After the treatment, the cells were treated with MTT (50 μl, 5 mg/ml). The Formazan crystals were solubilized in DMSO (100 μl). The optical density was measured at 540 nm in a microplate reader. The cell viability was compared relative to control. Cell proliferation was also compared in melanoma cells devoid any treatment.
To study the effect of Apigenin on cell survival, colony formation assay was done for which the melanoma cells were transferred in complete media. The cells were seeded after transferring them to wells (2 × 105 cells/well). After 4 days, the cells were exposed to various concentrations of Apigenin (20 and 40 µM) for next 2 weeks. The culture cells were incubated at room temperature conditions with 5% CO2.
Cell cycle studies
Flow cytometry study was done for cell cycle analysis following the propidium iodide staining as described earlier [19]. The cells after 48 h transfection with miR or exposure to Apigenin were submitted to cell cycle analysis Briefly, about 1 × 106 cells were submitted to trypsinisation, rinsed with PBS and resuspended in PBS (500 µl) and RNAse (50 µl) followed by incubation for 30 min at room temperature. After RNAse treatment the cells were added with propidium iodide (500 μl) followed by incubation of 1 h. The cell cycle analysis was done using Flow cytometer (ThermoFisher USA).
Immuno-blot analysis
The immunoblot analysis was carried on cell lysates of melanoma cell lines following respective treatments as described earlier [20]. For analysing the expression of proteins in tumour tissues, lysates were prepared as described earlier [7]. The proteins were separated on SDS-PAGE gels (10%) using nitrocellulose membranes. The membranes were blocked and were incubated with primary antibodies for 12 h at 4 °C, followed by incubation with respective secondary antibodies. Enhanced chemiluminescence was done for visualizing the bands. Actin was used as loading control.
miR-512-3p transfection
The melanoma cell lines WM1361B and WM983A transfected with miR-512-3p inhibitor (RiboBio) for functional analysis following the supplied instructions. Briefly, 1 × 105 cells were transferred to 6 well plates for seeding. The WM1361B and WM983A cell lines were transfected after they reached confluence of 70% in a serum free media along with anti-miR inhibitor or control at final concentration 60 nM using Lipofectamine 2000 reagent (ThermoFisher USA) as per supplied instructions. After 24 h following transfection, the cells were incubated in culture media 2% FBS for 48 h. The cells were collected and were submitted for functional analysis, cell cycle analysis and immunoblot analysis. The scramble and un-transfected probes were selected as controls for transfecting the WM1361B and WM983A cell lines.
Tumour xenograft model (in vivo study)
For in vivo studies we selected female athymic nude mice aging between 3 to 4 weeks. The mice were supplied by the animal centre of Xiangyang Central Hospital, China. Prior to this, the animal studies were approved by the animal ethical review board of Xiangyang Central Hospital China. The mice were housed under controlled conditions with temperature of 25 °C, provided free access to water and AIN-93G diet. The mice were injected with WM1361B melanoma cells (4 × 105 cells in 100 µl PBS for each mouse) under the skin in the right flank of each animal. After 24 h of injecting the cells, the mice were separated into two groups randomly with 10 mice in each. Group 1 received control AIN-93G diet whereas the group 2 received Apigenin (0.5%) supplemented AIN-93G diet throughout the experimental period. The study was concluded after 28 days after injecting the melanoma cells. The tumour size and volume were measured after every 7 days using a calliper. At the end of study, the mice were sacrificed and tumours were removed and processed to isolate miRs, immunostaining and for preparing lysates for immunoblot analysis.
Immunohistochemical analysis for detecting PCNA-positive cells
The isolated tumour tissues were processed for producing tumour sections of 5 μm thickness, the sections were deparaffinized using xylene and rehydrated using ethanol followed by retrieving of antigens as reported earlier [21]. Briefly, the tissue sections were blocked for non-specific sites followed by incubation with PCNA primary antibodies. The sections were rinsed and incubated with secondary antibody (biotinylated) and then with horseradish peroxidase-conjugated streptavidin. After this the tissue sections were exposed to 2,4-diaminobenzidine substrate followed by counterstaining of hematoxylin. The sections were observed under microscope (Olympus, BX40F4, Japan) for counting the PCNA positive cells in about 3 to 4 fields selected randomly.
