
Abstract
Diffuse large B-cell lymphoma (DLBCL) is the most prevalent subtype of non-Hodgkin lymphoma and is a very aggressive malignancy with tumor growing rapidly in organs like lymph nodes. The pathogenesis of DLBCL is not clear and the prognosis of DLBCL requires improvement. Here, we investigated the mechanisms of DLBCL, with the focus on lncRNA PVT1/miR-34b-5p/Foxp1 axis. Human DLBCL tissues from diagnosed DLBCL patients and four human DLBCL cell lines, one normal human B lymphoblastoid cell line were used. qRT-PCR and western blotting were employed to measure expression levels of lncRNA PVT1, Foxp1, miR-34b-5p, β-catenin, and proliferation-related proteins. MTT assay and colony formation assay were performed to determine cell proliferation. Flow cytometry was used to examine cell apoptosis. ChIP and Dual-luciferase assay were utilized to validate interactions of Foxp1/promoters, PVT1/miR-34b-5p and miR-34b-5p/Foxp1. Mouse tumor xenograft model was used to determine the effect of sh-PVT1 on tumor growth in vivo. In this study, we found PVT1 and Foxp1 were elevated in DLBCL tissues and cells while miR-34b-5p was decreased. Knockdown of PVT1, overexpression of miR-34b-5p, or Foxp1 knockdown repressed DLBCL cell proliferation but enhanced cell apoptosis. PVT1 directly bound miR-34b-5p to disinhibit Foxp1/β-catenin signaling. Foxp1 regulated CDK4, CyclinD1, and p53 expression via binding with their promoters. Knockdown of Foxp1 partially reversed the effects of miR-34b-5p inhibitor on DLBCL cell proliferation and apoptosis. Inhibition of PVT1 through shRNA suppressed DLBCL tumor growth in vivo. All in all, lncRNA PVT1 promotes DLBCL progression via acting as a miR-34b-5p sponge to disinhibit Foxp1/β-catenin signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most prevalent subtype of non-Hodgkin lymphoma (NHL) characterized by malignant hyperplasia of B lymphocytes, accounting for 40% of cases around the world [1, 2]. It is a heterogeneous tumor with distinct genetic, phenotypic, and clinical features of B cells and can be roughly divided into germinal center B-cell-like (GCB) subtype of DLBCL and non-GCB DLBCL based on the gene expression profiling [3]. Although more than half of the DLBCL patients can be cured by the current treatments including rituximab-based chemotherapy, about 30% patients will relapse later and 20% suffer from primary refractory [1, 4, 5]. Despite tremendous advances in research and clinical work, the prognosis of DLBCL remains further improvement. Understanding the pathogenesis and mechanisms of DLBCL is very necessary for better treatments.
Long non-coding RNAs (LncRNAs) are a well-studied class of endogenous non-coding RNAs that are longer than 200 nucleotides [6]. Although they used to be considered as cellular junk, emerging evidence shows that they are important regulators of gene expression and thus have crucial roles in many cellular processes, such as development and growth [7]. Moreover, dysregulated lncRNAs have been implicated in varieties of diseases including cancers [8, 9]. In lymphoma or DLBCL, aberrant expressions of lncRNAs have been observed, such as lncRNA p21, lncRNA PANDA and SNHG16 [10,11,12]. Recently, it is reported that lncRNA PVT1 predicts poor prognosis in patients with DLBCL [13]. However, the exact function of lncRNA PVT1 in DLBCL and the underlying molecular mechanisms remain largely unknown.
One key mechanism by which lncRNAs regulate gene expression is that they interact with microRNAs (miRNAs), a well-known class of non-coding RNAs that suppress gene expression by binding with target mRNAs [14], and disinhibit expression of miRNA targets [7]. In DLBCL, numerous miRNAs have been involved. For example, miR-34b-5p was diminished in DLBCL cells and loss of miR-34b-5p function accelerated the progression of DLBCL [15]. In addition, the transcription factor Forkhead box protein P1 (Foxp1), a conserved transcription factor with critical roles in numerous cellular processes including cell proliferation and development, has been shown largely involved in DLBCL as well [16]. Its expression is highly increased in a subset of DLBCL and this increase promotes cancer cell survival [16]. Mechanistically, it has been indicated that Foxp1 transcriptionally silences sphingosine-1-phsphate receptor 2 (SIPR2), but potentiates Wnt/β-catenin signaling [17, 18]. In our preliminary studies, through bioinformatic analysis, we found binding sites between PVT1 and miR-34b-5p, as well as between miR-34b-5p and Foxp1. Thus, we hypothesized that PVT1 might participate in DLBCL development by targeting miR-34b-5p/Foxp1 pathway.
In the present study, we fully investigated the function of PVT1/miR-34b-5p/Foxp1 axis in DLBCL. Using both in vivo and in vitro models of DLBCL, we found that PVT1 and Foxp1 were increased while miR-34b-5p was decreased in DLBCL human samples and DLBCL cells. Knockdown of PVT1 or overexpression of miR-34b-5p suppressed DLBCL cell proliferation but enhanced cell apoptosis. miR-34b-5p inhibitor reversed the effects of PVT knockdown while Foxp1 knockdown partially blocked the effects of miR-34b-5p inhibitor, indicating that PVT1 exerts its function through targeting miR-34b-5p and that miR-34b-5p functions via Foxp1. More importantly, we showed that inhibition of PVT1 suppressed DLBCL tumor growth in vivo. Our study reveals a critical role of PVT1/miR-34b-5p/Foxp1 axis in DLBCL, sheds light on the mechanisms of DLBCL, and provides avenues to develop therapeutic strategies for DLBCL.
