
Abstract
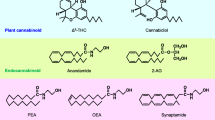
Anandamide is an endocannabinoid derived from arachidonic acid-containing membrane lipids and has numerous biological functions. Its effects are primarily mediated by the cannabinoid receptors CB1 and CB2, and the vanilloid TRPV1 receptor. Anandamide is known to be involved in sleeping and eating patterns as well as pleasure enhancement and pain relief. This manuscript provides a review of anandamide synthesis, degradation, and storage and hence the homeostasis of the anandamide signaling system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The endocannabinoid anandamide (AEA) is an ethanolamide derivative of arachidonic acid (AA) that serves to activate primarily cannabinoid and vanilloid receptors. The resulting G-protein signaling initiates a number of biological pathways. The name given to it by its discoverers [1] is derived from the Sanskrit word ananda meaning bliss or joy in reference to their finding that this molecule competes with exogenous cannabinoids for their specific receptors in the brain, the first endogenous molecule found to do so. Subsequent research reveals that anandamide activates both CB1 in the central nervous system, CB2 in the peripheral tissues, including immune cells, and TRPV1, a non-selective cation channel ubiquitously expressed in all tissues. The resulting signaling involves a diverse set of biological results including sleep and eating patterns, short-term memory, mood, as well as modulation of the sensation of heat, acid, and proinflammatory stimulants. There are a number of excellent reviews on these subjects [2,3,4,5].
As is typical for signaling molecules, the in vivo concentration of AEA is maintained through the relative rates of synthesis and degradation, but also through a less common storage system, designed to meet high demand on short notice. This manuscript discusses the key enzymes in AEA homeostasis, in terms of structure, reaction specificity, enzymatic activity, regulation, and tissue and cellular expression patterns with a focus on the human isoforms involved.
Anandamide biosynthesis
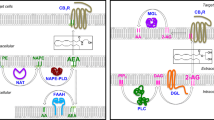
AEA is synthesized through ligation of a previously membrane bound arachidonic acid and a membrane bound phosphatidylethanolamine forming an N-arachadonoyl phosphatidylethanolamine (NArPE). Release of AEA from the composite molecule is accomplished by a series of phospholipases (Fig. 1).

Reactions involved in the formation of Anandamide Metabolites: AEA anandamide, glycero-p-AEA 1-acyl-sn-glycero-3-(N-arachidonyl) phosphoethanolamine, and NArPE N-acyl phosphatidylethanolamine, Enzymes: ABHD4 (lyso)-N-Acylphosphatidylethanolamine lipase, GDE1 Glycerophosphodiester phosphodiesterase 1, GDPD1,3 Lysophospholipase D isoforms 1 and 3, NAPE-PLD N-Acyl-Phosphatidylethanolamine-hydrolyzing Phospholipase D, PLAAT1,2,3,4,5 phospholipase A-Acyltransferases isoforms 1, 2, 3, 4, and 5, PLC phospholipase C, PTPN22 tyrosine-protein phosphatase non-receptor type 22, SHIP1 phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1, and sPLA2 secreted Phospholipase A2
Synthesis of N-acyl phosphatidylethanolamines (NAPEs)
Overview
The synthesis of NAPEs, including NArPE, is accomplished through the transfer of a fatty acyl group from a glycerophospholipid, phosphatidylcholine (PC) in particular, to the primary amine of a phosphatidylethanolamine (PE) by one of a series of N-acyltransferases that are either calcium-independent or calcium-dependent.
The calcium-independent group of N-acyltransferases are represented by the H-RAS-like suppressor (HRASLS) subfamilies of proteins and are members of the H-rev107 class of proteins. These proteins (PLA/AT subgroup) were originally classified as H-RAS-like tumor suppressors and later found to catalyze phospholipid hydrolysis and exhibit N-acyltransferase activity. All but PLAAT5 contains a putative transmembrane helix but is known to be membrane associated, nonetheless. Each of the enzymes catalyzes the hydrolysis of fatty acids from either the sn-1 or sn-2 position of glycerophospholipids, the sn-2 position being particularly important for anandamide synthesis, as arachidonic acid is typically found esterified to the sn-2 position in glycerophospholipids. Once the fatty acid is removed it is then a substrate for one of two secondary reactions, O-acyl transfer of the fatty acid to the free hydroxyl of a lysophospholipid or an N-acyl transfer to the primary amine on a phosphatidylethanolamine. These enzymes share a similar catalytic mechanism involving a thioester intermediate formed between an active site Cys and the fatty acid prior to transfer [6].
Phospholipase A and acyltransferase 1
There are two known splice isoforms of human phospholipase A/acyltransferase 1 (hPLAAT1, PLA/AT-1, UniprotKB-Q9HDD0) that are products of alternative splicing, Isoform 1 (Q9HDD0-1, PLAAT-1S, HRASLS, A-C1) and isoform 2 (Q9HDD0-2, PLAAT-1L) that contains an additional 105 residues—corresponding to exon 1—at the N-terminal [7]. Physical properties for all isoforms are given in the supplement Table S1. No coding single nucleotide polymorphism (SNP) variants are reported [8, 9] (https://genecards.org; https://www.ncbi.nlm.nih.gov/clinvar). Phobius web server predictions [10] (http://phobius.sbc.su.se/) suggest that both isoform 1 and isoform 2 are single-pass transmembrane proteins with large cytoplasmic N-terminal extracellular domains and short 8-residue lumenal C-terminal domains. There is some confusion in the literature regarding the expression of the two isoforms. Early work [11] suggests that Isoform 1 is the predominant species produced and is expressed in testes, muscle, brain, and heart. Later work suggests that only the longer Isoform 2 is the endogenous isoform in human tissue and is expressed in these same tissues, whereas both isoforms are known to coexist in murine tissues [7, 12]. The highest expression is found in the testis and skeletal muscle and it is ubiquitously expressed in most other tissues albeit at very low levels [13]. One difference between the two isoforms is the cellular distribution, where Isoform 1 is found in the cytosol, whereas Isoform 2 is found in the both the cytosol and the nucleus. The latter has been shown to be primarily associated with focal adhesion sites and in the cytosol to a lesser extent [13].
No X-ray structures have been reported for either isoform. There are, however, two potential structural model templates for both isoform 1 and 2 posted on the SWISS-MODEL website [14] (https://swissmodel.expasy.org/), template 4q95.1.A and template 2kyt.1.A. Predicted posttranslational modifications (PTMs) are given in the supplement Table S1 [15,16,17] and no confirmed sites have been reported.
Originally designated as a class II tumor suppressor protein, hPLAAT1 was later found to be involved in calcium-independent phospholipid metabolism with both lipase and acyltransferase activity [13]. The enzymatic activity of both isoforms is essentially the same [7]; however, most of the reported data refer to Isoform 1 (Table 1) [7, 11, 18]. There are three enzymatic activities to consider for this enzyme: 1) hydrolysis at the sn-1 position, 2) hydrolysis at the sn-2 position, and 3) the N-acyltransferase activity. Hydrolysis at the sn-1 position occurs at 7–9 times the rate for the sn-2 position and the hydrolysis rate of the arachidonic acid ester is slightly slower than observed for the palmitic acid ester (15–35% slower) [11]. The N-acyltransferase rate is 1.6–2.5 times faster than the overall phospholipase activity. Sequence comparison to other family members indicate that C119 is likely the active site nucleophile.
Regulation of the hPLAAT1 phospholipase or acyltransferase activity by expression or by posttranslational modification has not been reported. However, it has been shown that this protein is significantly downregulated in tumor cells by unknown processes [19].
Phospholipase A and acyltransferase 2
Human phospholipase A and acyltransferase 2(hPLAAT2, PLA/AT-2, HRASLS2, UniprotKB-Q9NWW9) presents as a single isoform with a sequence homology to PLAAT1 isoform 1 of 47.6%. Physical properties are given in the supplement Table S1. No coding SNP variants are reported [8, 9]. As expected, structure predictions [10] suggest that it is a single-pass membrane protein with a large cytoplasmic N-terminal domain (residues 1-129) and a small lumenal C-terminal domain (residues 156-162). One partial X-ray structure is reported (PDB entry DPZ). hPLAAT2 is expressed primarily in the GI tract and blood but is also found ubiquitously expressed in most other tissues [13]. The cellular distribution is localized to mitochondria. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Also designated as a tumor suppressor protein, hPLAAT2 exhibits a calcium-independent N-acyltransferase reaction that is about 25% of the combined sn-1 and sn-2 phospholipase (PLA1/2) reaction rate in whole cells, whereas in cell homogenates the opposite is true. Hydrolysis rates for the sn-1 position compared to the sn-2 position vary from 0.7 to 1.9 (Table 1) [18, 20, 21]. Structural analysis reveals that C113 is likely the active site nucleophile [20]. Regulation of phospholipase or acyltransferase activity by expression or by posttranslational modification has not been reported.