In situ hybridization for detecting miR-512-3p in tumour tissues
We performed FISH assay for detecting the levels of miR-512-3p in tumour tissues as reported earlier [22]. Briefly, the tumour tissues sections priorly fixed in formalin were deparaffinized using xylene and rehydrated with ethanol, the sections were exposed to proteinase kinase (20 μg/ml) in tris-buffer (ThermoFisher USA) for 10 min at room temperature followed fixing in p-formaldehyde (4%) at 4 °C. The sections were then treated with hydrogen peroxide (1%) for 30 min for blocking the peroxidases. The tissues sections were then treated with prehybridization buffer for 30 min at room temperature. Then, the sections were treated with hybridization buffer along with DIG labelled probes for 24 h at room temperature. After 24 h the sections were rinsed using phosphate buffered saline followed by exposure to FITC conjugated antibody for 2 h at room temperature, the nucleus in the cells were stained for DAPI and observed under fluorescence microscope.
In silico docking studies for target confirmation
Looking into the in vivo and in vitro outcomes we further performed in silico docking analysis for confirming the potential target of APG. We made an effort to virtually study the binding affinity of Ligand (APG) with target protein p27 Kip1. The study was done by using MGL tools with the help of Autodock Vina (Scrip’s research institute, USA). The crystal 3D structure of p27 Kip1 was downloaded from global protein data base and structure of APG was obtained from FlavoDB data base (http://bioinfo.net.in/flavodb/home.html). The structures were processed for converting them to pdb formats, the water molecules were deleted followed by addition of Kollman charges and minimization of energy. For initiating docking the grid box was prepared. The docking was done in Autodock Vina, after completion of docking the output parameters were analysed and binding energies were recorded. The 3D interaction between the binding sites of p27 Kip1 and APG were observed using Pymol software (Schrodinger, USA), the binding energies were also reported.
Statistics
The results were presented as mean ± standard deviation. The significance was established with t test or ANOVA. All the analysis was performed with GraphPad Prism software (San Diego CA). The value P < 0.05 were considered significant. All the experiments were done in triplicates.
Results
miR-512-3p is overexpressed in melanoma cell lines and is associated with increased cell viability
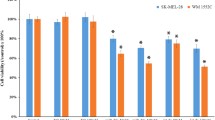
RT-PCR analysis was performed to study the expression of miR-512-3p in seven melanoma cell lines (WM1361B, WM983A, WM1341D, SK-MEL-3, SH-4, SK-MEL-24 and RPMI-7951) and NHEMs. The results suggested that all the melanoma cell lines showed high levels of miR-512-3p compared to NHEM (Fig. 1A). The miR-512-3p levels were varied in all the melanoma cells with WM1361B and WM983A showing the highest levels with about 3 to 5 times higher compared to NHEqMs (Fig. 1B). Further we assessed the involvement of miR-512-3p on the progression of cancerous cells, cell proliferation studies were carried by MTT assay. The results suggested that, over-expression of miR-512-3p was associated with high cell viability (Fig. 1C).

Expression of miR-512-3p and viability among various melanoma cell lines compared with NHEMs. A RT-PCR analysis for relative expression of miR-512-3p in various melanoma cell lines compared to NHEM. B Cell viability study suggested increased cell viability with overexpression of miR-512-3p suggesting involvement of miR-512-3p with cell proliferation. The cell viability studies were done by MTT assay, results were fold change of NHEM cells used as control
Inhibition of miR-512-3p supresses cell proliferation
To study the role of miR-512-3p in the process of cell proliferation of melanoma cell lines, we selected WM1361B and WM983A melanoma cells which showed high levels of miR-512-3p. The cells WM1361B and WM983A received transfection of anti-miR-512-3p with the help of lipofectamine reagent. The results suggested that the transfection with anti-miR-512-3p suppressed the expression of miR-512-3p in both the cell lines against the scrambled miR or control transfected (Fig. 2A). Further, we studied the effect of miR-512-3p inhibition on proliferation of both WM1361B and WM983A, it was observed that inhibition of miR-512-3p caused about 45 and 55% suppression in proliferation respectively (Fig. 2B). The results clearly suggested that inhibition of miR-512-3p suppressed cell proliferation in melanoma cell lines.