Materials and methods
Human DLBCL samples
Human DLBCL tissues were collected from 33 diagnosed DLBCL patients (18 ABC-DLBCL and 15 GCB-DLBCL) during surgical resection from the First Affiliated Hospital of Hainan Medical University. The non-tumor lymphoid tissues near the cancers were collected simultaneously from same patients. Patients did not receive preoperative treatments. All patients have consented to the study. The study was reviewed and received approval from the Ethics Committee of the First Affiliated Hospital of Hainan Medical University. All specimens were put the liquid nitrogen immediately after collection and then stored at the freezer (− 80 °C).
Cell culture and transfection
Four human DLBCL cell lines (U-2932 [ABC-DLBCL], OCI-Ly1 [GCB-DLBCL], OCI-Ly8 [ABC/GCB intermediate], SU-DHL-6 [GCB-DLBCL]), and one normal human B-cell line were used for the study. All cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were seeded and grown in 10% fetal bovine serum (FBS, Thermo-Fisher Scientific, China) containing Dulbecco’s Modified Eagle Medium (DMEM, Sigma-Aldrich, China). 1% penicillin–streptomycin was included into the medium. The cells were cultured in the cell culture CO2 incubator at 37 °C.
Lipofectamine 3000 (Invitrogen, USA) reagent was employed for cell transfection as the manufacturer’s protocol described. Briefly, cells were grown up to 70–80% confluence and about 1 μg construct together with 1 μL Lipofectamine 3000 were added into the media. Cells were harvested for further analysis at 48 h post transfection.
RNA extraction and RT-qPCR
Trizol (Invitrogen, China) was employed to extract total RNAs from DLBCL human tissues or cultured cells as the manufacturer’s instructions described. For miRNA analysis, total RNAs were isolated with the miRNeasy Advanced Mini Kit (QIAGEN, Hilden, Germany). DNaseI was included into the lysis buffer to avoid the contamination of DNA. Commercial kit (cDNA synthesis kits, Thermo-Fisher, China) was utilized to generate cDNAs through reverse transcription. SYBR Green Master Mix (Invitrogen, China) was used for the quantitative PCR. Relative expression levels of PVT1, miR-34b-5p or Foxp1 mRNA were normalized to U6 snRNA or GAPDH mRNA, respectively, as internal controls. The relative expression was calculated by 2 − ΔΔCt method. The primers listed as follows were from Guangzhou RiboBio Co., Ltd (Guangdong, China).
PVT1 forward primer (FP): 5′-AAAACGGCAGCAGGAAATGT-3′;
PVT1 reverse primer (RP): 5′-ATTCCCATAGAAGGGGCAGG-3′;
miR-34b-5p FP: 5′-CCCTGAAAGGTGCCTTCCTTG-3′;
miR-34b-5p RP: 5′–GCTTGTTCTTAGGGTTGCTGTTG-3′;
Foxp1 FP: 5′-CGAATGTTTGCTTACTTCCGACGC-3′;
Foxp1 RP: 5′-ACTTCATCCACTGTCCATACTGCC-3′;
U6 FP: 5′-GCTTCGGCAGCACATATACTAAAAT-3′;
U6 RP: 5′-CGCTTCACGAATTTGCGTGTCAT-3′;
GAPDH FP: 5′-GAGTCAACGGATTTGGTCGTT-3′;
GAPDH RP: 5′-TTGATTTTGGAGGGATCTCG-3′;
Chromatin immunoprecipitation (ChIP) assay
ChIP kit (Abcam, USA) was used as the manufacturer’s protocol described. Briefly, formaldehyde was used to cross-link proteins/DNA and cells were washed with PBS followed by harvest via micrococcal nuclease. Cell debris was removed through centrifugation and the supernatant was collected. To pull down chromatin fragments, 10 μg of anti-Foxp1 or IgG antibody was added to incubate with the lysate for 1 h at 4 °C. Protein G beads were incubated with all samples overnight at 4 °C. The next day, the beads were washed by wash buffer and eluted by elution buffer. The elution was proceeded for DNA purification and PCR was performed to detect promoter regions.
MTT cell proliferation assay
Transfected cells were plated in individual wells of 96-well plates with a density of 6000 cells per well and grew overnight followed by MTT incubation. 10 μL of 3-(4,5-Dimethyltjiazol-2-yl)-2,5-diphenltetrazolium bromide (MTT, 5 mg/mL) was incubated with cells for 3 h at 37 °C. Afterwards, 150 μL dimethyl sulfoxide (DMSO, Sigma-Aldrich, MO, USA) was added to end the reaction. The absorbance in each condition was analyzed by a 490 nm light.
Colony formation assay
Transfected cells were plated and cultured in the 12-well culture plate for 1 week in the incubator. 4% PFA was added to fix the observed colonies at room temperature for 13–15 min. PFA was washed out with PBS. Colonies were incubated with crystal violet (1%) for 30 min for staining followed by imaging. The number of colonies was quantified by ImageJ software.
Flow cytometry apoptosis assay
Transfected cells were plated and cultured in the 6-well culture plate until 70–80% confluence. Cells were harvested with lysis buffer and then incubated with Annexin-V-FITC/PI (ThermoFisher, USA) for 15 min at 4 °C for 12–15 min. Flow cytometry was used to analyze the relative number of positive and negative cells.