Phospholipase A and acyltransferase 3
Human phospholipase A and acyltransferase 3 (hPLAAT3, PLA/AT-3, HRASLS3, AdPLA, PLA2G16, H-Rev107, UniprotKB-P53816) presents as a single isoform with a sequence homology to PLAAT1 isoform 1 of 46.8%. Physical properties are given in the supplement Table S1. No coding SNP variants are reported [8, 9]. Structure predictions [10] suggest that it is a single-pass membrane protein with a larger cytoplasmic N-terminal domain (residues 1-132) and a small lumenal C-terminal domain (residues (155-162). Several partial X-ray structures are reported (e.g., PDB entry 4DOT). PLAAT3 is expressed primarily in the brain, adipose tissue, and blood, but is also found ubiquitously expressed in most other tissues [13]. Its cellular distribution appears to be primarily localized to the mitochondria [13] but has also been reported as cytosolic [22] and possibly plasma membrane and peroxisome membrane associated by comparison to the rat and mouse counterparts [21]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Also designated as a tumor suppressor protein, hPLAAT3 exhibits a calcium-independent activity. PLA1/2 reaction rates (Table 1) are faster than the N-acylation reactions by 10–200-fold [18, 20, 21, 23]. There is a preference for the sn-1 site that varies from 1.2 to 14 fold (Table 1) and it exhibits tenfold faster reactions with PC substrates as compared to PE substrates [23]. Mutagenesis studies reveal that C113, H23, and H35 are necessary for activity [24]. Regulation of phospholipase or acyltransferase activity by expression or by posttranslational modification have not been reported. However, although PLAAT3 is active in the absence of intracellular calcium [Ca2+]i, Vmax for lipase activity increases up to two fold in the presence of calcium, suggesting that this enzyme is a special kind of calcium-dependent lipase [25]. Downregulation of the PLAAT3 gene through promotor methylation in cancer cells is well documented [6]. Regulation of phospholipase or acyltransferase activity by expression other than in cancer or by posttranslational modification has not been reported.
Phospholipase A and acyltransferase 4
Human phospholipase A and acyltransferase 4 (hPLAAT4, PLA/AT-4, HRSL4, RARRES3, HRALSL4, TIG3, UniprotKB-Q9UL19) presents as two isoforms with a sequence homology to PLAAT1 isoform 1 of 56.6% for Isoform 1 (Q9UL19-1) and 51.2% for isoform 2 (Q9UL19-2). Physical properties for all isoforms are given in the supplement Table S1. Two coding SNP variants are reported for the canonical isoform, V69L and A162V [8] but no biological ramifications are noted. Phobius web server predictions [10] indicate that Isoform 1 is a single-pass transmembrane protein with a large cytoplasmic N-terminal domain (residues 1-136) and a short 8-residue lumenal C-terminal domain, and Isoform 2 shows no transmembrane domains. Two NMR structures are reported for residues 1-125 of Isoform 1 (PDB entry 2LKT and 2MY9). The SWISS-MODEL website [14] selects the same model protein for both isoforms (2LKT), suggesting similar structures. PLAAT4 is highly expressed in the kidney, thyroid gland, GI tract, and blood, but is also found ubiquitously expressed in most other tissues, including the brain [13]. Its cellular distribution appears to be primarily localized to intracellular membranes [13]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally. PLA1/2 reaction rates for purified protein are 50-fold faster than that for the N-acylation reactions with a threefold preference for the sn-1 site [18, 20, 23] (Table 1) and exhibits twofold faster reactions with PC substrates as compared to PE substrates [23]. Regulation of phospholipase or acyltransferase activity by expression or by posttranslational modification has not been reported. However, upregulation of hPLAAT4 mRNA indirectly in tumor cells by all-trans retinoic acid has been documented [26].
Phospholipase A and acyltransferase 5
Human phospholipase A and acyltransferase 5 (hPLAAT5, PLA/AT-5, HRSL5, HRALSL5, HRLP5, iNAT, UniprotKB-Q96KN8) present as three very similar isoforms with homologies to Isoform 1 of 96.7% and 96.4% for Isoform 2 and Isoform 3, respectively, with only 22.3% homology to PLAAT1 Isoform 1. Physical properties for all isoforms are given in the supplement Table S1. Four coding SNP variants are reported for isoform 1, S31G, A93P, Q214R, and A258V [8] but the clinical significance of these coding SNPs is unknown. Phobius web server predictions [10] indicate that no transmembrane domains exist. No X-ray structures are available, however, there are two potential structural model templates on the SWISS-MODEL website [14], template 2kyt.1.A and template 4q95.1.B. hPLAAT5 is highly expressed in the testis, granulocytes, pancreas, and expressed at lower amounts throughout the brain, and appears to be localized to the plasma membrane [13]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
PLA1/2 reaction rates for the purified protein are about 25% of that for the N-acylation reactions [22, 27] (Table 1). Comparison of positional preferences for deacylation have not been reported. Regulation of phospholipase of acyltransferase reactions by expression or by posttranslational modification has not been reported.
Cytosolic phospholipase A2 epsilon
Human cytosolic phospholipase A2 epsilon (hPLA2G4E, Phospholipase A2 group IVE, UniprotKB-Q3MJ16) is a phospholipase with a calcium-requiring N-acyl transferase activity [28, 29]. As reported by Hussain et al. [28] hPLA2G4E is expressed in two isoforms. Isoform 2 is identical to Isoform 1 apart from the first 376 residues that are missing in this isoform, including a membrane associating C2 domain. Physical properties for all isoforms are given in the supplement Table S1. There are four coding SNP variants reported for isoform 1: R81Q, H213Y, K292N, N400S, M573V and A693T [8, 9]. All coding SNPs are reported to be benign. Phobius web server [10] analysis indicates a 70–80% likelihood for a single transmembrane sequence in each of the isoforms. No X-ray structures are available; however, there is one potential structural model template suggested on the SWISS-MODEL website [14] is template 5iz5.1.A. hPLA2G4E is expressed primarily in the skin and although found in other mammal brain tissue it has only been identified in the human pituitary tissue [13]. Sequence comparison to the murine enzyme indicates that S412 is the likely active site nucleophile. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Catalytic activity for both purified recombinant and recombinant cell lysates have been reported in terms of overall production of N-acyl fatty acid products [28, 29]. Catalytic activity is enhanced in the presence of phosphatidylserine (PS) by 25-fold in the presence of saturating amounts of calcium ion and the EC50 for calcium is increased greater than eightfold in the presence of PS [28]. Other anionic phospholipids enhance the activity in the following order PS > phosphatidylinositol > phosphatidylglycerol > phosphatidic acid > phosphatidylcholine > phosphatidylinositol-4,5-bisphosphate [29]. Regulation by expression or by posttranslational modification has not been reported; however, phosphorylation of S800 is predicted by sequence comparison to the murine isoform, suggesting the possibility of regulation by phosphorylation.
The reaction has been characterized in the greatest detail for the recombinant murine isoform (UniProtK-Q50L42) with a 77.4% sequence homology to the human isoform. The brain protein expressed in HEK293 cells shows a strong preference (3.3:1 in mouse brain lysate) for transferring the sn-1 O-acyl chain to PE [30] and exhibits little PC-lipase activity in the absence of PE. The enzyme is strongly activated with increasing Ca2+, over 200-fold in cell lysates with recombinant protein and fourfold in murine brain tissue with maximal activity at 10 mM Ca2+. Catalytic activity is nearly fourfold higher in membrane fractions than cytosolic fractions obtained from transfected HEK293 cells suggesting that it is membrane associated. The N-terminal region of the murine protein does contain a C2 domain which upon calcium binding is known to target the protein to phospholipid membranes [31, 32]. However, unusual for a PLA2-class enzyme, the murine protein appears to utilize an alternative method for membrane targeting where a region of the C-terminus containing a number of positively charges amino acid residues bind to the phosphate of several membrane phosphoinositides [33]. Characterization of a S420A mutant shows that S420 is the active site nucleophile. Phobius web server [10] analysis shows a 70% likelihood for a single transmembrane helix near the C-terminal end. The fact that the murine enzyme shows a 77.4% sequence homology with the human form strongly suggests similar structure and enzymatic activity.
Converting N-acylphosphatidylethanolamines (NAPEs) to N-acylethanolamines (NAEs)
Overview
Converting NAPEs to N-acylethanolamides (NAEs) such as AEA is accomplished through several pathways (Fig. 1). The dominant pathway (75%) [34] is catalyzed by a phospholipase D (NAPE-PLD) where a NAPE is converted directly into the corresponding NAE and a phosphatidic acid.
N-Acyl-phosphatidylethanolamine-hydrolyzing phospholipase D
Human N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D (hNAPE-PLD, UniprotKB-Q6IQ20) presents as a single isoform and is a member of the metallo-β-lactamase family. Physical properties are given in the supplement Table S1. Two coding SNP variants are reported: S152A and D389N [8]. Clinical ramifications for these SNPs are not reported. hNAPE-PLD is a cytosolic, zinc-requiring protein that is ubiquitously expressed in most tissues with particularly high expression in the brain, GI tract, kidney, and bladder [13]. One X-ray structure is available (PDB entry 4QN9) [22]. Acetylation on the N-terminal methionine has been reported [35]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Enzymatic activity of recombinant proteins for mouse, rat and human proteins have been reported for a variety of NAPE substrates (Table 2) [36, 37]. The mouse and rat proteins have specific activities that are an order of magnitude higher than the human enzyme against N-palmitoyl-phosphatidylethanolamine substrates. The active enzyme acts as a homodimer and requires the presence of a hydrophobic cofactor, a bile salt or n-octyl-glucoside [22, 37, 38]. Different bile acids enhance the reaction rate to different degrees depending on the location of hydroxyl groups on the steroid structure [38], suggesting that the enzyme activity may be modulated in vivo by the presence of specific bile acids. The enzyme also hydrolyzes N-arachidonoyl-phosphatidylethanolamine (NArPE) substrate 7 times faster than N-palmitoyl- phosphatidylethanolamine substrate in the presence of cholic acid (Table 2). Recently it was reported that a murine NAPE-PLD hepatocyte deficient strain develops a high-fat diet-like phenotype and that is much more sensitive to liver inflammation, likely due to the associated reduction in almost all endocannabinoid mediators [39]. The authors hypothesize that NAPE-PLD acts as a “hub” that controls a large array of liver lipids.
Regulation via phosphorylation has not been reported; however, there is one report indicating a 4–14 fold increase in hNAPE-PLD at the protein level in lymphocytes taken from malignant endometrial carcinoma (EC) patients in an advanced disease state when compared to peripheral lymphocytes and without significant change in the transcription level [46]. This correlates well with the fact that N-acylethanolamine concentrations are higher in post-menopausal women with EC compared when with controls. A specific mechanism has not been proposed.
