
Abstract
Previously, we reported that 3H-1,2-dithiole-3-thione (D3T), an Nrf2 activator, acted as a potential chemoprotectant against lipopolysaccharide (LPS)-induced mortality in mice. In view of the critical involvement of macrophages in the pathogenesis of LPS-induced endotoxemia, in the present study, we investigated the protective effects of D3T on LPS-induced proinflammatory responses in cultured murine RAW 264.7 macrophage cell line and primary peritoneal macrophages and the potential involvement of antioxidant induction, NF-κB, and Nrf2. We showed that treatment with D3T resulted in increased levels of a series of antioxidants in RAW 264.7 cells in a concentration-dependent manner. These included the reduced form of glutathione, glutathione peroxidase, glutathione reductase, glutathione S-transferase, and NADPH:quinone oxidoreductase 1. Catalase was also potently induced by D3T which, however, did not show a concentration dependency. Concurrent with the ability to induce the above cellular antioxidants, D3T pretreatment of RAW 264.7 cells also led to a concentration-dependent suppression of LPS-induced interleukin-1beta (IL-1β) production and nitric oxide release. LPS-stimulated tumor necrosis factor-alpha (TNF-α) production was also suppressed by D3T, but to a much lesser extent. Using NF-κB reporter gene-expressing RAW 264.7 cells, we further showed that D3T pretreatment also suppressed LPS-induced NF-κB activation. To investigate the potential involvement of Nrf2, a chief regulator of cellular antioxidant genes, we used peritoneal macrophages isolated from Nrf2+/+ and Nrf2−/− mice. Our results showed that D3T pretreatment suppressed LPS-induced proinflammatory responses in Nrf2+/+ macrophages, and this inhibitory effect of D3T was completely lost in Nrf2−/− macrophages. Collectively, the results of the present study demonstrated that D3T acted as a potent suppressor of LPS-induced proinflammatory responses in macrophages. Antioxidant induction, NF-κB suppression, and Nrf2 activation appeared to contribute to the anti-proinflammatory activity of D3T in macrophages.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Endotoxemia and sepsis remain a major cause of death worldwide, accounting for nearly 20% of all global deaths [1]. Although cytokine storm and dysregulated inflammation have long been recognized as a major pathophysiological mechanism of sepsis, effective therapies for sepsis remain lacking [2,3,4]. Hence, identification of critical molecular and cellular targets of sepsis and development of mechanistically based modalities for sepsis intervention are of paramount clinical significance. In this context, recent studies have demonstrated a critical role for oxidative stress, NF-κB, and Nrf2 signaling in the pathophysiology of endotoxemia and sepsis in animal models [5, 6].
Dithiolethiones are five-membered cyclic sulfur-containing compounds, some of which are constituents of cruciferous vegetables [7]. We and others have found that 3H-1,2-dithiole-3-thione (D3T) is the most potent member of dithiolethiones for the induction of endogenous antioxidative/anti-inflammatory and phase 2 enzymes in various types of tissues and cells, including hepatic tissue [7, 8], myocardium [9], bone marrow stromal cells [10], and macrophages [11], among others. As oxidative and inflammatory stress are intimately involved in the pathophysiology of various diseases, upregulation of these cellular defenses by D3T has been shown to protect against such disease processes as chemical carcinogenesis [12], neurodegeneration [13], and cardiovascular injury [14] in experimental models. Notably, we and others have also previously demonstrated that D3T-mediated upregulation of antioxidative/anti-inflammatory defenses occurs via activation of Nrf2, the chief regulator of cytoprotective genes [8, 14, 15]. Indeed, D3T is among the most potent activators of Nrf2 signaling and the activation of Nrf2 is indispensable for D3T-mediated cytoprotection against oxidative and electrophilic injury [10, 15, 16]. Recently, our work further suggested that D3T also protected against lipopolysaccharide (LPS)-induced mortality in mice in an Nrf2-dependent manner [17]. D3T, like other Nrf2 activators, may thus be developed as a potential chemoprotectant for sepsis intervention. To this end, further elucidation of the molecular processes underlying D3T-mediated anti-proinflammatory responses in cells is warranted. As macrophages are crucial inflammatory cells involved in cytokine storm and inflammatory syndrome [18], in the present study, we used murine macrophage line RAW 264.7 cells and primary peritoneal macrophages isolated from Nrf2+/+ and Nrf2−/− mice as in vitro models to investigate the protective effects of D3T on LPS-induced proinflammatory responses and the potential involvement of antioxidant induction, NF-κB, and Nrf2 in D3T-mediated anti-proinflammation.