Western blotting analysis
Proteins from tumor tissues or cultured cells were extracted by utilizing the RIPA lysis buffer (Abcam, China) according to standard protocol. DC Protein Assay Kit (Bio-Rad, China) was utilized to quantify the protein concentrations. Equal protein from each condition was loaded into SDS–polyacrylamide gels and separated through electrophoresis. Later proteins in the gels were transferred to PVDF membranes (Sigma-Aldrich, China). 3% BSA was added to block the membranes for 30–60 min at room temperature and then specific primary antibodies were added to incubate at 4 °C overnight. The antibodies were discarded and TBST was utilized to wash the membranes 3 times before incubation with specific secondary antibodies for 1–2 h at room temperature. Protein band intensities were detected using the standard ECL kit. Primary antibodies used in the study were: Anti-Foxp1 (1:2000; Cell Signaling Technology, USA); Anti-β-catenin (1:1500; Abcam, USA); Anti-CDK4 (1:1500; Abcam, USA); Anti-CyclinD1 (1:1000; Abcam, USA); Anti-p53 (1:2000; Abcam, USA); Anti-GAPDH (1: 5000, Abcam, USA).
Nude mice xenograft experiments
All animal experiments have been reviewed and received approval by the Animal Care and Use Committee of Nanjing Medical University. Adult nude mice (8-week-old) were purchased from SJA Laboratory Animal Co., Ltd. (Hunan, China) and raised in the standard animal facility room. 10-week-old nude mice were unilaterally subcutaneously injected with 5 × 106 transfected DLBCL cells (SU-DHL-6 and U-2932, control non-transfected cells, sh-NC-transfected cells, sh-PVT1-transfected cells) to induce tumors. Tumors were monitored on a daily base for 30 days. Tumor length (L) and width (W) were analyzed to quantify the tumor volume (V): V (mm3) = 0.5 × (W)2 × (L). In the end, tumors were dissected out to measure weight.
H&E, Ki-67, and TUNEL staining
The mice were perfused with 10% formalin and liver tissues were incubated in 4% PFA for fixation overnight at 4 °C and subsequently embedded in optimal cutting temperature compound. Embedded tissues were cut into 10 μm thick slices and stained with hematoxylin and eosin (H&E) or used for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining with an in situ cell death detection kit (Roche Applied Science, USA) as manufacturer’s instructions described. The stained sections were washed with PBS and then mounted with the mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI). For Ki-67 staining, the slices were blocked with 5% bovine serum albumin (BSA) for 1 h, followed by primary antibody staining (Anti-Ki-67; 1:500, Abcam, USA) overnight at 4 °C. After several washed in PBS, the sections were incubated with secondary antibodies for 1 h at room temperature. All slices were then incubated with substrates for Envision system-HRP using the standard kit (Dako REAL Ebision kit; DAKO, Denmark) as the manufacturer’s protocol described. Images were taken using a light microscope.
Statistical analysis
All experimental data were analyzed in the GraphPad Prism 7. Statistical details were calculated by unpaired Student t test (for two groups) or one-way ANOVA (for groups more than two) and indicated in figure legends. The Data were presented as Mean ± SD (standard deviation).
Results
PVT1 and Foxp1 were elevated while miR-34b-5p was reduced in DLBCL patient and cells
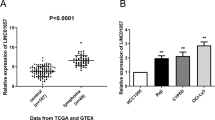
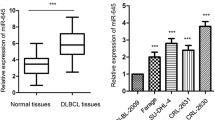
To investigate the functions of PVT1, Foxp1, and miR-34b-5p in DLBCL, we collected DLBCL samples (18 ABC-DLBCL and 15 GCB-DLBCL) from diagnosed DLBCL patients and measured expression levels of PVT1, Foxp1, and miR-34b-5p in those samples. Using qRT-PCR, we found that PVT1 and Foxp1 mRNA levels were greatly higher in DLBCL samples compared to normal lymphoid samples (Fig. 1A), while miR-34b-5p was significantly reduced (Fig. 1A). To further explore the mechanisms, we used four DLBCL cell lines and one normal human B lymphoblastoid cell line for subsequent studies. Consistent with our in vivo results, we observed a higher level of PVT1 and Foxp1 mRNA and a lower expression of miR-34b-5p in DLBCL cells compared to normal B lymphoblastoid cells (Fig. 1B). Western blotting results also showed a higher protein level of Foxp1 in DLBCL cells compared to normal B lymphoblastoid cells (Fig. 1C). Those changes were more robust in SU-DHL6 and U-2932 cells than in OCL-ly1 and OCL-ly8 cells and thus we employed SU-DHL6 and U-2932 cell lines for further experiments. Altogether, these results show that lncRNA PVT1 and Foxp1 are upregulated while miR-34b-5p is downregulated in DLBCL.

PVT1 and Foxp1 were elevated while miR-34b-5p was reduced in DLBCL patients and cells. A qRT-PCR analysis of PVT1, Foxp1 mRNA, and miR-34b-5p levels in DLBCL tissues (n = 33). B qRT-PCR to measure PVT1, Foxp1 mRNA, and miR-34b-5p levels in DLBCL cell lines (n = 4). C Foxp1 protein levels in DLBCL cell lines (n = 4). Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001
Knockdown of PVT1 suppressed DLBCL cell proliferation but promoted apoptosis
To further study the role of PVT1 in DLBCL, we manipulated its expression level and examine ensuing effects on cancer cell proliferation and apoptosis. Transfection with sh-PVT1 significantly decreased PVT1 level in DLBCL cells (Fig. 2A). Using MTT assay, we found that knockdown of PVT1 remarkably inhibited DLBCL cell proliferation (Fig. 2B). Similarly, the colony formation assay results indicated that the number of colonies formed in sh-PVT1 transfected cells was greatly smaller than that in sh-NC transfected cancer cells (Fig. 2C). In contrast, using the apoptosis assay, we observed that sh-PVT1 transfected cells had a higher percentage of cell apoptosis compared to sh-NC transfected cells (Fig. 2D). Using western blotting, we found that knockdown of PVT1 diminished Foxp1 protein, and β-catenin was reduced as well (Fig. 2E). Expression of β-catenin, proliferation-related proteins including CDK4 and Cyclin D1 were significantly suppressed while p53 was upregulated in sh-PVT1 transfected cells in comparison with sh-NC transfected cells (Fig. 2E). Taken together, we show that knockdown of PVT1 suppresses DLBCL proliferation but enhances cell apoptosis.