Inhibition of miR-512-3p in melanoma cell lines decreases cell viability. A RT-PCR analysis of expression of miR-512-3p in melanoma cell lines (WM1361B and WM983A) submitted to transfection of anti-miR-512-3p for 48 h followed by isolation of miR with Trizol reagent. B Results of cell viability after inhibition of miR-512-3p in melanoma cells (WM1361B and WM983A) compared to control. *P < 0.001 compared to control. C Cell cycle analysis of melanoma cells after transfection with anti-miR-512-3p and control. D Immunoblot analysis was done in cell lysates for expression of Cyclin D1, D2 and E, CDK 2, CDK 4, CDK 6 against control. Suppression of miR-512-3p decreased the expression of proteins of cell cycle in both WM1361B and WM983A cell lines
Inhibition of miR-512-3p causes arrest of G0/G1 in cell cycle and suppression of proteins responsible for cell cycle regulation
We studied whether suppression of cell viability after inhibition of miR-512-3p was associated with alterations in cell cycle in melanoma cells. For studying transfected the WM1361B and WM983A melanoma cell lines with anti-miR-512-3p for 48 h, the cells were submitted for cell cycle studies. It was evidenced that in WM1361B cells transfected with miR-512-3p showed arrest in G0/G1 phase (P < 0.01) of the cell cycle compared to control cells (Fig. 2C), in the WM983A cell lines also the results were similar. The process of cell division depends upon activation of cyclins which binds to cyclin dependent kinases for inducing the progression of cell cycle further leading to mitosis. We in our study hence evaluated the effect of G0/G1 cell cycle arrest on the levels of cell cycle regulatory proteins opting immunoblot analysis. The results of immunoblot analysis suggested that, suppression of miR-512-3p in WM1361B and WM983A cells inhibited the levels of cyclinD1, cyclinD2 and cyclinE and also decreased the expression of CDK 2, CDK 4 and CDK 6 in WM1361B and WM983A cells (Fig. 2D). Hence from the results it could be confirmed that upregulation of miR-512-3p may be involved in increased cell cycle progression whereas suppression of miR-512-3p is linked with cell cycle arrest in the G0/G1 phase of cell cycle along with inhibition of cyclin-dependent kinases and cyclins in the G0/G1 phase of cell cycle.
Inhibition of miR-512-3p after treatment of Apigenin supresses viability of melanoma cells
We next evaluated whether treatment of Apigenin could inhibit the increased levels of miR-512-3p in melanoma cells. The WM1361B and WM983A cells were exposed to Apigenin at various concentrations (0, 10, 20 and 40 µM) for 48 h. The cells after treatment were collected and levels of miR-512-3p were analysed with RT-PCR. The results suggested that treatment of Apigenin in melanoma cells inhibited the expression of miR-512-3p with increasing dose (Fig. 3A). The expression of miR-512-3p were significantly suppressed after exposing melanoma cells to Apigenin.

Effect Apigenin treatment of expression of miR-512-3p and cell viability in WM1361B and WM983A melanoma cells (in vitro). A Both WM1361B and WM983A melanoma cell lines were exposed to various concentrations of Apigenin (0, 10, 20, 40 µM) for 48 h. The expression of miR-512-3p was studied by RT-PCR, treatment of Apigenin suppressed the expression of miR-512-3p. B Percent viability after the WM1361B and WM983A melanoma cell lines were treated with Apigenin for 24 and 48 h. C Colony formation assay of WM1361B and WM983A cell lines after exposure to 20 and 40 µM of Apigenin
As we evidenced that Apigenin caused suppression in levels of miR-512-3p also the suppression of miR-512-3p decreased the cell viability, we also studied the consequences of Apigenin on the cell viability. For viability studies, the WM1361B and WM983A cells were exposed to various concentrations of Apigenin for 24 and 48 h followed by MTT assay to determine cell viability. The results (Fig. 3B), suggested that in WM1361B cells treatment of Apigenin decreased the viability of cells with increasing dose, the cells showing decrease in viability ranging from 12 to 35% after 24 h and 20 to 50% after 48 h of exposure. Under similar conditions almost same viability pattern was observed in WM983A cells (Fig. 3B). Further in our study we evaluated the cytotoxic and anti-carcinogenic activity of Apigenin in melanoma cells by performing colony formation assay. It was evidenced that treatment of Apigenin in WM1361B and WM983A cells caused inhibition in colony forming potential of both the cell lines (Fig. 3C). Apigenin treatment inhibited the colony forming potential of WM1361B cells by 45% and 70% at concentrations 20 and 40 µM respectively and in WM983A cells decreased the colony forming potential by 40% and 65% at 20 and 40 µM concentrations, respectively (Fig. 3C), all the results were compared with control (Not treated with Apigenin).