Materials and methods
Materials
D3T with a purity of 99.8% was generously provided by Dr. Mary Tanga at SRI International (Menlo Park, CA) and Dr. Linda Brady at National Institute of Mental Health (Bethesda, MD). RPMI-1640 medium, penicillin, streptomycin, fetal bovine serum (FBS), and Dulbecco’s phosphate buffered saline (PBS) were from Thermo Fisher Scientific (Waltham, MA). All other chemicals and agents were from Sigma-Aldrich (St. Louis, MO). Cell culture flasks and other plastic wares were purchased from Thermo Fisher Scientific (Waltham, MA).
Cell cultures
Regular RAW 264.7 cells were obtained from ATCC (Manassas, VA) and NF-κB reporter gene-expressing RAW 264.7 cells were purchased from Genlantis (San Diego, CA). Primary peritoneal macrophages were isolated from inhouse-bred male Nrf2+/+ and Nrf2−/− C57BL/6 mice as described previously following the procedures approved by Institutional Animal Care and Use Committee [11]. All cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 100 units/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C in a humidified atmosphere of 5% CO2. For measuring D3T-mediated induction of cellular antioxidants, the cells cultured on tissue culture flasks were incubated with D3T (0, 25, 50, and 100 μM) at 37 °C for 24 h. To determine the effects of D3T on LPS-induced cytokine and nitric oxide release, cells were pretreated with D3T for 24 h and then exposed to LPS (10 ng/ml) for another 24 h followed by the measurement of the endpoints released into culture media. On the other hand, activation of NF-κB was measured after a 6-h exposure to LPS. Cellular protein content was quantified with Bio-Rad protein assay dye (Hercules, CA) using bovine serum albumin as the standard.
Measurement of cellular antioxidants
Preparation of cell extracts and measurement of cellular levels/activities of reduced glutathione (GSH), glutathione peroxidase (GPx), glutathione reductase (GR), glutathione S-transferase (GST), catalase, and NADPH:quinone oxidoreductase 1 (NQO1) were performed according to the published procedures described by us before [10, 11, 19].
Measurement of IL-1β and TNF-α
The levels of IL-1β and TNF-α released into the culture media were measured using corresponding mouse Quantikine ELISA kits from R&D Systems (Minneapolis, MN) according to the manufacturer’s instruction. A concurrently run standard curve was used for quantification.
Measurement of nitric oxide
The production of nitric oxide was determined by measuring the total amount of nitrate/nitrite released into the culture media using a commercial kit from R&D Systems (Minneapolis, MN) according to the manufacture’s instruction. A concurrently run standard curve was used for quantification.
Determination of NF-κB activation
The cells expressing the NF-kB-luciferase report gene were lysed, and the luciferase activity was measured using a Berthold LB9505 multichannel luminometer (Wildbad, Germany). The intensity of the luminescence is proportional to the degree of NF-kB activation.
Statistical analysis
All data are expressed as means ± standard deviation (SD) from at least three separate experiments unless otherwise indicated. Differences between mean values of multiple groups were analyzed by one-way analysis of variance followed by Student–Newman–Keuls test. Differences between two groups were analyzed by Student’s t test. Statistical significance was considered at p < 0.05.