Knockdown of PVT1 suppressed DLBCL cell proliferation but promoted apoptosis. A qRT-PCR to measure PVT1 levels in DLBCL cells with the transfection of indicated constructs. B MTT analysis of cell proliferation in DLBCL cells with the transfection of indicated constructs. C Cell proliferation of transfected DLBCL cells were measured with colony formation assay. D Percentage of apoptotic cells in transfected DLBCL cells. E Western blotting to determine expression levels of Foxp1, β-catenin, and proliferation-related proteins (CDK4, CyclinD1, and p53) in transfected DLBCL cells. N = 4, Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001
Overexpression of miR-34b-5p suppressed DLBCL cell proliferation but enhanced apoptosis
As presented above, we found miR-34b-5p was reduced in DLBCL tissues and cells (Fig. 1). We wondered whether it was involved in DLBCL as well. Overexpression of miR-34b-5p greatly increased miR-34b-5p level in transfected cells (Fig. 3A). Similar to PVT1 knockdown, with MTT assay, colony formation assay, and flow cytometry, we observed that miR-34b-5p mimics suppressed DLBCL cell proliferation but enhanced cell apoptosis (Fig. 3B–D). Also, Foxp1 and β-catenin protein levels were reduced following ectopic expression of miR-34b-5p (Fig. 3E). Proliferation-related proteins such as CDK4 and CyclinD1 were declined while p53 was enhanced in miR-34b-5p mimics-transfected cells (Fig. 3E). These data indicate that overexpression of miR-34b-5p represses DLBCL cell proliferation but promotes apoptosis.

Overexpression of miR-34b-5p suppressed DLBCL cell proliferation but enhanced apoptosis. A miR-34b-5p levels in DLBCL cells with the overexpression of miR-NC or miR-34b-5p mimics. B MTT analysis of cell proliferation in transfected DLBCL cells. C Cell proliferation of transfected DLBCL cells were analyzed with colony formation assay. D Percentage of apoptotic cells in transfected DLBCL cells. E Western blotting to determine levels of Foxp1, β-catenin, and proliferation-related proteins (CDK4, CyclinD1, and p53) in transfected DLBCL cells. N = 4, Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001
PVT1 disinhibited Foxp1 expression via sponging miR-34b-5p
Given that miR-34b-5p exhibited opposite changes to PVT1 and Foxp1 in DLBCL cells and that overexpression of miR-34b-5p has similar effects to PVT1 knockdown, we were wondering that they might interact with each other. First, knockdown of PVT1 robustly upregulated miR-34b-5p level in DLBCL cells but downregulated Foxp1 level (Fig. 4A and B). miR-34b-5p inhibitor diminished miR-34b-5p expression but remarkably increased Foxp1 protein level (Fig. 4C and D). With Starbase bioinformatic analysis (http://starbase.sysu.edu.cn/), we observed some complementary binding sits between PVT1 and miR-34b-5p, as well as between Foxp1 mRNA and miR-34b-5p (Fig. 4E). Then we performed dual luciferase assay to examine this potential interaction. We found that miR-34b-5p mimics significantly diminished the relative luciferase activity of WT-PVT1 and WT-Foxp1, but not MUT-PVT1 and MUT-Foxp1 wherein the predicted binding sites with miR-34b-5p were mutated (Fig. 4F). Therefore, we conclude that PVT1 directly binds miR-34b-5p while miR-34b-5p targets Foxp1.

PVT1 disinhibited Foxp1 expression via sponging miR-34b-5p. A miR-34b-5p levels in DLBCL cells with overexpression of sh-NC or sh-PVT1. B Foxp1 protein levels in DLBCL cells with overexpression of sh-NC or sh-PVT1. C miR-34b-5p levels in DLBCL cells with overexpression of inhibitor-NC or miR-34b-5p inhibitor. D Foxp1 protein levels in DLBCL cells with overexpression of inhibitor-NC or miR-34b-5p inhibitor. E Predicted binding sites between PVT1 and miR-34b-5p, as well as between Foxp1 mRNA and miR-34b-5p. F Luciferase activities of WT-PVT1, MUT-PVT1, WT-Foxp1, and MUT-Foxp1 in cells with transfection of indicated constructs. G Western blotting to measure levels of Foxp1, β-catenin, and proliferation-related proteins including CDK4, CyclinD1, and p53 in transfected DLBCL cells. N = 4, Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001
We then assessed the role of PVT1/miR-34b-5p interaction in cancer cell proliferation. As shown in Fig. 4G, miR-34b-5p inhibitor reversed the effects of shPVT1 on expression of Foxp1/β-catenin and proliferation-related proteins, such as CDK4, CyclinD1 and p53, suggesting that PVT1 regulates DLBCL proliferation via sponging miR-34b-5p.
miR-34b-5p regulated DLBCL proliferation and apoptosis via Foxp1
Next, we studied the mechanisms of how miR-34b-5p regulated DLBCL. Using bioinformatic tools, we observed binding sites of Foxp1 on the promoter regions of CDK4, CyclinD1 and p53 (Fig. S1A, D, G). With ChIP, we confirmed that immunoprecipitation with Foxp1 significantly enriched the promoter regions of CDK4, CyclinD1, and p53 compared with control IgG (Fig. S1B, E, H). Moreover, the results from dual luciferase activity assay revealed that knockdown of Foxp1 greatly decreased the luciferase activities of CDK4-WT, CyclinD1-WT and increased luciferase activity of p53-WT, but did not affect the activities of mutants in which the binding sites were mutated (Fig. S1C, F, I). These data indicate that Foxp1 directly binds with their promoters and promote their expression. With Starbase, we also identified binding sites on CDK4 mRNA, but not p53 mRNA (Fig. S1J). However, the dual luciferase activity assay results showed that miR-34b-5p mimics did not affect the activities of CDK4-WT or CDK4-MUT (Fig. S1K), suggesting that miR-34-5p doesn’t directly targets CDK4 mRNA. These results show that Foxp1 binds with promoters of CDK4, CyclinD1 and p53, promoting CDK4 and CyclinD1 expression but inhibiting p53 expression.