Apigenin causes arrest of G1-phase of cell cycle in cancerous melanoma cells
Looking into the effects of Apigenin in cell viability of melanoma cells, we screened three concentrations i.e. 10, 20 and 40 µM of Apigenin for cell cycle experiments in melanoma cell lines. As it was observed that inhibition of miR-512-3p in melanoma cell caused G1 phase arrest of cell cycle (Fig. 2C), it was studied whether if suppression of cell viability mediated by Apigenin also results in arrest of G1 phase. Both WM1361B and WM983A cell lines were exposed to Apigenin for 48 h followed by analysis of cell cycle using BD FACSymphony™ Cell Analyzers (BD bioscience USA) analyser as reported earlier [18, 20]. The results showed that, treatment of Apigenin in WM1361B cells for 48 h resulted in high number of cells in the G1 phase with increasing concentrations and 40 µM compared to vehicle treated control group. However, it was found that the number of cells in G1 phase of cell cycle in control group (vehicle treated) of WM1361B cells was less (Fig. 4A) as compared to that reported earlier i.e. in Fig. 2. We believe that this variation may be due to differences created by passages of the melanoma WM1361B cells and some other variables of experiments. In case of WM983A cells similar effect of Apigenin were observed on cell cycle progression. These outcomes suggest that the Apigenin mediated decrease in cell viability and proliferation of melanoma cancer cells may be linked with initiation of G1 arrest by Apigenin and this can be related to suppression of miR-512-3p in melanoma cells upon treating with Apigenin.

Effect of Apigenin on cell cycle progression in melanoma cells. A The WM1361B and WM983A cell lines were exposed to various concentrations of Apigenin for 48 h and were submitted to cell cycle analysis by flow cytometer. B Immunoblot analysis was done on cell lysates for analysis of CDK2, CDK4, CDK6, Cyclin D1, Cyclin D2 and Cyclin E
Apigenin inhibits the expression of cyclins associated with G1-phase and cyclin dependent kinases in cancer cells
Looking into the results of cell-cycle progression, we studied the effects of Apigenin on expression levels of regulatory proteins of cell cycle in melanoma. The WM1361B and WM983A cell lines were exposed to Apigenin for 48 h, the cell-lysates were analysed for expression regulatory proteins of cell cycle by immunoblot analysis. The results suggested that in WM1361B and WM983A cells upon treatment with Apigenin for 48 h resulted in suppression of levels of cyclin D1, cyclin D2 and cyclin E with increasing concentrations of Apigenin (Fig. 4B). In addition, it was observed that the treatment of Apigenin caused significant decrease in expression of CDK2, CDK4 and CDK6 in WM1361B and WM983A melanoma cells (Fig. 4B).
miR-512-3p targets the tumour suppressor proteins p27 Kip1
To study whether p27 Kip1 was the potential target of miR-512-3p, the melanoma cells were exposed to anti- and scrambled miR-512-3p for 48 h. It was observed that exposure of melanoma cell lines with anti-miR-512-3p inhibited the expression of miR-512-3p and increased the levels of p27 Kip1 in both the WM1361B and WM983A melanoma cell lines compared to control group which do not received treatment of anti-miR-512-3p or treated with scrambled miR (Fig. 5A).

A Inhibition of miR-512-3p in melanoma cells increased the levels of p27 Kip1 protein in melanoma cells. B and C Apigenin restored or increased the expression of p27 Kip1 and suppressed the levels of E2F1 and E2F4 in dose-dependent manner
Apigenin restores the expression levels of p21 Kip1 protein
As it was evidenced that Apigenin treatment in melanoma cells caused inhibition of miR-512-3p (Fig. 3), we further studied whether Apigenin treatment could lead to overexpression or could restore the levels of p27 Kip1 in melanoma cells. The results of western blot analysis suggested that Apigenin restored the expression of p27 Kip1 in WM1361B and WM983A melanoma cells compared non-treated cells (Fig. 5B). It was also observed that Apigenin decreased the expression levels of pRb and E2F family member proteins E2F1 and E2F4 in melanoma cells which are the downstream target of p27 Kip1(Fig. 5C).