Results and discussion
D3T-mediated induction of antioxidants in RAW 264.7 cells
We previously showed that D3T-induced a host of cellular antioxidant defenses in primary peritoneal macrophages isolated from mice [11]. For the present study, we first determined if D3T also induced antioxidants in RAW 264.7 cells, a mouse macrophage cell line. As shown in Fig. 1, incubation of RAW 264.7 cells with D3T for 24 h resulted in significant induction in cellular GSH, GPx, GR, GST, catalase, and NQO1. The induction of the above antioxidants, except for catalase, showed a D3T-concentration dependency. It remains unclear why catalase induction was independent of D3T concentrations used (25–100 μM). It is possible that 25 μM D3T was enough to cause the maximal induction of catalase under the present experimental conditions.

D3T-mediated induction of antioxidants in RAW 264.7 cells. The cells were incubated with the indicated concentrations of D3T (0, 25, 50, and 100 µM) for 24 h followed by measurement of the levels or activities of various cellular antioxidants, including GSH (a), GPx (b), GR (c), GST (d), catalase (e), and NQO1 (f). Data represent mean ± SD (n = 4–8). *p < 0.05 versus 0 µM D3T; #p < 0.05 versus 25 and 50 µM D3T; $, p < 0.05 versus 25 µM D3T
GSH, GPx, GR, and GST are members of the glutathione system—a major cellular antioxidant defense system involved in the detoxification of various reactive oxygen and nitrogen species (ROS/RNS) [20]. On the other hand, catalase is an enzyme selective for decomposing hydrogen peroxide, whereas NQO1 acts as both a quinone-detoxifying enzyme and a superoxide scavenger [21]. As ROS/RNS are critical players in dysregulated inflammatory responses [22], induction of the above cellular antioxidants would render macrophages more resistant to oxidative stress and dampen macrophage’s proinflammatory responses. Thus, this antioxidant induction experiment would set a stage for the subsequent investigation of the effects of D3T on LPS-induced proinflammatory responses.
D3T-mediated suppression of LPS-induced proinflammatory responses in RAW 264.7 cells
Release of proinflammatory cytokines by macrophages has been shown to play a critical role in many inflammatory disorders, especially the acute phase of sepsis [21, 23]. We next determined if D3T pretreatment could suppress LPS-induced production of IL-1β and TNF-α, two major proinflammatory cytokines, in macrophages. As shown in Fig. 2, under the basal unstimulated condition, RAW 264.7 cells released minimal amounts of IL-1β and TNF-α; however, exposure of the cells to LPS led to dramatically increased release of both IL-1β and TNF-α into the culture media. Notably, pretreatment of the cells with D3T resulted in a marked concentration-dependent attenuation of LPS-induced IL-1β release. A 71%, 89%, and 94% reduction in LPS-induced IL-1β release was seen with 25, 50, and 100 μM D3T pretreatment, respectively (Fig. 2a). In contrast, the effect of D3T pretreatment on LPS-induced TNF-α release was much less remarkable; only the highest concentration of D3T caused a 28% reduction (Fig. 2b). This differential effect on IL-1β versus TNF-α suggests that different signaling pathways may be involved in the LPS-induced release of these two proinflammatory cytokines. Indeed, a previous study showed that blocking mitochondrial ROS production blunted LPS-induced IL-1β expression without affecting LPS-induced TNF-α expression [18].