We then investigated whether miR-34-5p regulated DLBCL cell proliferation and apoptosis via Foxp1. Transfection of DLBCL cells with sh-Foxp1 greatly decreased Foxp1 mRNA and protein level (Fig. S2A, B). Furthermore, knockdown of Foxp1 significantly decreased cell viability and proliferation but increased cell apoptosis, accompanied by reductions on β-catenin, CDK4, and CyclinD1 and elevation on p53 (Fig. S2C–F). These results demonstrate that knockdown of Foxp1 suppresses cell proliferation and promotes cell apoptosis, which is similar to the effects of miR-34-5p overexpression. To directly tackle the relationship between miR-34-5p and Foxp1 during DLBCL, we knocked down Foxp1 expression in miR-34-5p inhibitor-transfected DLBCL cells and measured the ensuing effects. As expected, miR-34-5p inhibitor decreased miR-34-5p level but increased Foxp1 expression that was reversed by sh-Foxp1 (Fig. S3A–D). Moreover, we found that miR-34-5p inhibitor enhanced cell proliferation but suppressed cell apoptosis while knockdown of Foxp1 partially blocked the effects. Consistently, the upregulations of Foxp1, β-catenin, CDK4, and CyclinD1 and the downregulation of p53 caused by miR-34-5p inhibitor were reversed by sh-Foxp1 (Fig. S3). Taken together, our results support that notion that miR-34-5p regulates DLBCL cell proliferation and apoptosis through targeting Foxp1.
Knockdown of PVT1 restrained DLBCL tumor growth in vivo
In the end, we evaluated the function of PVT1 in DLBCL in vivo. DLBCL cells (SU-DHL-6 and U-2932) were infected with sh-NC or sh-PVT1 lentivirus and were subcutaneously implanted to the nude mice. For the mice that injected with DLBCL cells or sh-NC transfected DLBCL cells, the tumor grew very fast, with tumor volume and weight progressively increasing with time (Fig. 5A). In contrast, in the mice injected with sh-PVT1-transfected cells, the tumor volume and weight were significantly reduced (Fig. 5A). We collected the tumor samples by the end of 4 weeks and performed H&E staining. As shown in Fig. 5B, there were obvious proliferating cells in the tumor tissues from the control mice. However, in the tissues from mice injected with sh-PVT1-transfected cells, we did not observe many cancer cells (Fig. 5B). Moreover, with Ki-67 and TUNEL staining, we found that knockdown of PVT1 significantly decreased the Ki-67 positive cells but increased the TUNEL positive cells (Fig. 5B), suggesting that knockdown of PVT1 inhibited tumor growth by inhibiting DLBCL cell proliferation and promoting cell apoptosis. At the molecular level, we found that knockdown of PVT1 significantly increased miR-34b-5p level (Fig. 5C). Western blotting data showed that Foxp1, β-catenin, and proliferation-related proteins (CDK4 and CyclinD1) were all reduced while p53 was increased in the shPVT1 group compared to control groups (Fig. 5D). These data suggest that knockdown of PVT1 restrains DLBCL tumor growth in vivo, most likely, through targeting miR-34b-5p.

Knockdown of PVT1 restrains DLBCL tumor growth in vivo. A Representative tumor images, tumor volume and weigh in mice injected with transfected DLBCL cells. B H&E, Ki-67, and TUNEL staining of the tumor tissues from mice injected with transfected DLBCL cells. C qRT-PCR to assess miR-34b-5p levels in the tumor tissues from each group of mice. D Western blotting to measure levels of Foxp1, β-catenin, and proliferation-related proteins including CDK4, CyclinD1, and p53 in tumors from mice injected with transfected DLBCL cells. N = 5, Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001
Discussion
As the most prevalent subtype of NHLs, DLBCL represents about 22% of newly diagnosed NHL in USA and more than 18,000 people new cases are reported each year [2, 19]. It is a very aggressive carcinoma with tumor growing rapidly in organs like lymph nodes, spleen [20]. Despite great advances made in the past decades, exploring new treatments for the disease is very necessary to improve the prognosis. In the present study, we investigated the mechanisms of DLBCL development, with the focus on PVT1/miR-34b-5p/Foxp1 pathway. We showed that PVT1 and Foxp1 were elevated in DLBCL tissues and cells while miR-34b-5p was reduced. PVT1 knockdown, miR-34b-5p overexpression, or Foxp1 knockdown suppressed DLBCL cell proliferation but enhanced cell apoptosis. Mechanistically, we showed that PVT1 directly interacted with miR-34b-5p and regulated DLBCL cell proliferation via miR-34b-5p, while miR-34b-5p targeted Foxp1. Moreover, inhibition of PVT1 significantly repressed DLBCL tumor growth in vivo. These results provide mechanistic insights into DLBCL development, as well as avenues for future therapy development.