Apigenin suppresses the growth of tumours of WM1361B cells in experimental mice
Further role of Apigenin on the growth of WM1361B cells induced xenograft in nude mice was analysed. As from the in vitro studies it was found that both the WM1361B and WM983A cell lines showed almost same effects, for the in vivo studies we used only one cell lines i.e. WM1361B for inducing tumour xenograft in nude mice. The experimental mice were given Apigenin (0.5%) supplemented AIN-93G diet. The nude mice were injected with WM1361B cells subcutaneously into the nude mice followed by observations of tumour growth regularly (Fig. 6A). It was found that the AIN-93G diet supplemented with Apigenin suppressed the growth of WM1361B induced tumours during the protocol and at concluding the treatment the inhibitory effect of Apigenin diet was about 60% against the tumour growth control having fed with AIN-93G diet without Apigenin (Fig. 6A and B). At the end of study, the tumour weight was calculated for each animal, the results (Fig. 6C) showed that diet supplemented with Apigenin significantly suppressed the tumour growth compared to control group, suggesting anti-tumour effect of Apigenin.

Inclusion of Apigenin in diet inhibits the growth of xenografts in vivo in nude mice. A Tumour volume/mouse as analysed on various days post inoculation of tumour cells. B Tumours were harvested from experimental rats of control and Apigenin treatment and were imaged C Tumour weight of control and Apigenin treated rats. **P < 0.05, ***P < 0.001 compared to control
Apigenin supress the expression of miR-512-3p in tumour tissues
RT-PCR study for levels of miR-512-3p in tumour tissues suggested that the levels of miR-512-3p were downregulated in mice treated with Apigenin compared to control group (Fig. 7A). To study the changes for miR-512-3p levels in tumour tissues, we performed fluorescent in situ hybridization assay (FISH) which marked the localization of miR-512-3p with the help of locked nucleic acid probe (Fig. 7B). In agreement to the RT-PCR results, the in situ study suggested that the expression of miR-512-3p (green color) was towards lower side in mice treated with Apigenin compared to the control mice.

Apigenin rich diet altered the levels of miR-512-3p, p27 Kip1 and PCNA in tumour tissues. A Expression of miR-512-3p in isolated tumour xenografts in rats fed with Apigenin rich and non-Apigenin diet. B FISH analysis of miR-512-3p in tumour tissues, miR-512-3p positive in situ hybridization signals are green and DAPI appears blue. C Immunoblot analysis showing expression of p24 Kip1 and PCNA in tumour tissue lysates. D immunohistochemical analysis showing PCNA positive cells in tumour tissues of rats fed with Apigenin rich and non-Apigenin diet. ***P < 0.001 compared to control group
Apigenin restores the expression of p27 Kip1 and inhibits proliferation of xenograft cells
The results of in vitro studies suggested that suppression of miR-512-3p restored the expression of p27 Kip1 indicating p27 Kip1 as the potential target of miR-512-3p. For studying the effects in vivo in tumour xenograft mice, we performed western blot analysis for studying expression of p27 Kip1 in tumour induced control and mice fed with Apigenin supplemented diet. The results of our study suggested that expression of p27 Kip1 were overexpressed in tumour tissues of mice fed with Apigenin supplemented diet compared to control fed with Apigenin free diet (Fig. 7C). We then studied the anti-proliferative activity of Apigenin by performing immunoblot study for expression of PCNA and immunohistochemistry for PCNA- positive cells. The results of immunoblot analysis suggested that the levels of PCNA decreased in the tumour tissue fed with Apigenin supplemented diet compared to control mice (Fig. 7C). The results of immunohistochemistry showed that the percent PCNA positive cells in tumour tissues were lower in mice receiving Apigenin supplemented diet compared to control mice (Fig. 7D).
Apigenin showed high binding affinity for P27 Kip1
The docking analysis was done looking into the outcomes of in vivo and in vitro studies. The results of docking as obtained in the form of binding energies suggested low binding energy for interaction between ligand APG and protein p27 Kip1 (Table 1). Low binding energies for interaction are indication of better affinity of ligand and protein. The 3D analysis of protein structure also suggested perfect encapsulation of ligand APG in the protein structure p27 Kip1 (Fig. 8) showing good protein ligand affinity. The outcomes of in silico docking study suggested potential binding affinity of Ligand APG with protein p27 Kip1.