(lyso)-N-acylphosphatidylethanolamine lipase (ABHD4)
Human (lyso)-N-Acyl-phosphatidylethanolamine lipase (hABHD4, alpha/beta- hydrolase 4, UniprotKB-Q8TB40) is expressed as two splice isoforms. Physical properties for all isoforms are given in the supplement Table S1. No coding SNP variants are reported [8, 9]. Only Isoform 1 (hABHD4) will be discussed here. hABHD4 is a member of the peptidase S33 family, ABHD4/ABHD5 subfamily. No X-ray structures or posttranslational modifications have been reported. There are two potential structural model templates suggested on the SWISS-MODEL website [14], template 3nwo.1.A and template 5mxp.1.B. hABHD4 is produced ubiquitously in all tissue types studied, including brain, and is localized to the nucleoplasm with cytoplasmic expression in some tissues [13]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally. However, the rat isoform (UniprotKB-Q6QA69) with 94.5% sequence homology is known to be phosphorylated at S124 which corresponds to S122 on the human isoform [47].
The enzymatic activity of hABHD4 has not been reported on to date. However, there are several studies available for the mouse counterpart which shares a 96.5% sequence identity to the human protein suggesting a similar value for the human isoform (Table 2) [34]. ABHD4 catalyzes the removal of fatty acids from NAPE and the product 1-acyl-sn-glycero-3-(N-acyl) phosphoethanolamine (lyso-NAPE) and does so using an active site serine as the nucleophile that reacts readily with methoxy arachidonyl fluorophosphonate (MAFP) inhibitor. Only the latter reaction has been examined directly and only as the combined activity of ABHD4 and glycerophosphodiester phosphodiesterases (GDEs) [40]. Comparison of data from NAPE-PLD−/− mice in the presence and absence of inhibitors of either the ABHD4/GDE arm or the PLC/phosphatase arm of the alternative pathways provides insight regarding the regulation of these two pathways [34]. By comparing results from 1 min incubation to 1 hr it is clear that the PLC/phosphatase pathway is responsible for a rapid production of AEA when the NAPE-PLD is blocked and that the ABHD2/GDE pathway is upregulated in response to low AEA. Comparison of data for the conversion of NAPE and lyso-NAPE to AEA [34, 40] also indicates that the ABHD2/GDE pathway exhibits a higher specific activity than the PLC/phosphatase pathway and is thus the dominant contributor to AEA production once upregulated.
Phospholipase C (PLC) and secreted phospholipase A2 (sPLA2)
Phospholipase C (PLC) represents a class of phospholipases that cleave the phospho-head group from phospholipids, the most studied of which involve the production of inositol 1,4,5-trisphosphate from a 1,2-diacyl-glycero-3-phospho-(1D-myo-inositol-4,5-bisphosphate). The production of phospho-AEA (pAEA) from NAPE has been observed in mouse brain homogenates, clearly involving a member of the PLC class [34, 45]. The identity of the particular isoform has yet to be reported. Secreted rat phospholipase A2 group IB (rat sPLA2-IB), secreted human calcium-dependent phospholipase A2 Group V (hPLA2G5) and human phospholipase A2 Group IIA (hPLA2G2A) all are known to produce lyso-NAPE from NAPE [48]. Specific activities for the human isoforms are given in Table 1.
Human phospholipase A2 Group IIA (hPLA2G2A, phospholipase A2, UniProtKB-P39877) is presented as a single isoform and is a secreted, calcium-dependent lipase. Physical properties are given in the supplement Table S1. There are 14 coding SNP variants reported [9]. Five of these are associated with familial fleck retina (G45C, G49S, Q128 frame shift, and two termination mutants W62 and R53) and the remaining with unknown significance (P6S, S14T, R73C, C103R, Y116C, R123Q, Q130C, Y131C and N134K). Its expression is limited but is found in the highest amounts in the liver followed by all muscle tissue, GI tract and female tissues [13]. There are numerous X-ray structures available (e.g., PBL entry 1DB4). Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Human phospholipase A2 Group V (hPLA2G5) is a secreted, calcium-dependent lipase. It is translated as 138 residue polypeptide that is processed to 118 residues following removal of the signal peptide. Physical properties are given in the supplement Table S1. There are 16 coding SNP variants reported [9]. Four of these are associated with familial fleck retina (G45C, Q128 frame shift, and two termination mutants W62 and R53), one is associated with late onset retinal degeneration (V99 frame shift), and the remaining with unknown significance (P6S, S14T, G45S, G49S, R73L, E76 deletion, C103R, Y116C, R123Q, Q130C, Y131C, and N134K). hPLA2G5 is expressed in most tissues but is highly expressed in the retina as well as heart and smooth muscle [13]. X-ray structures are not available, but a 3D model has been published (PDB entry 2GHN). Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Human phospholipase A2 Group IB (hPLA2G1B, PLA2, PLA2A, PPLA2, UniProtKB-P04054) is a secreted, calcium-dependent lipase. Four SNP variants are reported (R122H, D16A, N89T, and N89K) none of which has any known clinical significance [8, 9]. hPLA2G1B is expressed exclusively in the pancreas [13]. Two Xray (PDB entry 3ELO and 6Q42) and one model structure (PBD entry 1YSK) are available. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Specific activities have yet to be reported for purified hPLA2G2A, hPLA2G5 or hPLA2G1B. However, reaction rates of recombinant forms expressed in HEK293 cells have been reported as 0.37 nmol/min/106 cells for hPLA2G2A and 5.5 nmol/min/106 cells for PLA2G5 [48]. Regulation of hPLA2G5 expression is reduced in mast cells when treated with vitamin D (1,25-dihydroxyvitamin D3) a known modulator of the inflammatory response, whereas other PLA2 isoforms are upregulated [49]. Regulation involving anandamide metabolism by expression or by posttranslational modification for any of these isoforms has not been reported.
Lysophospholipase D GDPD1
Lysophospholipase D GDPD1 is known to catalyze two reactions leading to the formation of AEA and other NAEs: (1) hydrolysis of lyso-NAPE and (2) hydrolysis of glycerophospho-N-acylethanolamine, each producing the same amide product [42]. Human lysophospholipase D GDPD1 (hGDPD1, GDE4, glycerophosphodiester phosphodiesterase 4, UniprotKB-Q8N9F7) is expressed as three splice isoforms. Physical properties for all isoforms are given in the supplement Table S1. Only the canonical sequence, Isoform 1, will be discussed here. No coding SNP variants are reported [8, 9]. hGDPD1 is a metallo-membrane protein containing two transmembrane helices with a preference for Mg2+ or Mn2+ [42, 50] and is inhibited by both Ca2+ and EDTA [44]. hGDPD1 is widely expressed in many tissues with the highest expression in the brain followed by a lower expression in endocrine tissue, the GI tract, and male and female tissues [13]. X-ray structures are not available, but a 3D model has been published [50]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Specific activities have been reported for both overexpressed mouse and human recombinant GDPD1 obtained from cell lysates (Table 2) [42]. These two enzymes share a 91.9% sequence homology. Both enzymes show a preference for N-acyl-phosphatidylethanolamine substrates over phosphatidylcholine substrates. However, for mGDPD1 the opposite is true for non-N-acylated substates [41]. Specific activities for both enzymes for the same substrates follow the following order: glycerophospho-N-palmitoylethanolamine (GP-NPE) > N-palmitoyl-lysophosphatidylethanolamine (N-Pal-lyso-PE) ≈ N-oleoyl-lysophosphatidylethanolamine (N-Ole-lyso-PE) > N-arachidonoyl-lysophosphatidylethanolamine (N-Ara-lyso-PE), all of which are orders of magnitude slower than the corresponding NAPE-PLD pathway competitor. Purified recombinant mGDPD1 shows a 5000-fold increase in specific activity for GP-NPE over that obtained from cell lysates with similar specific activities for lyso-PC, lyso-PE, and N-Pal-lyso-PE [44].
Although a specific catalytic mechanism has not been proposed, modeling reveals that D72, E74, and H87 are likely to be required for activity [50]. Further, a proteinase K protection assay reveals that the catalytic domain faces the lumen/extracellular space. Regulation involving anandamide metabolism by expression or by posttranslational modification has not been reported.
Lysophospholipase D GDPD3
Lysophospholipase D GDPD3 is known to convert lyso-NAPEs to AEA or other NAEs in competition with GDPD1 [41, 42]. Human lysophospholipase D GDPD3 (hGDPD3, GDE7, glycerophosphodiester phosphodiesterase 7, UniprotKB-Q7L5L3) is expressed as two splice isoforms. Isoform 2 is identical to Isoform 1 with the exception of missing residues 1-63. Physical properties for all isoforms are given in the supplement Table S1. Only the canonical sequence, Isoform 1, will be discussed here. No coding SNP variants are reported [8, 9]. hGDPD3 is a metallo-membrane protein containing two transmembrane helices with a preference for Ca2+ and is partially or completely inhibited by other divalent cations [41, 42]. No X-ray structures are available. There is, however, one potential structural model template suggested on the SWISS-MODEL website [14], template 5vug.1.B. hGDPD3 is widely expressed with the highest expression in tonsils and appendix, tongue, and esophagus, as well as being expressed in all areas of the brain [13]. Intracellular distribution is found on membrane structures throughout the cytosol and nucleoplasm. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Specific activities have been reported for both mouse and human recombinant GDPD3 [42] which share a 79.1% sequence homology. In contrast to GDPD1, both GDPD3 enzymes show a preference for phosphatidylcholine substrates over N-acyl-phosphatidylethanolamine substrates. The same is true for mGDPD1 hydrolyzing non-N-acylated substates [41]. Specific activities for both enzymes for the same substrates show the following order: N-Pal-lyso-PE ≈ N-Ole-lyso-PE > > N-Ara-lyso-PE > GP-NPE, an order different than observed for GDPD1, and the reactions are orders of magnitude slower than the corresponding NAPE-PLD pathway competitor. Regulation of activity involving anandamide metabolism by expression or by posttranslational modification has not been reported.