D3T-mediated suppression of LPS-induced proinflammatory cytokine release in RAW 264.7 cells. The cells were pretreated with the indicated concentrations of D3T (0, 25, 50, and 100 µM) for 24 h and then exposed to LPS (10 ng/ml) for another 24 h, followed by measurement of IL-1β (a) and TNF-α (b) release into the culture media. Data represent mean ± SD (n = 4–6). *p < 0.05 versus Control (0 µM D3T); #p < 0.05 versus LPS only; $p < 0.05 versus D3T 25 µM/LPS; &p < 0.05 versus D3T 50 µM/LPS
To investigate the effect of D3T pretreatment on ROS/RNS production, we measured the total nitric oxide production by RAW 264.7 cells. In line with the effects on the proinflammatory cytokines, LPS exposure dramatically increased the production of total nitric oxide (as assessed by measuring nitrate/nitrite formation) in the cells. The LPS-induced RNS production was significantly blunted by D3T pretreatment in a concentration-dependent manner (Fig. 3). It remains unclear how D3T pretreatment reduced LPS-induced nitric oxide production. It is well-established that LPS activates inducible nitric oxide synthase (iNOS), leading to increased production of nitric oxide in macrophages [24, 25]. The suppression by D3T on LPS-induced nitric oxide production in RAW 264.7 cells might result from two potential mechanisms: (i) scavenging nitric oxide by the D3T-induced cellular antioxidants (e.g., GSH) and (ii) suppression of LPS-induced NF-κB activation (see Sect. 3.3). In this context, LPS-induced iNOS expression occurs via an NF-κB-dependent mechanism [26].

D3T-mediated suppression of LPS-induced nitric oxide release in RAW 264.7 cells. The cells were pretreated with the indicated concentrations of D3T (0, 25, 50, and 100 µM) for 24 h and then exposed to LPS (10 ng/ml) for another 24 h, followed by measurement of nitric oxide (in the form of nitrate/nitrite) release into the culture media. Data represent mean ± SD (n = 3). *p < 0.05 versus Control (0 µM D3T); #p < 0.05 versus LPS only; $p < 0.05 versus D3T 25 µM/LPS; &p < 0.05 versus D3T 50 µM/LPS
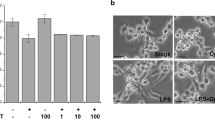
D3T-mediated suppression of LPS-induced NF-κB activation in RAW 264.7 cells
Since NF-κB is a chief regulator of the gene expression of proinflammatory cytokines (including IL-1β and TNF-α) as well as iNOS [27], we determined if D3T pretreatment could also suppress LPS-induced NF-κB activation. As shown in Fig. 4, the LPS-stimulated NF-κB activation was significantly suppressed by D3T pretreatment. However, the degree of NF-κB suppression by D3T was not in line with the extent of D3T-mediated IL-1β suppression (Fig. 2a). In this regard, the suppression of LPS-induced NF-κB activation by D3T did not show a concentration dependency and only a 25–35% inhibition was seen with D3T (25–100 μM) pretreatment (Fig. 4). On the other hand, LPS-induced IL-1β production was suppressed by 71–94% following D3T pretreatment. This discrepancy suggests that inhibition of NF-κB signaling is only one of the mechanisms of D3T-mediated blockage of LPS-induced IL-1β production in macrophages.

D3T-mediated suppression of LPS-induced NF-κB activation in RAW 264.7 cells. The cells were pretreated with the indicated concentrations of D3T (0, 25, 50, and 100 µM) for 24 h and then exposed to LPS (10 ng/ml) for another 6 h, followed by detection of NF-κB activation. Data represent mean ± SD (n = 4). *p < 0.05 versus LPS only
D3T-mediated suppression of LPS-induced proinflammatory responses in Nrf2+/+ and Nrf2−/− peritoneal macrophages
We previously showed that D3T-mediated induction of antioxidants in macrophages was dependent on Nrf2 status [11] and D3T is a potent activator of Nrf2 as well as an inducer of Nrf2 expression [17]. Accordingly, we determined if Nrf2 status could also affect the ability of D3T to suppress LPS-induced proinflammatory cytokine release. To this end, we used peritoneal macrophages isolated from Nrf2-null (Nrf2−/−) mice and the wild-type (Nrf2+/+) mice. As shown in Fig. 5, the suppressing effect of D3T pretreatment on LPS-induced IL-1β and TNF-α release was completely lost in the Nrf2−/− macrophages, suggesting that Nrf2 signaling is indispensable for the anti-proinflammatory activity of D3T in macrophages. Notably, the absence of Nrf2 resulted in a drastic augmentation of the LPS-induced release of TNF-α (Fig. 5B); there was also an increasing trend in LPS-induced IL-1β release, but it did not reach statistical significance due to large sample variations (Fig. 5a). Nevertheless, this finding suggests that the inducible express of proinflammatory cytokines, especially TNF-α, could readily become out of control in the absence of functional Nrf2 signaling, causing overt inflammatory stress. Indeed, targeted disruption of Nrf2 gene has been shown to dramatically sensitize animals to inflammatory tissue injury in an experimental sepsis animal model [17, 28].