Docking studies showing potential binding of Apigenin and protein structure of p27Kip1
Discussion
MiRNAs are small non-coding RNAs which are responsible for regulating the expression of proteins and multiple genes [23]. miRs have been reported to be involved in number of pathological conditions such as cancer. Role of miR-512-3p is evidenced in many types of cancers [6, 7, 24], however, its involvement in melanoma remains poorly explored. Here we evaluated the expression levels of miR-512-3p in eight melanoma cell lines and compared them against the normal human epidermal melanocytes by performing RT-PCR analysis. The findings of our study suggested that expression levels of miR-512-3p was at least 3 to 4 times higher in melanoma cell lines compared to their normal counterparts. miRs regulate various processes in biological system such as angiogenesis, apoptosis, metastasis and cell proliferation in addition to this miRs can behave both as tumour suppressors as well as oncogenes [25]. In our study, the cell proliferation data suggested that the selected eight melanoma cell lines showed at least 5 to 6 times higher cell proliferation compared to normal human epidermal melanoma cells, in addition to this the melanoma cell lines showed higher expression of miR-512-3p against the normal epidermal melanoma cell lines.
To confirm role of miR-512-3p overexpression in increased proliferation of melanoma cells, the WM1361B and WM983A cell lines were exposed to miR-512-3p inhibitor and the cell viability was studied. It was observed that, suppression of miR-512-3p resulted in inhibition of viability of melanoma cells indicating prime involvement of overexpressing miR-512-3p in cell proliferation. The results were in agreement to earlier study, which suggested that increased levels of miR-512-3p in prostate carcinoma encourages cell proliferation and progression of cell cycle [24]. The present study also showed that, p27Kip1 was the favourable target of miR-512-3p and its inhibition plays a prime role in miR-512-3p mediated cell cycle progression. The analysis of cell cycle suggested that upon treating the WM1361B and WM983A cell lines with anti-miR-512-3p (inhibitor) significantly induced G1-phase arrest in both the cell lines suggesting cell cycle progression as the mechanism involved. It has been reported that uncontrolled division or proliferation of cells is governed by activation of CDKs and cyclins in the G1 phase, which further leads the cell cycle progression to the S phase [6]. CDK play a major role in progression of cancer, CDK activity is negatively regulated by number of inhibitory proteins such as p27Kip1 [26]. Decreased p27Kip1 levels may lead to tumour development by either decreasing the apoptosis and increasing the proliferation of cells [27]. In the present study, arrest of G1 phase in melanoma cells after they were treated to anti-miR-512-3p resulted in inhibition of CDKs (i.e. CDK2, CDK4 and CDK6) along with reactivation of p27Kip1. The findings suggested potential of miR-512-3p inhibitor for blocking the uncontrolled progression of cell cycle in melanoma cells and trigger G1-phase arrest via suppressing the levels of CDKs and cyclins, and reactivating the p27Kip1 protein. In a pathway, property of miRs to target multiple genes is an important phenomenon, therapeutic inhibition of miRs may be a potential approach.
Apigenin is a member of phyto active compounds flavonoids. It has been reported to possess important pharmacological activities including anti-cancer activity [28]. Apigenin have been reported to induce apoptosis in lymphoma cells via AKT/PKB pathway involving the p27Kip1 proteins [29]. Apigenin also have been demonstrated to modulate the expressions of number of miRs which include miR-155, miR-34a-5p, miR-152-5p, miR-423-5p [30,31,32,33]. In an attempt develop a potential miR-512-3p inhibitor for treating melanoma, we screened Apigenin on the levels of miR-512-3p in melanoma. The findings of our study showed that exposure of melanoma cells to Apigenin significantly decreased the expression of miR-512-3p resulting in suppression of viability and their property of forming colonies during colony formation assay. Further the treatment of Apigenin resulted in activation of p27Kip1 which plays an important part in suppressing the carcinogenic property of melanoma cells. The findings were in clear agreement to activity of Apigenin which resulted in activation of p27Kip1 proteins in lymphoma [29] and in prostate cancer [34]. These findings suggest that, in melanoma cells Apigenin showed inhibitory action on miR-512-3p and its treatment activates p27Kip1 protein as shown by anti-miR-512-3p in melanoma cells. In addition to the activation of p27Kip1 by Apigenin, it was found that Apigenin also impacted the downstream signalling pathway of p27Kip1 as shown by knocking of Apigenin on the expression of pRb and E2F family member E2F1 and E2F4.
The results of in vivo experiments done in nude mice suggested that Apigenin showed inhibitory effect on the development of myeloma tumours in tumour induced xenograft mice without any noticeable signs of toxicity. The knocking effect of Apigenin in the tumour growth was linked with suppression of miR-512-3p levels as well as overexpression of p27Kip1 protein, causing inhibition of tumour cell proliferation in tumour tissues.