Glycerophosphodiester phosphodiesterase 1
Glycerophosphodiester phosphodiesterase 1 (GDE1) is known to convert GP-NAEs to NAEs as well as exhibiting glycerophosphoinositol phosphodiesterase activity [32] and is a member of the glycerophosphoryldiester phosphodiesterase family (GDE phosphodiesterase family). Human glycerophosphodiester phosphodiesterase 1 (hGDE1, MIR16, RGS16-interacting membrane protein, UniprotKB-Q9NZC3) is expressed as a single isoform. Physical properties are given in the supplement Table S1. There are two reported coding SNP variants, R208Q and D328K [9]. Neither variant has a known clinical significance. hGDE1 is a metallo-membrane protein containing two transmembrane helices with a preference for Mg2+ and is inhibited by both Ca2+ and EDTA [44]. hGDE1 is widely expressed with the highest expression in the brain, endocrine tissues, GI tract, and muscle tissue. Intracellular distribution is found on membrane in the nucleoli, nucleoplasm, and vesicles throughout the cytosol. No X-ray structures are available for hGDE1, but 3D model has been proposed [51]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally. However, endo H treatment of rat GDE1 (87.9% sequence homology to the human isoform) confirms that 5–6 kDa of N-glycosylation is present, but specific sites of attachment are unknown [52].
Specific activities have not been reported for human GDE1 but have been reported for both rat and mouse [43, 44]. These enzymes have an 87.9% and 87.3% sequence homology to the human protein, respectively, and thus these specific activities can serve as an estimate for human values. Recombinant rat GDE1 shows a pH-dependent substrate specificity where lyso-phosphatidylcholine (lyso-PC) is the preferred substrate at pH 7.5 and lyso-phosphatidylinositol (lyso-PI) is the preferred substrate at pH 8.5 [43]. Specific activities for both enzymes for the same substrates follow the following order: GP-NPE > lyso-PE > N-Pal-lyso-PE > lyso-PC.
Although a specific catalytic mechanism for hGDE1 has not been proposed, site specific mutagenesis reveals that E97, D99, and H112 are required for activity [43]. Further detail has been gleaned from a 3D model study [52]. A crude mechanism has been proposed for rat GDE1 where H356 and E341 act as a general acid/base catalyst, generating a cyclic phosphate intermediate that is then hydrolyzed to form the respective products [43]. The catalytic domain has been shown to face the lumenal/extracellular space [43].
Murine GDE1 (mGDE1) was originally identified as a RGS16-interacting membrane protein, where RGSs are known GTPase activating proteins. This fact led to the discovery that the catalytic activity of mGDE1 is regulated by G-protein signaling. When HEK293T cells are transfected with mGDE1 they respond to isoproterenol, a known Gαs coupled β-androgenic receptor agonist, with an ≈ 50% increase in GDE1 activity. When treated with phenylephrine, a known Gαi/q coupled α-androgenic receptor agonist, however, GDE1 activity decreases by ≈ 30% [43]. These results indicate that GDE1 activity is both positively and negatively regulated by G-protein signaling. PRAF2 has also been shown to interact with RGS16, which in turn is known to interact with CCR5, a G-protein coupled chemokine receptor [51]. It has been proposed that a GDE1, RGS16, PRAF2, CCR5 complex may be involved in cell chemotaxis or perhaps desensitization, internalization, and recycling of CCR5 [52]. Immunoprecipitation studies and size exclusion chromatography reveals that both Gαq/11 and Gβ proteins associate with rat GDE1 [53, 54]. In addition, Gαq/11 knockdown Neuro2A cells exhibit lysoPLD activity that is reduced by about 50% compared to wildtype and the endogenous activity is partially rescued by overexpression of Gαq in these cells. Taken together all of these results strongly suggest G-protein modulation of GDE1 activity. Regulation of activity involving anandamide metabolism by expression or by posttranslational modification has not been reported.
Tyrosine-protein phosphatase non-receptor type 22 (PTPN22)
As the name suggests, tyrosine-protein phosphatase non-receptor type 22 is known primarily for its ability to remove phosphates from tyrosines [55]. However, several reports clearly show that PTPN22 also catalyzes the dephosphorylation of pAEA to AEA [34, 45]. Human PTPN22 (hPTPN22, PEP, LyP, 70Z-PEP, PTPN8, UniprotKB-Q9Y2R2) is expressed as six splice isoforms. Sequence alignment reveals that Isoforms 5 and 6 lack both the known active site nucleophile [56] and key sections of the active site structure of the canonical sequence and can thus be discounted as biologically functional in terms of phosphatase activity. Isoform 3 lacks a key portion of the substrate binding site and thus is also likely not to be biologically competent for phosphatase activity. It is unclear from the literature which or if any combination of the three remaining isoforms is responsible for the phosphatase activity for substrate pAEA. Phobius web server [10] analysis indicates a 50% likelihood for a single transmembrane sequence in each of the three phosphatase competent isoforms noted above. However, hPTPN22 is known to be membrane associated rather than an integral membrane protein. The proposed transmembrane helix may instead represent a helical structure that is partially imbedded in the membrane. Physical properties for all isoforms are given in the supplement Table S1. There are 26 reported coding SNP variants of which only one has known clinical significance [8, 9]. The variant R620W is implicated in susceptibility to insulin dependence, diabetes, rheumatoid arthritis, and other disorders. hPTPN22 is widely expressed with the highest expression in bone marrow, lymphoid tissues and blood as well as being expressed in all areas of the brain. Intracellular distribution is found in the nucleoplasm, vesicles, and on the plasma membrane [13]. Numerous Xray structures are available for the first 300 or so residues which contains the N-terminal through the active site regions for all three of the isoforms noted above (e.g., PDB entry 2P6X). One report suggests the presence of second catalytic site could account for the diversity of substrate, lipid vs. peptide [57]. Predicted PTMs are given in the supplement Table S1, of which only phosphorylation at S751 [58] and S35 [59, 60] on isoform 1 have been confirmed experimentally. Comparison of the human to murine PTPN22 (UniProtKB-P29352; 70.7% sequence homology) sequences suggest potential phosphorylation at S449, S635, S684, and S692 as well.
Specific activities have only been reported for mPTPN22 but since there is a 70.7% sequence homology between the human and murine protein, the reported value should serve as a rough estimate of the human enzyme activity (Table 2). Only two studies have reported specific activities related to this enzyme. One study examined mouse brain homogenates from NAPE-PLD−/− mice in the presence of an inhibitor for ABHD4 and EDTA to knock out any GDE or GDPD activity [34]. The resulting specific activity (Table 2) can thus be attributed to a combination of a yet-to-be identified PLC, PTPN22, and SHIP1 (see below) with values in the low pmol/mg/min range (discussed below). A second study examined mouse macrophages (RAW264.7) in culture, comparing the conversion of pAEA to AEA by wildtype to PTPN22−/− knockout strains. The difference in specific activity between the two represents the combined activity of PTPN22 and SHIP1 (see below) [45].
Regulation of this protein is multifold. One of the first descriptions of the involvement of PTPN22 in anandamide metabolism was obtained from a study in which mouse macrophages were treated with the inflammation initiator lipopolysaccharide (LPS) [45]. Here it was found that LPS upregulates anandamide synthesis and PTPN22 expression while at the same time downregulates NAPE-PLD expression. Further, treatment of these cells with phosphatase inhibitors or the specific PLC inhibitor neomycin reduces but does not eliminate LPS-induced pAEA dephosphorylation. These results indicate that the NAPE-PLD pathway is likely the constitutive pathway to AEA production, whereas the PLC pathway is invoked during inflammation. A second regulatory pathway for PTPN22 involves the active site C227 and two other Cys residues where C129 protects C227 from irreversible oxidation in an oxidizing environment through the formation of a disulfide bond and concomitant reversible loss of activity [56]. On the other hand, formation of a disulfide with the local C231 also results in inactivation of the enzyme but suppresses reactivation in a reducing environment, providing negative regulation. A third regulatory pathway involves phosphorylation of specific Ser residues. Phosphorylation is known to prolong the half-life of hPTPN22 by inhibiting K48-linked ubiquitination and also inhibits its recruitment to the plasma membrane, a requirement for its inhibition of T cell receptor signaling [58]. Phosphorylation of hPTPN22 at S35 by protein kinase C has been shown to occur in vivo and in vitro and inhibits the ability to downregulate T cell receptor signaling [48]. Further, phosphorylation impairs hPTPN22’s ability to inactivate Src family kinases. Similar studies on AEA production have not been reported.
Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1 (SHIP1)
As the name suggests, the primary function of Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1 (SHIP1) is the removal of the 5′-phosphate from phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3). However, its involvement in AEA metabolism has been observed where SHIP1 competes with PTPN22 for the hydrolysis of pAEA. Human SHIP1 (hSHIP1, INPP5D, SIP-45p, 150Ship, hp51CN, UniprotKB-Q92835) is expressed as three alternative splicing isoforms. Isoform 2 lacks V117 and is otherwise identical to Isoform 1 and thus is very likely to be involved in the AEA pathway. Isoform 3, however, lacks both the SH2 and SH3 binding domains essential for its PtdIns(3,4,5)P3 activity and for this reason is not likely to be a biologically competent isoform for this process. Physical properties for all isoforms are given in the supplement Table S1. Two coding SNP variants for the canonical isoform have been reported: H1169Y and V685D [8]. None of the variants have any known clinical significance. One X-ray structure for residues 397-857 (PDB entry 6IBD) and one nmr structure for residues 1-112 (PDB entry 2YSX) are available. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
The only direct reference to the pAEA activity for SHIP1 comes from a comparison of murine control and mSHIP1−/− knockout macrophages (Table 2) [45]. The difference in specific activities between the knockout and controls is 0.77 nmol/mg/min which is quite comparable to the 1.0 nmol/mg/min determined for PTPN22. Like mPTPN22, mSHIP1 is also upregulated in response to LPS and is tyrosine phosphorylated [61]. Although specific phosphorylation sites stimulating upregulation were not determined, there are three known mSHIP1 tyrosine phosphorylation sites, Y918, Y945, and Y1021 [62], corresponding to Y915, Y944, and Y1022 in hSHIP1. There are also five known mSHIP1 serine/threonine phosphorylation sites, S246, S935, T964, S967, and S972 [63, 64], corresponding to S243, S934, T963, and S971 in hSHIP1. There is no human counterpart for S967. The 87.4% sequence homology suggests a similar likelihood of phosphorylation in human cells. Regulation activity involving anandamide metabolism by expression has not been reported.