LncRNAs play important roles in varieties of cellular processes, including physiological processes and pathological processes [7, 8]. In DLBCL, thousands of lncRNAs have been implicated, including lncRNA PVT1 [13, 21, 22]. PVT1 has been shown to function as an oncogene that promotes tumor development and progression [23,24,25]. For example, it can promote proliferation and migration of gallbladder cancer cells via miR-143 [26]. PVT1 also contributes to the development of colon cancer via sponging miR-26b [27]. Here, in DLBCL, we observed a similar oncogene function of PVT1. Its level was elevated in both DLBCL tissues from patients and DLBCL cells. Further, knockdown of its level could greatly inhibit cell proliferation and promote cell apoptosis. Our study, together with previous studies, confirms that PVT1 usually functions as a tumor-promoter. Interestingly, in DLBCL, we found that PVT1 exerted its function by acting as a miR-34b-5p sponge. We validated the direct interaction between PVT1 and miR-34b-5p in DLBCL cells also showed that miR-34b-5p inhibitor blocked the effects of sh-PVT1. PVT1 has been shown to function by sponging many miRNAs [24], but miR-34b-5p has not been reported. It might be interesting to examine the role of this novel interaction in other types of cancers. In addition, it could be possible that PVT1 has other downstream targets besides miR-34b-5p in DLBCL. Indeed, we observed that many miRNAs were upregulated, such as miR-21-5p, miR-17-5p, in sh-PVT1 transfected DLBCL cells compared to control transfected cells (Supplemental Material), and observed that miR-34b-5p exhibited the biggest change compared to other miRNAs. Therefore, we selected miR-34b-5p for further characterization.
Foxp1 belongs to the FOX transcription factor family and has crucial roles in embryonic development [28, 29]. Aberrant expression of Foxp1 has been implicated in many diseases, such as neurological disorders and cardiac diseases [30,31,32]. In cancers, Foxp1 has dual biological functions [33, 34]. It can act as a tumor suppressor or an oncogene depending on specific types of cancers. For example, in breast cancer, it functions as a tumor suppressor and loss of Foxp1 function contributes to the cancer development [35]. In hepatocellular carcinoma, Foxp1 facilitated the cancer progression [36]. In DLBCL, it is well acknowledged that Foxp1 is overexpressed and that the high abundance of Foxp1 predicts a poor prognosis of DLBCL [16]. The increased expression could be partially caused by copy number amplifications and chromosomal translocations, as indicated in a subset of DLBCL patients [16]. Whether other mechanisms are involved is not clear. Here, we showed that Foxp1 was elevated in both types of DLBCL tissues, indicating that Foxp1 is a key downstream signaling. Next, we found that the increase of Foxp1 level resulted from the reduced expression of miR-34b-5p, which directly targeted Foxp1 mRNA. PVT1 disinhibited Foxp1 expression via sponging miR-34b-5p. Therefore, multiple mechanisms lead to the upregulation of Foxp1 in DLBCL. Notably, knockdown of Foxp1 only partially blocked the effects of miR-34b-5p inhibitor on DLBCL cell proliferation and apoptosis, suggesting that other targets of miR-34b-5p are involved in the regulation. Future studies are required to examine other downstream targets. Foxp1 has been shown to activate Wnt/β-catenin [18], which promotes cancer cell proliferation. Consistently, we observed similar changes of β-catenin. It might be interesting to study whether other molecular mechanisms that contribute to the increase of Foxp1 in DLBCL.
In summary, we provide strong evidence that PVT1/miR-34b-5p/Foxp1 plays a critical role in DLBCL development. Inhibition of PVT1 or Foxp1 or rescue the level of miR-34b-5p could be a very useful way to repress DLBCL progression and treat the disease.
Data availability
All data generated or analyzed during this study are included in this published article.
Code availability
Not applicable.
References
Horvat M, Zadnik V, Juznic Setina T, Boltezar L, Pahole Golicnik J, Novakovic S, Jezersek Novakovic B (2018) Diffuse large B-cell lymphoma: 10 years’ real-world clinical experience with rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisolone. Oncol Lett 15:3602–3609. https://doi.org/10.3892/ol.2018.7774
Yin X, Xu A, Fan F, Huang Z, Cheng Q, Zhang L, Sun C, Hu Y (2019) Incidence and mortality trends and risk prediction nomogram for extranodal diffuse large B-cell lymphoma: an analysis of the surveillance, epidemiology, and end results database. Front Oncol 9:1198. https://doi.org/10.3389/fonc.2019.01198
Nowakowski GS, Czuczman MS (2015) ABC, GCB, and double-hit diffuse large B-cell lymphoma: does subtype make a difference in therapy selection? Am Soc Clin Oncol Educ Book. https://doi.org/10.14694/EdBook_AM.2015.35.e449
Rovira J, Valera A, Colomo L, Setoain X, Rodriguez S, Martinez-Trillos A, Gine E, Dlouhy I, Magnano L, Gaya A, Martinez D, Martinez A, Campo E, Lopez-Guillermo A (2015) Prognosis of patients with diffuse large B cell lymphoma not reaching complete response or relapsing after frontline chemotherapy or immunochemotherapy. Ann Hematol 94:803–812. https://doi.org/10.1007/s00277-014-2271-1