Based on the findings of in vivo and in vitro studies, we performed in silico docking analysis for confirming the target affinity of Apigenin with p27Kip1. Docking studies indicate the extent of interaction the ligand shows with the target proteins. Also, in-silico docking analysis are usually done for confirming the binding affinity of ligand with target protein which in turn governs the expression of protein in cellular pathway. The results of in vitro and in vivo studies suggested p27Kip1 was the protein whose expression was altered, hence possible interaction between ligand Apigenin and p27Kip1 was calculated. The docking analysis between Apigenin and p27Kip1 showed potential interaction between them, lower binding energies suggested high binding affinity of ligand Apigenin and protein p27Kip1. Also, the ligand was well encapsulated in the protein structure further confirming potential interaction.
The present work demonstrated that miR-512-3p was significantly overexpressed in melanoma cell lines (WM1361B and WM983A) and acted as oncogene via modulating the proliferation and progression of cell cycle. In addition, the study suggested that Apigenin could be a potential molecule which could inhibit the proliferation of melanoma cells and inhibit the progression of cell cycle by exerting inhibitory effect on the expression of miR-512-3p. Our study demonstrated that, miR-512-3p could be an important therapeutic target for treating melanoma. Apigenin showed potential of inducing p27Kip1 as observed by in vivo and in vitro studies. The findings were further made concrete by in silico docking studies which suggested that Apigenin bonded potentially with p27Kip1 protein with lower energy. Further, more studies are needed for confirming role of Apigenin by testing some more cell lines obtained at various stages of melanoma.
Data availability
All data generated or analysed during this study are included in this article. Further any enquiries can be directed to the corresponding author.
References
American Cancer Society Cancer facts and figures. http://www.cancer.org/. Accessed 11 July 2014
Maddodi N, Setaluri V (2008) Role of UV in cutaneous melanoma. Photochem Photobiol 84:528–536
Strouse JJ, Fears TR, Tucker MA, Wayne AS (2005) Pediatric melanoma: risk factor and survival analysis of the surveillance, epidemiology and end results database. J Clin Oncol 23:4735–4741
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435(7043):834–838
Esquela-Kerscher A, Slack FJ (2006) Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer 6:259–269
Chen F, Zhu HH, Zhou LF, Wu SS, Wang J, Chen Z (2010) Inhibition of c-FLIP expression by miR-512-3p contributes to taxol-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep 23:1457–1462
Zhu X, Gao G, Chu K, Yang X, Ren S, Li Y, Wu H, Huang Y, Zhou C (2015) Inhibition of RAC1-GEF DOCK3 by miR-512-3p contributes to suppression of metastasis in non-small cell lung cancer. Int J Biochem Cell Biol 61:103–114
Mohamadzade Z, Mahjoubi F, Soltani BM (2021) Introduction of hsa-miR-512-3p as a new regulator of HER2 signaling pathway in breast cancer. Breast Cancer Res Treat 185(1):95–106
Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:1501–1512
Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M (1997) Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 3:231–234
Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ (1998) The murine gene p27Kip1 is haplo-insufficient for tumor suppression. Nature 396:177–180
Tanner FC, Yang ZY, Duckers E, Gordon D, Nabel GJ, Nabel EG (1998) Expression of cyclin-dependent kinase inhibitors in vascular disease. Circ Res 82:396–403
van den Heuvel S, Dyson NJ (2008) Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 9:713–724
Jones V, Katiyar SK (2013) Emerging phytochemicals for prevention of melanoma invasion. Cancer Lett 335:251–258
Ali F, Naz F, Jyoti S, Siddique YH (2017) Health functionality of apigenin: a review. Int J Food Prop 20:1197–1238
Lotha R, Sivasubramanian A (2018) Flavonoids nutraceuticals in prevention and treatment of cancer: a review. Asian J Pharm Clin Res 11:42–47
Yan X, Qi M, Li P, Zhan Y, Shao H (2017) Apigenin in cancer therapy: anti-cancer effects and mechanisms of action. Cell Biosci 7:50