Anandamide loss
Anandamide concentrations are reduced through three general pathways: (1) degradation to AA and ethanolamine, (2) conversion to other molecules, and (3) sequestration through transport to intracellular lipid microvesicles. Figure 2 depicts the known reaction pathways, the enzymes, and other proteins known to be involved.

Reactions involved in the degradation of anandamide Metabolites: 5,6-EET-EA N-(2-hydroxyethyl)-5,6-epoxyeicosatrienamide, 11,12-EET-EA N-(2-hydroxyethyl)-11,12-epoxyeicosatrienamide, 12S-HPETE-EA N-(2-hydroxyethyl)-12S-Hydroperoxyicosatetraenamide, 14,15-EET-EA N-(2-hydroxyethyl)-14,15-epoxyeicosatrienamide, 15S-HETE-EA N-(2-hydroxyethyl)-15S-Hydroxyicosatetraenamide, 15S-HPETE-EA N-(2-hydroxyethyl)-15S-Hydroperoxyicosatetraenamide, 20-HETE-EA N-(2-hydroxyethyl)-20-hydroxyeicosatetraenamide, 8,9-EET-EA N-(2-hydroxyethyl)-8,9-epoxyeicosatrienamide, AA arachidonic acid, AEA anandamide, EA ethanolamine, EXA4-EA eoxin A4 ethanolamide, EXC4-EA eoxin C4 ethanolamide, EXD4-EA eoxin D4 ethanolamide, EXE4-EA eoxin E4 ethanolamide, PGD2-EA prostaglandin D2 ethanolamide, PGE2-EA prostaglandin E2 ethanolamide, PGF2a-EA prostaglandin F2a ethanolamide, PGH2-EA prostaglandin H2 ethanolamide, PGI2-EA prostaglandin I2 ethanolamide, TXA2-EA thromboxane A2 ethanolamide, TXB2-EA thromboxane B2 ethanolamide. Enzymes: AKR1C3 aldo–keto reductase family 1 member C3, ALOX 15 arachidonate 15-Lipoxygenase, ALOX12 arachidonate 12-Lipoxygenase, COX-2 cyclooxygenase-2, CYP450s cytochrome P450s (see text for specific isoforms), FAAH-1,2 fatty-acid amide hydrolase isoforms 1 and 2, GPX glutathione peroxidase, NAAA N-acylethanolamine-hydrolyzing acid amidase, PRXL2B prostamide/prostaglandin F synthase, PTGDS prostaglandin D2 synthase, PTGES1,2,3 prostaglandin E2 synthase isoforms 1, 2, and 3, PTGIs prostaglandin I2 synthase, and TBXAS1 thromboxane A synthase 1
Degradation to free AA and ethanolamine
Fatty-acid amide hydrolase 1 (FAAH-1)
The hydrolysis of fatty-acid amides to their corresponding fatty acid and amine is facilitated by fatty-acid amide hydrolase 1 (FAAH-1). Human fatty-acid amide hydrolase 1 (hFAAH-1, FAAH, anandamide amino hydrolase 1, oleamide hydrolase 1, UniprotKB-O00519) is expressed as a single isoform. Physical properties are given in the supplement Table S1. There are 6 reported coding SNP variants: G110E, P129T, R295Q, S356V, A476G, and A345D [8, 9]. The P129T isoform is known to have reduced cellular stability and has been linked to problem drug use [65]. A345D may also have clinical significance, as it was obtained from a breast cancer sample. There are no crystal structures for hFAAH-1; however, the rat counterpart (rFAAH-1) with an 82.7% sequence homology has numerous published structures to serve as a model. The crystal structure of a truncated rFAAH-1 (residue 30-579, PDB entry 1MT5) indicates that it is a dimeric enzyme, each monomer containing two intramembrane hydrophobic helices that are thought to provide a hydrophobic membrane cap (residues 404-433). Phobius web server predictions [10] indicate a single transmembrane helix located at the N-terminus (residue 9-29) and a potential second intramembrane helical structure (residue 404-433) for both hFAAH-1 and rFAAH-1 the former is more likely an intramembrane structure. PSORT analysis [66] predicts that the catalytic domain is located in the cytosolic compartment. Further analysis of rFAAH-1 reveals a unique catalytic triad in the active site consisting of S241-S217-K142 for which S241 is the catalytic nucleophile. Sequence comparison shows that the same residues apply to the human isoform. hFAAH-1 is widely expressed with the highest expression in the brain as well as high expression in male and endocrine tissues [13]. Intracellular distribution includes its presence on membrane structures as well as throughout the cytosol. Treatment with PNGaseF does not alter gel migration, indicating the absence of N-linked glycosylation [66]. Predicted PTMs are given in the supplement Table S1.
Specific activities have been reported for both human and rat isoforms with the latter being ≈10 times larger (Table 3) [66,67,68]. FAAH-1 has a catalytic preference for the particular fatty acid present where anandamide > oleamide > N-oleoyl ethanolamine > N-palmitoyl ethanolamine and also has the ability to hydrolyze N-acyltaurines (NATs) but at lower rates than ethanolamides [66].
Regulation of FAAH-1 occurs at both transcriptional and post-transcriptional levels. Physiological levels of progesterone have been shown to upregulate hFAAH-1 at both the transcriptional and translational levels in T-lymphocytes, up to a 270% increase over controls resulting in a 60% decrease in anandamide concentrations [69]. It was subsequently shown that the increase in hFAAH-1 results from an increase in the transcription factor Ikaros that subsequently binds to the FAAH-1 promotor site. These findings are consistent with a role for FAAH-1 in modulating immunoendocrine interactions in human pregnancy. Membrane bound non-raft cholesterol in AEA/cholesterol rich membranes such as the ER have been shown to stabilize dimeric FAAH-1 and directly enhances the enzymatic activity by increasing the accessibility of the membrane port to AEA [70]. Regulation of FAAH-1 by phosphorylation has not been reported.
Fatty-acid amide hydrolase 2 (FAAH-2)
The hydrolysis of fatty-acid amides to their corresponding fatty acid and amine is also facilitated by fatty-acid amide hydrolase 2, but with different substrate preferences than FAAH-1. Human fatty-acid amide hydrolase 2 (hFAAH-2, AMDD, anandamide amino hydrolase 2, oleamide hydrolase 2, UniprotKB-Q6GMR7) is expressed as a single isoform. Physical properties are given in the supplement Table S1. There are 7 reported coding SNP variants (R42W, R192Q, G342R, A458S, A388S, and A418S) of which none have known clinical significance [8, 9]. There are no crystal structures for hFAAH-2 and only a 32.1% sequence homology to hFAAH-1. However, a threaded model for hFAAH-2 based on rFAAH-1 has been reported and clearly shows the expected Ser-Ser-Lys catalytic triad involving S206, S230, and K131 [71] in which S230 appears to be the active site nucleophile. Interestingly, Phobius web server predictions [10] indicate a single transmembrane helix located near the C-terminus (residue 394-414) rather than at the N-terminal as observed for hFAAH-1 but in the same region where a potential intramembrane helix is predicted for hFAAH-1. In addition, a potential N-terminal intramembrane helix is predicted for hFAAH-2, albeit with low probability, in the same region the transmembrane helix is predicted for hFAAH-1. PSORT analysis predicts that the catalytic domain is located in the lumenal compartment [66]. hFFAH2 is widely expressed throughout most tissues and is particularly abundant in the pancreas and endocrine tissues but is also expressed in the brain with highest amounts in the cerebellum [13]. Intracellular distribution appears to be localized to lipid droplet membranes [72]. Predicted PTMs are given in the supplement Table S1, none of which have been confirmed experimentally.
Specific activity has been reported for hFAAH-2 with a value for AEA hydrolysis that is only 1–2% of that exhibited by FAAH-1, but comparable activity for hydrolysis of oleamide (Table 3). In addition, FAAH-2 has a catalytic preference for the fatty acid present where oleamide >> N-oleoyl ethanolamine > anandamide > N-palmitoyl ethanolamine and is unable to hydrolyze NATs [66]. These data suggest that FAAH-1 is primarily involved in anandamide and NAT hydrolysis and both FAAH-2 and FAAH-1 contribute to the hydrolysis of monosaturated lipid amides [66]. Regulation of FAAH-2 at any level has yet to be reported.