Kubuschok B, Held G, Pfreundschuh M (2015) Management of diffuse large B-cell lymphoma (DLBCL). Cancer Treat Res 165:271–288. https://doi.org/10.1007/978-3-319-13150-4_11
Kung JT, Colognori D, Lee JT (2013) Long noncoding RNAs: past, present, and future. Genetics 193:651–669. https://doi.org/10.1534/genetics.112.146704
Yao RW, Wang Y, Chen LL (2019) Cellular functions of long noncoding RNAs. Nat Cell Biol 21:542–551. https://doi.org/10.1038/s41556-019-0311-8
DiStefano JK (2018) The emerging role of long noncoding RNAs in human disease. Methods Mol Biol 1706:91–110. https://doi.org/10.1007/978-1-4939-7471-9_6
Schmitt AM, Chang HY (2016) Long noncoding RNAs in cancer pathways. Cancer Cell 29:452–463. https://doi.org/10.1016/j.ccell.2016.03.010
Nobili L, Ronchetti D, Taiana E, Neri A (2017) Long non-coding RNAs in B-cell malignancies: a comprehensive overview. Oncotarget 8:60605–60623. https://doi.org/10.18632/oncotarget.17303
Wang Y, Zhang M, Xu H, Wang Y, Li Z, Chang Y, Wang X, Fu X, Zhou Z, Yang S, Wang B, Shang Y (2017) Discovery and validation of the tumor-suppressive function of long noncoding RNA PANDA in human diffuse large B-cell lymphoma through the inactivation of MAPK/ERK signaling pathway. Oncotarget 8:72182–72196. https://doi.org/10.18632/oncotarget.20053
Zhu Q, Li Y, Guo Y, Hu L, Xiao Z, Liu X, Wang J, Xu Q, Tong X (2019) Long non-coding RNA SNHG16 promotes proliferation and inhibits apoptosis of diffuse large B-cell lymphoma cells by targeting miR-497-5p/PIM1 axis. J Cell Mol Med 23:7395–7405. https://doi.org/10.1111/jcmm.14601
Yang R, Shao T, Long M, Shi Y, Liu Q, Yang L, Zhan M (2020) Long noncoding RNA PVT1 promotes tumor growth and predicts poor prognosis in patients with diffuse large B-cell lymphoma. Cancer Commun (Lond). https://doi.org/10.1002/cac2.12073
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) 9:402. https://doi.org/10.3389/fendo.2018.00402
Qian CS, Li LJ, Huang HW, Yang HF, Wu DP (2020) MYC-regulated lncRNA NEAT1 promotes B cell proliferation and lymphomagenesis via the miR-34b-5p-GLI1 pathway in diffuse large B-cell lymphoma. Cancer Cell Int 20:87. https://doi.org/10.1186/s12935-020-1158-6
Gascoyne DM, Banham AH (2017) The significance of FOXP1 in diffuse large B-cell lymphoma. Leuk Lymphoma 58:1037–1051. https://doi.org/10.1080/10428194.2016.1228932
Flori M, Schmid CA, Sumrall ET, Tzankov A, Law CW, Robinson MD, Müller A (2016) The hematopoietic oncoprotein FOXP1 promotes tumor cell survival in diffuse large B-cell lymphoma by repressing S1PR2 signaling. Blood 127:1438–1448. https://doi.org/10.1182/blood-2015-08-662635
Walker MP, Stopford CM, Cederlund M, Fang F, Jahn C, Rabinowitz AD, Goldfarb D, Graham DM, Yan F, Deal AM, Fedoriw Y, Richards KL, Davis IJ, Weidinger G, Damania B, Major MB (2015) FOXP1 potentiates Wnt/β-catenin signaling in diffuse large B cell lymphoma. Sci Signal 8:ra12. https://doi.org/10.1126/scisignal.2005654
Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR (2016) 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 66:443–459. https://doi.org/10.3322/caac.21357
Schneider C, Pasqualucci L, Dalla-Favera R (2011) Molecular pathogenesis of diffuse large B-cell lymphoma. Semin Diagn Pathol 28:167–177. https://doi.org/10.1053/j.semdp.2011.04.001
Huang X, Qian W, Ye X (2020) Long noncoding RNAs in diffuse large B-cell lymphoma: current advances and perspectives. Onco Targets Ther 13:4295–4303. https://doi.org/10.2147/ott.S253330
Verma A, Jiang Y, Du W, Fairchild L, Melnick A, Elemento O (2015) Transcriptome sequencing reveals thousands of novel long non-coding RNAs in B cell lymphoma. Genome Med 7:110. https://doi.org/10.1186/s13073-015-0230-7
Onagoruwa OT, Pal G, Ochu C, Ogunwobi OO (2020) Oncogenic role of PVT1 and therapeutic implications. Front Oncol 10:17. https://doi.org/10.3389/fonc.2020.00017
Wang W, Zhou R, Wu Y, Liu Y, Su W, Xiong W, Zeng Z (2019) PVT1 Promotes cancer progression via MicroRNAs. Front Oncol 9:609. https://doi.org/10.3389/fonc.2019.00609
Derderian C, Orunmuyi AT, Olapade-Olaopa EO, Ogunwobi OO (2019) PVT1 signaling is a mediator of cancer progression. Front Oncol 9:502. https://doi.org/10.3389/fonc.2019.00502
Chen J, Yu Y, Li H, Hu Q, Chen X, He Y, Xue C, Ren F, Ren Z, Li J, Liu L, Duan Z, Cui G, Sun R (2019) Long non-coding RNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer. Mol Cancer 18:33. https://doi.org/10.1186/s12943-019-0947-9
Zhang R, Li J, Yan X, Jin K, Li W, Liu X, Zhao J, Shang W, Liu Y (2018) Long noncoding RNA plasmacytoma variant translocation 1 (PVT1) promotes colon cancer progression via endogenous sponging miR-26b. Med Sci Monit 24:8685–8692. https://doi.org/10.12659/msm.910955
Laissue P (2019) The forkhead-box family of transcription factors: key molecular players in colorectal cancer pathogenesis. Mol Cancer 18:5. https://doi.org/10.1186/s12943-019-0938-x
Golson ML, Kaestner KH (2016) Fox transcription factors: from development to disease. Development 143:4558–4570. https://doi.org/10.1242/dev.112672
Siper PM, De Rubeis S, Trelles MDP, Durkin A, Di Marino D, Muratet F, Frank Y, Lozano R, Eichler EE, Kelly M, Beighley J, Gerdts J, Wallace AS, Mefford HC, Bernier RA, Kolevzon A, Buxbaum JD (2017) Prospective investigation of FOXP1 syndrome. Mol Autism 8:57. https://doi.org/10.1186/s13229-017-0172-6