D3T-mediated suppression of LPS-induced proinflammatory responses in Nrf2+/+ and Nrf2−/− peritoneal macrophages. Peritoneal macrophages of Nrf2+/+ and Nrf2−/− genotypes were pretreated with 0 (control) and 100 µM D3T for 24 h and then exposed to LPS (10 ng/ml) for another 24 h, followed by measurement of IL-1β and TNF-α release into the culture media. Data represent mean ± SD (n = 3). *p < 0.05 versus respective Control; #p < 0.05 versus LPS only; $p < 0.05 versus LPS only in Nrf2+/+ cells
In conclusion, the results of the present study demonstrated that D3T—a cruciferous dithiolethione-related compound, is effective in attenuating LPS-induced proinflammatory stress in macrophages, and Nrf2-dependent antioxidant induction as well as suppression of NF-κB activation may collectively contribute to the ability of D3T to protect against experimental sepsis observed previously in mice [17] (Fig. 6). It should be noted, however, that the present study did not focus on delineating the detailed signaling pathways on which D3T may act to suppress LPS-induced proinflammatory responses. In fact, the exact signaling pathways involved in LPS-induced cytokine (e.g., IL-1β) expression remain to be defined. In this context, recent studies by others showed that LPS-induced IL-1β production was dependent, at least partly, on mitochondrial ROS production [18, 29]. Hence, the ability of D3T to increase intracellular and intramitochondrial antioxidants [30] may account, at least partly, for the attenuation of LPS-induced IL-1β production.

Schematic illustration of the potential involvement of Nrf2 activation (antioxidant induction) and NF-κB inhibition in D3T-mediated suppression of oxidative and inflammatory stress underlying endotoxemia and sepsis. Nox denotes NADPH oxidase, a well-established ROS-producing enzyme stimulated by LPS
It should also be noted that D3T may exert its effects independent of Nrf2 activation. In this regard, D3T has been shown to react with sulfhydryl groups [31] and may thus directly modify cellular proteins including those involved in LPS receptor signaling. Nevertheless, the dramatic abolishment of D3T-mediated protective effects on LPS-induced proinflammatory responses in Nrf2-null macrophages suggests that Nrf2 activation is a major underlying mechanism.
A final notion about the findings of the present study is that we previously demonstrated that D3T induces a battery of antioxidant and phase 2 enzymes in macrophages via upregulating specific isozyme gene expression as evidenced by increased mRNA and protein levels of the specific isozymes [11]. As such, in the present study, we did not determine the mRNA or protein expression of the antioxidant isoenzymes. Moreover, the exact contribution of the individual Nrf2-regulated antioxidant enzymes to D3T-mediated suppression of LPS-induced proinflammatory responses in macrophages remains to be determined. In this context, an early study suggested that selective overexpression of NQO1 in human monocytes attenuated LPS-induced TNF and IL-1β expression [32]. As efficient detoxification of ROS/RNS requires the complementary actions of various cellular antioxidant enzymes, we propose that the coordinated upregulation of diverse cellular antioxidants by D3T via an Nrf2-dependent mechanism may represent a much more effective strategy than overexpression of a single antioxidant enzyme in protecting against oxidative and inflammatory stress underlying endotoxemia and sepsis (Fig. 6).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, Fleischmann-Struzek C, Machado FR, Reinhart KK, Rowan K, Seymour CW, Watson RS, West TE, Marinho F, Hay SI, Lozano R, Lopez AD, Angus DC, Murray CJL, Naghavi M (2020) Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395:200–211. https://doi.org/10.1016/S0140-6736(19)32989-7