N-acylethanolamine-hydrolyzing acid amidase (NAAA)
The hydrolysis of fatty-acid amides to their corresponding fatty acid and amine is also facilitated by an N-acylethanolamine-hydrolyzing acid amidase which exhibits a preference for small saturated fatty acids. Human N-acylethanolamine-hydrolyzing acid amidase (hNAAA, ASAHL, PLT, acid ceramidase-like protein, UniprotKB-Q02083) is expressed as three isoforms where isoform 2 is missing the last 35 residues present in isoform 1 and isoform 3 is missing the last 159 residues of isoform 1. Physical properties for all isoforms are given in the supplement Table S1. Only Isoform 1 exhibits demonstrated activity [82] and thus will be the only isoform discussed further. There are 4 reported coding SNP variants (K75R, N107K, V151I, and F334L) none of which has a known clinical significance [8, 9]. Isoform 1 is processed further to remove the signal peptide, the first 28 residues and a single cleavage between residues 125 and 126 forming a hetero-dimer consisting of the α-subunit (residue 29-125, 14.6 kDa) and the β-subunit (residue 126-359, 33.3 kDa) [74]. There are two crystal structures for hNAAA, one without the 125–126 cleavage (residue 29-359, PDB entry 6DXW) and one with the 125–126 cleavage (PDB entry 6DXX). hNAAA is a glycoprotein for which N37, N107, N309, N315 and N333 are confirmed sites of glycosylation [73,74,75, 83]. hNAAA is widely expressed in most tissues with highest expression in blood, monocytes in particular, lymphoid tissues, spleen, and lymph nodes, and to a lesser extent in the gastrointestinal tract and brain [13]. Intracellular distribution favors lysosomes [75, 84]. Predicted PTMs are given in the supplement Table S1, but only the N-glycosylations noted above have been confirmed experimentally.
hNAAA is a cysteine amidase where the active site C126 is responsible for not only substrate hydrolysis, but also facilitates auto-cleavage of the 125–126 bond [83] along with residues R142, D145, N287 [84], and L325 and T335[82]. Although NAAA is considered to be a cytosolic protein, it must also penetrate the lipid bilayer in order to access substrate. It is thought that helix a3 on the α-subunit and helix a6 from the β-subunit are attracted to the anionic surface of lysosomal vesicles through their associated cationic residues leading to imbedding of the associated hydrophobic residues [83]. In addition, its activity is stimulated by both DTT and the detergent Nonidet P-40, the latter shown to initiate a conformational change in the structure leading to the formation of the active site.
Specific activity has been reported for hNAAA with a value for AEA hydrolysis that is only 30–50% of that exhibited by FAAH-2 and only 3% of that for PEA hydrolysis (Table 3). hNAAA has a catalytic preference for the smaller fatty-acid amides where PEA > > N-myristylethanolamine > > N-steroylethanolamine ≈ N-laurylethanolamine > AEA [73]. Clearly the primary biological function of this enzyme is the hydrolysis of PEA but can readily compete with FAAH-2 for AEA.
Regulation of NAAA by lipid and thiol compounds has been reported [85]. The presence of phosphatidylcholine (PC), phosphatidylethanolamine, or sphingomyelin increases the hydrolysis rate of PEA by fivefold at 100 µM and increases with phospholipid concentration based on PC results. This compares to the sixfold increase with a similar amount of the nonionic detergent Nonidet P-40. Numerous endogenous and non-endogenous thiol compounds were tested where DTT and the endogenous dihydrolipoic acid have the greatest effect. These compounds presumably help keep the catalysis C126 in the reduced state that is required for activity. Neither additive influences substrate preference.
Regulation of NAAA expression involving anandamide metabolism has not been reported. N-glycosylation is necessary for transport of hNAAA from the Golgi, but not required for catalytic activity [86].
Conversion of anandamide to other compounds
There are numerous pathways involved in the conversion of anandamide to other compounds that display a wide variety of different biological effects. The reactions center on the enzyme catalyzed oxidation of the AA moiety itself. The reactions discussed below examine the first steps in such conversions which involve enzymes that are also associated with metabolism of AA along numerous eicosanoid metabolic pathways (Fig. 2). Greater detail on these enzymes and the pathways are found elsewhere [87, 88].
Prostaglandin G/H synthase 2 (COX-2)
The primary biological role for prostaglandin G/H Synthase 2 (COX-2) and prostaglandin G/H Synthase 1 (COX1) is the conversion of AA to prostaglandin H2 (PGH2) via prostaglandin G2 (PGG2) through the reaction of substrate with molecular oxygen. PGH2 is then rapidly converted enzymatically to prostaglandin D2 (PGD2), prostaglandin E2 (PGE2) and prostaglandin F2a (PGF2a), prostaglandin I2 (PGI2) and thromboxane A2 (TXA2) [87]. PGH2 is quite unstable and is also rapidly converted to PGD2, PGE2, levuglandin E2 (LGE2) and levuglandin D2 (LGD2) non enzymatically in aqueous solution [88, 89]. COX-2 and not COX1 oxygenates anandamide to PGH2 ethanolamide (PGH2-EA) which is then rapidly converted to PGD2 ethanolamide (PGD2-EA), PGE2 ethanolamide (PGE2-EA), PGF2a ethanolamide (PGF2a-EA), PGI2 ethanolamide (PGI2-EA), and TXA2 ethanolamide (TXA2-EA) enzymatically (Fig. 2) [76, 90,91,92,93].
Human cyclooxygenase-2 (COX-2, PTGS2, PGHS-2, cyclooxygenase-2, UniProtKB-P35354) is a heme-requiring enzyme that is expressed as a single isoform. Its physical properties and PTMs have been discussed in detail elsewhere [87, 94,95,96]. During inflammation, COX-2 is significantly upregulated in the affected tissue with the exception of brain, testes, and the macula densa of the kidney where it is constitutively expressed [97]. Intracellular distribution favors the endoplasmic reticulum.
Specific activity for partially purified hCOX-2 towards AEA has been reported and found to be 70% of that observed for the more typical substrate AA (Table 3). Clearly hCOX-2 is a formidable contributor to AEA degradation when expressed in appropriate quantities, for example, during inflammation. Regulation of COX-2 at the transcriptional level has been reported where a variety of cytokines and growth factors have been shown to stimulate induction [98]. Tyrosine phosphorylation at Y120 and Y446 are known to enhance the activity of hCOX-2 towards AA substrate [95, 99]. Nitrosylation of C526 by an inducible nitric oxide synthase, a major mediator of inflammation, results in a twofold enhancement of activity towards AA [96, 100].
Cytochrome P450s
The primary biological role for cytochrome P450 enzymes is the oxidation of steroids, lipids, and various xenobiotics. Cytochrome P450 (CYP) represents a large class of heme-requiring monooxygenases, too vast to discuss in detail here. However, it is worthwhile to examine a few that are known to be involved in anandamide catabolism. There are five major products (Fig. 2) [77]—10 in total—for anandamide metabolism by CYP enzymes: N-(2-hydroxyethyl)-20-hydroxyeicosatetraenamide (20-HETE-EA), N-(2-hydroxyethyl)-5,6-epoxyeicosatrienamide (5,6-EET-EA), N-(2-hydroxyethyl)-8,9-epoxyeicosatrienamide (8,9-EET-EA), N-(2-hydroxyethyl)-11,12-epoxyeicosatrienamide (11,12-EET-EA), N-(2-hydroxyethyl)-14,15-epoxyeicosatrienamide (14,15-EET-EA), each of which is formed at different rates by different CYP isoforms (Table 3). Isoforms CYP2D6, CYP3A4, CYP2J2, CYP4F4 are known anandamide monooxygenases and are found in the human brain as well as other tissues [13, 77,78,79, 101]. hCYP2D6 and hCYP3A4 have been shown to be major contributors to anandamide metabolism in the brain where incubation with isoform-specific inhibitory antibodies reduces AEA metabolism by 88% and 42–66%, respectively [77]. Interestingly, one of the metabolites, 5,6-EET-EA, is a potent endocannabinoid CB2 receptor agonist with comparable binding efficiency to AEA as well as being more stable than AEA [101], the production of which thus increases the potency of the original anandamide signaling. In addition, 5,6-EET-EA is also an agonist for the endocannabinoid CB1 receptor, albeit with > 1000-fold weaker binding. 20-HETE-EA and 14,15-EET-EA also bind to the CB1 receptor with 3.6-fold and 5.7-fold lower efficiency than anandamide, respectively, and are more rapidly degraded in the brain than anandamide [80]. The biological impact of these and the other P450 metabolites of AEA needs to be clarified.
Lipoxygenases
Lipoxygenases represent a class of non-heme iron-requiring enzymes responsible for oxygenation of a wide array of polyunsaturated fatty acids (PUFAs). The primary biological role is to oxygenate PUFAs at specific positions in a stereospecific manner, producing the corresponding mono-hydroperoxy products that are readily converted to mono-hydroxy derivatives by ubiquitous glutathione peroxidases (GPX). Individual lipoxygenases are responsible for hydroperoxidation at specific positions and associated stereochemistry. For example, of the 12 possible peroxy positions on arachidonic acid, 10 are found in nature and 5 found in humans. Both 12S and 15S peroxidations of AEA catalyzed by 12S-lipoxygenase and 15S-lipoxygenase, respectively, have been reported [81, 102, 103]. However, 5S and 12R peroxidation, and 15R hydroxylation catalyzed by 5S-lipoxygenase, 12R-lipoxygenase, and aspirin-acetylated COX-2, respectively, have been observed for AA substrate, but not for AEA.
12S-lipoxygenase (ALOX12)
12S-lipoxygenase catalyzes the oxygenation of AEA to 12(S)-hydroperoxyarachadonoylethanolamide (12(S)-HPETE-EA) which is rapidly converted to 12(S)-hydroxyarachadonoylethanolamide (12(S)-HETE-EA) as well as numerous other products (Fig. 2) [103, 104]. Human12S-lipoxygenase (hALOX12, 12S-LOX, 12LO, LOG12, UniprotKB-P18054) is translated as a single isoform. Physical properties and PTMs are discussed elsewhere [104]. The specific activity for hALOX12 with AEA was determined for solubilized protein precipitates obtained from human leukocyte/platelet homogenates and found to be ≈ 70% of that found for AA [81] (Table 3).