Liu J, Zhuang T, Pi J, Chen X, Zhang Q, Li Y, Wang H, Shen Y, Tomlinson B, Chan P, Yu Z, Cheng Y, Zheng X, Reilly M, Morrisey E, Zhang L, Liu Z, Zhang Y (2019) Endothelial forkhead box transcription factor P1 regulates pathological cardiac remodeling through transforming growth factor-β1-endothelin-1 signal pathway. Circulation 140:665–680. https://doi.org/10.1161/circulationaha.119.039767
Chang SW, Mislankar M, Misra C, Huang N, Dajusta DG, Harrison SM, McBride KL, Baker LA, Garg V (2013) Genetic abnormalities in FOXP1 are associated with congenital heart defects. Hum Mutat 34:1226–1230. https://doi.org/10.1002/humu.22366
Kim JH, Hwang J, Jung JH, Lee HJ, Lee DY, Kim SH (2019) Molecular networks of FOXP family: dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol Cancer 18:180. https://doi.org/10.1186/s12943-019-1110-3
Koon HB, Ippolito GC, Banham AH, Tucker PW (2007) FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets 11:955–965. https://doi.org/10.1517/14728222.11.7.955
Ijichi N, Ikeda K, Horie-Inoue K, Inoue S (2013) FOXP1 and estrogen signaling in breast cancer. Vitam Horm 93:203–212. https://doi.org/10.1016/b978-0-12-416673-8.00006-x
Wang X, Sun J, Cui M, Zhao F, Ge C, Chen T, Yao M, Li J (2016) Downregulation of FOXP1 inhibits cell proliferation in hepatocellular carcinoma by inducing G1/S phase cell cycle arrest. Int J Mol Sci. https://doi.org/10.3390/ijms17091501
Acknowledgements
Thanks to the members of our laboratory for their contributions.
Funding
None.
Author information
Authors and Affiliations
Contributions
ST: supervision, concepts, design, experimental studies, data acquisition, data analysis, preparation, editing; YC: literature research; MH: experimental studies, clinical studies; LX: data analysis, editing; C-BF: clinical studies; X-BH: design, review; All the authors approved for the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Ethical approval
The study was reviewed and received approval from the Ethics Committee of the First Affiliated Hospital of Hainan Medical University. All animal experiments have been reviewed and received approval by the Animal Care and Use Committee of Nanjing Medical University.
Consent to participate
All patients have consented to the study.
Consent for publication
The informed consent obtained from study participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material. Supplemental material—Knockdown of PVT1 induced changes of many miRNAs. Relative fold changes of miRNAs in sh-PVT1 transfected U-2932 cells compared to sh-NC transfected cells. N=4, Data are expressed as the mean ± SD.
11010_2021_4335_MOESM1_ESM.tif
Fig. S1 Foxp1 regulated CDK4, CyclinD1, and p53 expression. (A) Predicted binding sites of Foxp1 on CDK4 promoter. (B) Relative enrichment of CDK4 promoter assessed by ChIP. (C) Relative luciferase activities in transfected cells measured by dual luciferase assay. (D) Predicted binding sites of Foxp1 on CyclinD1 promoter. (E) Relative enrichment of CyclinD1 promoter assessed by ChIP. (F) Relative luciferase activities in transfected cells measured by dual luciferase assay. (G) Predicted binding sites of Foxp1 on p53 promoter. (H) Relative enrichment of p53 promoter assessed by ChIP. (I) Relative luciferase activities in transfected cells measured by dual luciferase assay. (J) Predicted binding sites between miR-34b-5p and CDK4 mRNA. (K) Relative luciferase activities in transfected cells measured by dual luciferase assay. N=4, Data are expressed as the mean ± SD. *P <0.05; **P <0.01, ***P<0.001. Supplementary file1 (TIF 596 KB)
11010_2021_4335_MOESM2_ESM.tif
Fig. S2 Knockdown of Foxp1 suppressed DLBCL cell proliferation and increased cell apoptosis. (A-B) Foxp1 mRNA and protein levels in non-transfected control DLBCL cells and cells transfected with shNC or shFoxp1. (C) MTT analysis of cell proliferation in transfected DLBCL cells. (D) Cell proliferation of transfected DLBCL cells were analyzed with colony formation assay. (E) Percentage of apoptotic cells in transfected DLBCL cells. (F) Western blotting to determine levels of Foxp1, β-catenin, and proliferation-related proteins (CDK4, CyclinD1, and p53) in transfected DLBCL cells. N=4, Data are expressed as the mean ± SD. *P <0.05; **P <0.01, ***P<0.001. Supplementary file2 (TIF 3428 KB)
11010_2021_4335_MOESM3_ESM.tif
Fig. S3 miR-34b-5p regulated DLBCL cell proliferation and apoptosis via Foxp1. (A) MTT analysis of cell proliferation in transfected DLBCL cells. (B) Cell proliferation of transfected DLBCL cells were analyzed with colony formation assay. (C) Percentage of apoptotic cells in transfected DLBCL cells. (D) Western blotting to determine levels of Foxp1, β-catenin, and proliferation-related proteins (CDK4, CyclinD1, and p53) in transfected DLBCL cells. N=4, Data are expressed as the mean ± SD. *P <0.05; **P <0.01, ***P<0.001. Supplementary file3 (TIF 4218 KB)
Rights and permissions
About this article

Cite this article
Tao, S., Chen, Y., Hu, M. et al. LncRNA PVT1 facilitates DLBCL development via miR-34b-5p/Foxp1 pathway. Mol Cell Biochem 477, 951–963 (2022). https://doi.org/10.1007/s11010-021-04335-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-021-04335-7