Cohen J, Vincent JL, Adhikari NK, Machado FR, Angus DC, Calandra T, Jaton K, Giulieri S, Delaloye J, Opal S, Tracey K, van der Poll T, Pelfrene E (2015) Sepsis: a roadmap for future research. Lancet Infect Dis 15:581–614. https://doi.org/10.1016/S1473-3099(15)70112-X
Seymour CW, Rosengart MR (2015) Septic shock: advances in diagnosis and treatment. JAMA 314:708–717. https://doi.org/10.1001/jama.2015.7885
Cecconi M, Evans L, Levy M, Rhodes A (2018) Sepsis and septic shock. Lancet 392:75–87. https://doi.org/10.1016/S0140-6736(18)30696-2
Liu SF, Malik AB (2006) NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 290:L622–L645. https://doi.org/10.1152/ajplung.00477.2005
Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, Biswal S (2011) Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med 184:928–938. https://doi.org/10.1164/rccm.201102-0271OC
Kwak MK, Egner PA, Dolan PM, Ramos-Gomez M, Groopman JD, Itoh K, Yamamoto M, Kensler TW (2001) Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat Res 480–481:305–315. https://doi.org/10.1016/s0027-5107(01)00190-7
Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW (2003) Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem 278:8135–8145. https://doi.org/10.1074/jbc.M211898200
Cao Z, Li Y (2004) The chemical inducibility of mouse cardiac antioxidants and phase 2 enzymes in vivo. Biochem Biophys Res Commun 317:1080–1088. https://doi.org/10.1016/j.bbrc.2004.03.156
Zhu H, Zhang L, Itoh K, Yamamoto M, Ross D, Trush MA, Zweier JL, Li Y (2006) Nrf2 controls bone marrow stromal cell susceptibility to oxidative and electrophilic stress. Free Radic Biol Med 41:132–143. https://doi.org/10.1016/j.freeradbiomed.2006.03.020
Zhu H, Jia Z, Zhang L, Yamamoto M, Misra HP, Trush MA, Li Y (2008) Antioxidants and phase 2 enzymes in macrophages: regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp Biol Med (Maywood) 233:463–474. https://doi.org/10.3181/0711-RM-304
Otieno MA, Kensler TW, Guyton KZ (2000) Chemoprotective 3H–1,2-dithiole-3-thione induces antioxidant genes in vivo. Free Radic Biol Med 28:944–952. https://doi.org/10.1016/s0891-5849(00)00175-1
Burton NC, Kensler TW, Guilarte TR (2006) In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology 27:1094–1100. https://doi.org/10.1016/j.neuro.2006.07.019
Cao Z, Zhu H, Zhang L, Zhao X, Zweier JL, Li Y (2006) Antioxidants and phase 2 enzymes in cardiomyocytes: Chemical inducibility and chemoprotection against oxidant and simulated ischemia-reperfusion injury. Exp Biol Med (Maywood) 231:1353–1364. https://doi.org/10.1177/153537020623100809
Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y (2005) Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett 579:3029–3036. https://doi.org/10.1016/j.febslet.2005.04.058
Zhu H, Jia Z, Misra BR, Zhang L, Cao Z, Yamamoto M, Trush MA, Misra HP, Li Y (2008) Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol 8:71–85. https://doi.org/10.1007/s12012-008-9016-0
Li YR, Jia Z, Zhu H (2019) 3H–1,2-dithiole-3-thione as a potentially novel therapeutic compound for sepsis intervention. React Oxyg Species 8:202–212
Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167(457–470):e13. https://doi.org/10.1016/j.cell.2016.08.064
Zhu H, Cao Z, Zhang L, Trush MA, Li Y (2007) Glutathione and glutathione-linked enzymes in normal human aortic smooth muscle cells: chemical inducibility and protection against reactive oxygen and nitrogen species-induced injury. Mol Cell Biochem 301:47–59. https://doi.org/10.1007/s11010-006-9396-z
Hopkins RZLY (2020) Essentials of antioxidant biology and medicine. Cell Med Press, Raleigh, NC