As observed for CYP enzymes, the hydroxylated product also binds to the endocannabinoid receptors CB1 and CB2. 12(S)-HETE-EA binds to rat brain membranes containing CB1 with lower efficiency (≈ 53%) than AEA and human CB2 expressed in CHO cells also binds with lower efficiency than AEA (≈ 60%) [103]. A second report indicates that 12(S)-HETE-EA binds to both CB1 in rat brain membranes and CB2 in rat spleen membranes with lower efficiency than AEA, 60% and 72%, respectively [105]. The binding of 12(S)-HETE-EA to mixed CB1 and CB2 receptors has been shown to be agonistic but less effective than AEA with an EC50 that is 2.5 times that observed for AEA for the inhibition of electrically initiated twitch response in murine deferens [81].
The catalytic activity of hALOX12 is sensitive to the redox conditions [106] within the cell and may be regulated by phosphorylation at S246 based on the known phosphorylation of the rat isoform [47] for which it shares an 84.5% sequence homology. Upregulation of ALOX12 in porcine aortic vascular smooth muscle cells by the proinflammatory cytokines IL-1β, IL-4, and IL-8 has also been reported [107].
15S-lipoxygenase (ALOX15)
15S-lipoxygenase catalyzes the oxygenation of AEA to 15(S)-peroxyarachadonoylethanolamide (15(S)-HPETE-EA) which is rapidly converted to 15(S)-hydroxyarachadonoylethanolamide (15(S)-HETE-EA) by ubiquitous GPXs (Fig. 2). ALOX15 can also convert 15(S)-HPETE-EA to eoxin A4 ethanolamide (EXA4-EA) which is then readily converted to the ethanolamides of eoxin C4 (EXC4-EA) and eoxin D4 (EXD4-EA) [108] through the action of leukotriene C4 synthase (LTC4S) and gamma-glutamyl transaminase (GTT1) in a sequential manner (Fig. 2) [104]. Like hALOX12, human 15S-lipoxygenase (hALOX15, 15A-LOX, 15-LOX, 15-LOX-1, 12/15-lipoxygenase, UniProtKB-P16050) is a non-heme, iron-requiring cytosolic protein that becomes membrane associated in the presence of calcium. It is transcribed as two isoforms, but only the canonical form, Isoform 1, has been observed at the protein level. The physical properties and PTMs are discussed elsewhere [104, 109].
Specific activity for hALOX15 with AEA has not been reported; however, one publication gives a Km for AEA of 108 µM [108]. It should be noted that hALOX15 also produces 12S-HETE from AA in a 1:9 12S:15S ratio [110]. The production of 12S-HETE-EA by this enzyme is to be expected but has yet to be reported. 15(S)-HETE-EA binds to rat brain CB1 with lower efficiency than AEA (14%) and does not bind to human CB2 expressed in CHO cells [103]. A second report indicates that 15(S)-HETE-EA binds to CB1 in rat brain membranes with a lower efficiency than AEA (15%) and does not bind to CB2 in rat spleen membranes [105]. The binding of 15(S)-HETE-EA to mixed CB1 and CB2 receptors has been shown to be agonistic but less effective than AEA with an EC50 that is 4.9 times that for AEA for the inhibition of electrically initiated twitch response in murine deferens [81].
Regulation via posttranslational modification has not been reported. However, upregulation of hALOX15 expression by the proinflammatory cytokines IL-4 and IL-13 in human monocytes is well documented [111].
Anandamide transport and storage
AEA is a well-known paracrine and autocrine signaling molecule. The fact that it is uncharged and hydrophobic necessitates that AEA be transported both extracellularly and intracellularly by carrier molecules. Further, both the biosynthetic and degradation machinery are located within the cell, necessitating the existence of some process or processes to move AEA out of the cell for extracellular signaling and import for degradation. Until recently this process was thought to operate solely on a supply and demand mode. However, it has become clear that AEA is also stored in lipid droplets, thus complicating the supply demand model with a “buffer” system that can modulate available AEA. The following sections outline each of the processes noted above, including characterizations of the proteins involved.
Extracellular AEA transport proteins (AETs)
Transport of hydrophobic molecules in aqueous solution in living systems is typically facilitated by specific binding proteins. In plasma, albumin, and members of the lipocalin family are involved in transporting lipophilic molecules in plasma and may possibly be involved in extracellular AEA transport. Proteins historically shown to be involved in intracellular transport [112] have more recently been shown to exist extracellularly and one is known to be involved in extracellular AEA transport and others may be involved as well [113, 114]. Several fatty acid binding proteins, FABP3, FABP5, and FABP7 are known to be involved in intracellular trafficking of AEA [115]. FABP5 was recently shown to mediate retrograde endocannabinoid transport, suggesting that it may serve as a synaptic carrier for AEA [113]. heat shock proteins (HSPs) are also involved in intracellular transport of AEA [112]. HSP70, HSP90, HSP60 as well as several other HSPs (e.g., Grp75, Grp 80) have been identified extracellularly [114]. All of these species have been shown to bind lipids and thus may potentially act as additional AETs.
Microvesicles and exosomes are also well documented as purveyors of both intracellular and extracellular signaling through their transport of proteins, peptides, nucleic acids, and lipids [116], the latter making them potential candidates for both intracellular an extracellular transport of AEA. One report supporting vesicular transport of AEA shows that microglia produce extracellular vesicles with AEA on their surface and in turn stimulate type-1 endocannabinoid receptors (CB1), facilitating inhibition of presynaptic transmission [117].
Transport of AEA across plasma membranes
Endocannabinoids are readily transported across plasma membranes. The mode for transport of AEA has been a hotly debated subject for many years. Model proponents fall into three camps, diffusion, facilitated transport, or an endocytotic pathway. It is clear, however, that AEA does not pass through membranes by active transport [118].
Support for diffusion control of AEA transport is multifold. Some cite the lack of direct evidence for a proteinaceous transporter and a simpler model where sequestration of cytosolic AEA by proteins or vesicles necessarily leads to the AEA gradient necessary for diffusion across the plasma membrane [119]. Indeed, external addition of AEA to lipid vesicles containing internalized FAAH results in rapid AEA transport and hydrolysis [115]. Further, this transport is enhanced by cholesterol and coprostanol but is not affected by cholesterol sulfate. The equality of cholesterol and coprostanol transport indicates that the sterol ring enhances transport and coprostanol facilitated transport indicates that lipid rafts are not necessary for this process to occur. The lack of enhancement with cholesterol sulfate supports a mechanism whereby sterol ring flip-flopping may be involved in the transport process. Earlier work shows that both AEA insertion into lipid membranes and transport across them is cholesterol-dependent [120]. In related work, AEA present in resealed red blood cell membranes (ghosts) rapidly exits to external solutions containing bovine serum albumin (BSA), a known AEA binder, in a BSA-dependent manner [118]. Similarly, treatment of HeLa cells expressing mouse FABPs, known AEA binders, with FABP inhibitors reduces uptake of [14C] AEA as do FABP knockout cells in the absence of inhibitors [121].
Much of the support for AEA membrane transporter proteins (AMT) comes from observations that a variety of pharmaceuticals can reduce AEA transport presumably by interacting with an undiscovered AMT. On the other hand, these inhibitors may instead be interacting with intracellular proteins involved in sequestration or destruction of AEA. A more recent study clearly shows the existence of an AMT that is capable of bi-directional trafficking of AEA [122]. Using specific inhibitors of both AMT and FAAH they show that inhibition of FAAH results in an increase in intracellular AEA and inhibition of the putative AMT results in a decrease in intracellular AEA, clearly showing the existence of an AMT. In addition, they also show that the same AMT inhibitors produce a concentration-dependent decrease in AEA export, clearly supporting the existence of a yet-to-be identified AMT.
A third possibility for transmembrane trafficking involves endocytosis. Treatment of rat basophil cells (RBL-2H3) with inhibitors of caveola-related (clatherin-independent) endocytosis reduces AEA transport by ≈ 50% compared to controls [123]. Further, treatment of these cells with inhibitors of clatherin-dependent processes has no effect on AEA transport, indicating that only the caveola-related transport is involved. The same study found that AEA is concentrated in caveolin-rich membranes following internalization, further supporting the clatherin-independent pathway.
Storage of AEA
The long-held belief that AEA is synthesized on demand has been seriously challenged by more recent reports (see review [2]). It has been shown that once AEA is taken up by cells it is rapidly targeted to adiposomes [112, 124]. Further, it is also observed that larger adiposome compartments have an increased capacity to both store and metabolize AEA [124]. Adiposomes not only house intracellular AEA, but also contain degradation enzymes FAAH-1&2 [72, 112] and thus these lipid bodies serve as modulators of intracellular accumulation and degradation of AEA.
Both mobilization and degradation of AEA stored in liposomes requires a mechanism by which to move the internalized AEA from within the liposome to the surface. One mode for this transport has been shown to involve sterols inserted into the liposome membrane [115]. Both lipid-raft associating cholesterol and lipid-raft avoiding coprostanol enhance transport of AEA across the liposome bilayer indicating that lipid rafts are not necessary for this process to occur. The mode of action is thought to involve a competition for shielding the small headgroups from solvent water resulting in increased surface exposure of AEA to FAAH [115] or to specific transport proteins.
Conclusions
Anandamide homeostasis is achieved through the rates of synthesis, degradation, and storage of this molecule. This manuscript discusses the known enzymes and other proteins that are major contributors to this system. Biosynthesis occurs along four parallel pathways, each of which is catalyzed by its own set of enzymes and under independent control. Degradation can be simple, involving a hydrolysis of the amide by enzymes, or complex involving the eicosanoid biosynthesis machinery. Lastly, there is a storage system involving lipid microvesicles that modulate available AEA through sequestration, regulated release, and degradation. One additional item for consideration is the fact that AEA can be converted to other molecules that are known to activate the same CB receptors that AEA stimulates, thus continuing the signaling in the absence of AEA itself.