Prasad R, Vaid M, Katiyar SK (2012) Grape proanthocyanidin inhibit pancreatic cancer cell growth in vitro and in vivo through induction of apoptosis and by targeting the PI3K/Akt pathway. PLoS ONE 7:e43064
Albino AP, Juan G, Traganos F, Reinhart L, Connolly J, Rose DP, Darzynkiewicz Z (2000) Cell cycle arrest and apoptosis of melanoma cells by docosahexaenoic acid: association with decreased pRb phosphorylation. Cancer Res 60(15):4139–4145
Singh T, Sharma SD, Katiyar SK (2011) Grape seed proanthocyanidins induce apoptosis by loss of mitochondrial membrane potential of human non-small cell lung cancer cells in vitro and in vivo. PLoS ONE 6:e27444
Prasad R, Katiyar SK (2012) Bioactive phytochemical proanthocyanidins inhibit growth of head and neck squamous cell carcinoma cells by targeting multiple signaling molecules. PLoS ONE 7:e46404
Wang X, Zhang H, Zhang A, Han L, Wang K, Liu R, Yang S, Pu P, Shen C, Kang C, Yu C (2012) Upregulation of miR-20a and miR-106b is involved in the acquisition of malignancy of pediatric brainstem gliomas. Oncol Rep 28:1293–1300
Lawler S, Chiocca EA (2009) Emerging functions of microRNAs in glioblastoma. J Neurooncol 92:297–306
Rao Z, He Z, He Y, Guo Z, Kong D, Liu J (2018) MicroRNA-512-3p is upregulated, and promotes proliferation and cell cycle progression, in prostate cancer cells. Mol Med Rep 17:586–593
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM (2005) A microRNA polycistron as a potential human oncogene. Nature 435:828–833
Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, Yu M, Lin J, Cui Q (2020) The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci 21(6):1960
Zheng JY, Wang WZ, Li KZ, Guan WX, Yan W (2005) Effect of p27(KIP1) on cell cycle and apoptosis in gastric cancer cells. World J Gastroenterol 11(45):7072–7077
Imran M, AslamGondal T, Atif M, Shahbaz M, BatoolQaisarani T, Hanif Mughal M, Salehi B, Martorell M, Sharifi-Rad J (2020) Apigenin as an anticancer agent. Phytother Res 34(8):1812–1828
Hussain AR, Khan AS, Ahmed SO, Ahmed M, Platanias LC, Al-Kuraya KS, Uddin S (2010) Apigenin induces apoptosis via downregulation of S-phase kinase-associated protein 2-mediated induction of p27Kip1 in primary effusion lymphoma cells. Cell Prolif 43(2):170–183
Arango D, Diosa-Toro M, Rojas-Hernandez LS, Cooperstone JL, Schwartz SJ, Mo X, Jiang J, Schmittgen TD, Doseff AI (2015) Dietary apigenin reduces LPS-induced expression of miR-155 restoring immune balance during inflammation. Mol Nutr Food Res 59(4):763–772
Aida R, Hagiwara K, Okano K, Nakata K, Obata Y, Yamashita T, Yoshida K, Hagiwara H (2021) miR-34a-5p might have an important role for inducing apoptosis by down-regulation of SNAI1 in apigenin-treated lung cancer cells. Mol Biol Rep 48(3):2291–2297
Zhao X, Zhou HB, Liu J, Xie J, Hu R (2021) Apigenin suppresses proliferation, invasion, and epithelial-mesenchymal transition of cervical carcinoma cells by regulation of miR-152/BRD4 axis. Kaohsiung J Med Sci 37(7):583–593
Javed Z, Sadia H, Iqbal MJ, Shamas S, Malik K, Ahmed R, Raza S, Butnariu M, Cruz-Martins N, Sharifi-Rad J (2021) Apigenin role as cell-signaling pathways modulator: implications in cancer prevention and treatment. Cancer Cell Int 21(1):189
Shukla S, Gupta S (2006) Molecular targets for apigenin-induced cell cycle arrest and apoptosis in prostate cancer cell xenograft. Mol Cancer Ther 5(4):843–852
Acknowledgements
The authors are thankful to the staff and management of Xiangyang Central Hospital for providing the required support.
Funding
The work received no external funding.
Author information
Authors and Affiliations
Contributions
QX and RZ contributed equally to this work. JY, DL, QH, CS, XL along with QX and RZ designed and performed experiments. All the authors contributed in preparing the manuscript and read the paper before submission.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval
The animal experiments were approved by the Xiangyang Central Hospital, China.
Consent to publication
The university is aware about the work has consent for publication. The authors declare consent for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xie, Q., Zhang, R., Liu, D. et al. Apigenin inhibits growth of melanoma by suppressing miR-512-3p and promoting the G1 phase of cell cycle involving the p27 Kip1 protein. Mol Cell Biochem 477, 1569–1582 (2022). https://doi.org/10.1007/s11010-022-04363-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04363-x