Zhu H, Jia Z, Mahaney JE, Ross D, Misra HP, Trush MA, Li Y (2007) The highly expressed and inducible endogenous NAD(P)H:quinone oxidoreductase 1 in cardiovascular cells acts as a potential superoxide scavenger. Cardiovasc Toxicol 7:202–211. https://doi.org/10.1007/s12012-007-9001-z
Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20:1126–1167. https://doi.org/10.1089/ars.2012.5149
Johnson DW, Kalil AC (2016) Is interleukin-1 receptor blockade ready for prime time in patients with severe sepsis and macrophage activation syndrome? Crit Care Med 44:443–444. https://doi.org/10.1097/CCM.0000000000001460
Xie QW, Whisnant R, Nathan C (1993) Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med 177:1779–1784. https://doi.org/10.1084/jem.177.6.1779
Weinberg JB, Misukonis MA, Shami PJ, Mason SN, Sauls DL, Dittman WA, Wood ER, Smith GK, McDonald B, Bachus KE et al (1995) Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 86:1184–1195
Liu Y, Fang S, Li X, Feng J, Du J, Guo L, Su Y, Zhou J, Ding G, Bai Y, Wang S, Wang H, Liu Y (2017) Aspirin inhibits LPS-induced macrophage activation via the NF-kappaB pathway. Sci Rep 7:11549. https://doi.org/10.1038/s41598-017-10720-4
Zhang Q, Lenardo MJ, Baltimore D (2017) 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168:37–57. https://doi.org/10.1016/j.cell.2016.12.012
Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S (2006) Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 116:984–995. https://doi.org/10.1172/JCI25790
Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, Zang QS (2015) Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE 10:e0139416. https://doi.org/10.1371/journal.pone.0139416
Zhu H, Jia Z, Zhou K, Misra HP, Santo A, Gabrielson KL, Li Y (2009) Cruciferous dithiolethione-mediated coordinated induction of total cellular and mitochondrial antioxidants and phase 2 enzymes in human primary cardiomyocytes: cytoprotection against oxidative/electrophilic stress and doxorubicin toxicity. Exp Biol Med 234:418–429. https://doi.org/10.3181/0811-RM-340
Jia Z, Zhu H, Trush MA, Misra HP, Li Y (2008) Generation of superoxide from reaction of 3H–1,2-dithiole-3-thione with thiols: implications for dithiolethione chemoprotection. Mol Cell Biochem 307:185–191. https://doi.org/10.1007/s11010-007-9598-z
Rushworth SA, MacEwan DJ, O’Connell MA (2008) Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol 181:6730–6737. https://doi.org/10.4049/jimmunol.181.10.6730
Acknowledgements
This study was supported in part by a grant from the National Institutes of Health (GM124652). The Berthold LB9505 luminometer was provided by Dr. Michael A. Trush from The Johns Hopkins University (Baltimore, MD).
Funding
This study was supported in part by a Grant (GM124652) from the National Institutes of Health, Bethesda, MD.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception, design, performance, data analysis, and writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interest to disclose.
Ethical approval
This work does not directly involve intact animals or human subjects. The experimental protocol for the isolation of the primary peritoneal macrophages from Nrf2+/+ and Nrf2−/− mice (on C57BL/6 background) was approved by the Institutional Animal Care and Use Committee as described in Ref [9], and the cells has been kept in liquid nitrogen ever since and were used in the present study for generating the data presented in Fig. 5.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhu, H., Bui, A., Santo, A. et al. 3H-1,2-dithiole-3-thione suppresses LPS-induced proinflammatory responses in macrophages: potential involvement of antioxidant induction, NF-κB, and Nrf2. Mol Cell Biochem 477, 1499–1506 (2022). https://doi.org/10.1007/s11010-021-04331-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-021-04331-x