There are more than a few missing aspects that have yet to be uncovered in the laboratory and are required to provide a comprehensive picture of anandamide homeostasis. For example, a relative comparison of the synthesis degradation enzyme rates within particular tissues under defined metabolic conditions (e.g., healthy vs. inflammatory state) would be most enlightening. In addition, exploration of potentially new endocannabinoid products formed by oxidation catalyzed by eicosanoid enzymes other than ALOX12 and ALOX15 is in order. Lastly, a thorough investigation of how each pathway enzyme is regulated would help to better define the condition-dependent roles of each arm of the multi-armed pathways.
References
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949. https://doi.org/10.1126/science
Maccarrone M (2017) Metabolism of the endocannabinoid anandamide: open questions after 25 years. Front Mol Neurosci 10:166. https://doi.org/10.3389/fnmol.2017.00166
Marsicano G, Chaouloff F (2011) Moving bliss: a new anandamide transporter. Nat Neurosci 15:5–6. https://doi.org/10.1038/nn.3011
Mechoulam R, Parker LA (2013) The endocannabinoid system and the brain. Annu Rev Psychol 64:21–47. https://doi.org/10.1146/annurev-psych-113011-143739
Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140:790–801. https://doi.org/10.1038/sj.bjp.0705467
Mardian EB, Bradley RM, Duncan RE (2015) The HRASLS (PLA/AT) subfamily of enzymes. J Biomed Sci 22:99. https://doi.org/10.1186/s12929-015-0210-7
Hussain Z, Uyama T, Kawai K, Rahman IA, Tsuboi K, Araki N, Ueda N (2016) Comparative analyses of isoforms of the calcium-independent phosphatidylethanolamine N-acyltransferase PLAAT-1 in humans and mice. J Lipid Res 57:2051–2060. https://doi.org/10.1194/jlr.M071290
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, Kaplan S, Dahary D, Warshawsky D, Guan-Golan Y, Kohn A, Rappaport N, Safran M, Lancet D (2016) The genecards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform 54(1):1–30. https://doi.org/10.1002/cpbi.5
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44(D1):D862-868. https://doi.org/10.1093/nar/gkv1222
Käll L, Krogh A, Sonnhammer EL (2007) Advantages of combined transmembrane topology and signal peptide prediction–the phobius web server. Nucleic Acids Res 35:W429–W432. https://doi.org/10.1093/nar/gkm256
Shinohara N, Uyama T, Jin XH, Tsuboi K, Tonai T, Houchi H, Ueda N (2011) Enzymological analysis of the tumor suppressor A-C1 reveals a novel group of phospholipid-metabolizing enzymes. J Lipid Res 52:1927–1935. https://doi.org/10.1194/jlr.M015081
Uyama T, Tsuboi K, Ueda N (2017) An involvement of phospholipase A/acyltransferase family proteins in peroxisome regulation and plasmalogen metabolism. FEBS Lett 591:2745–2760. https://doi.org/10.1002/1873-3468.12787
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F (2015) Tissue-based map of the human proteome. Science 347:1260419. https://doi.org/10.1126/science
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. https://doi.org/10.1093/nar/gky427
Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4:1633–1649. https://doi.org/10.1002/pmic.200300771
Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos-Silva L, Gupta R, Bennett EP, Mandel U, Brunak S, Wandall HH, Levery SB, Clausen H (2013) Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 32:1478–1488. https://doi.org/10.1038/emboj.2013.79
Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294:1351–1362. https://doi.org/10.1006/jmbi.1999.3310
Uyama T, Ikematsu N, Inoue M, Shinohara N, Jin XH, Tsuboi K, Tonai T, Tokumura A, Ueda N (2012) Generation of N-acylphosphatidylethanolamine by members of the phospholipase A/acyltransferase (PLA/AT) family. J Biol Chem 287:31905–31919. https://doi.org/10.1074/jbc.M112.368712
Sers C, Emmenegger U, Husmann K, Bucher K, Andres AC, Schäfer R (1997) Growth-inhibitory activity and downregulation of the class II tumor-suppressor gene H-rev107 in tumor cell lines and experimental tumors. J Cell Biol 136:935–944. https://doi.org/10.1083/jcb.136.4.935
Golczak M, Kiser PD, Sears AE, Lodowski DT, Blaner WS, Palczewski K (2012) Structural basis for the acyltransferase activity of lecithin:retinol acyltransferase-like proteins. J Biol Chem 287:23790–23807. https://doi.org/10.1074/jbc.M112.361550
Uyama T, Jin XH, Tsuboi K, Tonai T, Ueda N (2009) Characterization of the human tumor suppressors TIG3 and HRASLS2 as phospholipid-metabolizing enzymes. Biochim Biophys Acta 1791:1114–1124. https://doi.org/10.1016/j.bbalip.2009.07.001
Magotti P, Bauer I, Igarashi M, Babagoli M, Marotta R, Piomelli D, Garau G (2015) Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 23:598–604. https://doi.org/10.1016/j.str.2014.12.018
Uyama T, Morishita J, Jin XH, Okamoto Y, Tsuboi K, Ueda N (2009) The tumor suppressor gene H-Rev107 functions as a novel Ca2+-independent cytosolic phospholipase A1/2 of the thiol hydrolase type. J Lipid Res 50:685–693. https://doi.org/10.1194/jlr.M800453-JLR200
Pang XY, Cao J, Addington L, Lovell S, Battaile KP, Zhang N, Rao JL, Dennis EA, Moise AR (2012) Structure/function relationships of adipose phospholipase A2 containing a cys-his-his catalytic triad. J Biol Chem 287:35260–35274. https://doi.org/10.1074/jbc.M112.398859
Duncan RE, Sarkadi-Nagy E, Jaworski K, Ahmadian M, Sul HS (2008) Identification and functional characterization of adipose-specific phospholipase A2 (AdPLA). J Biol Chem 283:25428–32546. https://doi.org/10.1074/jbc.M804146200
Higuchi E, Chandraratna RA, Hong WK, Lotan R (2003) Induction of TIG3, a putative class II tumor suppressor gene, by retinoic acid in head and neck and lung carcinoma cells and its association with suppression of the transformed phenotype. Oncogene 22:4627–4635. https://doi.org/10.1038/sj.onc.1206235
Jin XH, Uyama T, Wang J, Okamoto Y, Tonai T, Ueda N (2009) cDNA cloning and characterization of human and mouse Ca(2+)-independent phosphatidylethanolamine N-acyltransferases. Biochim Biophys Acta 1791:32–38. https://doi.org/10.1016/j.bbalip.2008.09.006
Hussain Z, Uyama T, Kawai K, Binte Mustafiz SS, Tsuboi K, Araki N, Ueda N (2018) Phosphatidylserine-stimulated production of N-acyl-phosphatidylethanolamines by Ca2+-dependent N-acyltransferase. Biochim Biophys Acta Mol Cell Biol Lipids 1863:493–502. https://doi.org/10.1016/j.bbalip.2018.02.002
Binte Mustafiz SS, Uyama T, Hussain Z, Kawai K, Tsuboi K, Araki N, Ueda N (2019) The role of intracellular anionic phospholipids in the production of N-acyl-phosphatidylethanolamines by cytosolic phospholipase A2e. J Biochem 165:343–352. https://doi.org/10.1093/jb/mvy104
Ogura Y, Parsons WH, Kamat SS, Cravatt BF (2016) A calcium-dependent acyltransferase that produces N-acyl phosphatidylethanolamines. Nat Chem Biol 12:669–671. https://doi.org/10.1038/nchembio.2127
Murray D, Honig B (2002) Electrostatic control of the membrane targeting of C2 domains. Mol Cell 9:145–154. https://doi.org/10.1016/s1097-2765(01)00426-9
Hussain Z, Uyama T, Tsuboi K, Ueda N (2017) Mammalian enzymes responsible for the biosynthesis of N-acylethanolamines. Biochim Biophys Acta Mol Cell Biol Lipids 1862:1546–1561. https://doi.org/10.1016/j.bbalip.2017.08.006
Capestrano M, Mariggio S, Perinetti G, Egorova AV, Iacobacci S, Santoro M, Di Pentima A, Iurisci C, Egorov MV, Di Tullio G, Buccione R, Luini A, Polishchuk RS (2014) Cytosolic phospholipase A2e drives recycling through the clathrin-independent endocytic route. J Cell Sci 127:977–993. https://doi.org/10.1242/jcs.136598
Liu J, Wang L, Harvey-White J, Huang BX, Kim HY, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, Razdan RK, Kunos G (2008) Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 54:1–7. https://doi.org/10.1016/j.neuropharm.2007.05.020
Van Damme P, Lasa M, Polevoda B, Gazquez C, Elosegui-Artola A, Kim DS, De Juan-Pardo E, Demeyer K, Hole K, Larrea E, Timmerman E, Prieto J, Arnesen T, Sherman F, Gevaert K, Aldabe R (2012) N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc Natl Acad Sci USA 109:12449–12454. https://doi.org/10.1073/pnas.1210303109
Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279:5298–5305. https://doi.org/10.1074/jbc.M306642200
Wang J, Okamoto Y, Morishita J, Tsuboi K, Miyatake A, Ueda N (2006) Functional analysis of the purified anandamide-generating phospholipase D as a member of the metallo-beta-lactamase family. J Biol Chem 281:12325–12335. https://doi.org/10.1074/jbc.M512359200
Margheritis E, Castellani B, Magotti P, Peruzzi S, Romeo E, Natali F, Mostarda S, Gioiello A, Piomelli D, Garau G (2016) Bile acid recognition by NAPE-PLD. ACS Chem Biol 11:2908–2914. https://doi.org/10.1021/acschembio.6b00624
Lefort C, Roumain M, Van Hul M, Rastelli M, Manco R, Leclercq I, Delzenne NM, Marzo VD, Flamand N, Luquet S, Silvestri C, Muccioli GG, Cani PD (2020) Hepatic NAPE-PLD Is a key regulator of liver lipid metabolism. Cells 9(1247):1. https://doi.org/10.3390/cells9051247
Simon GM, Cravatt BF (2006) Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem 281:26465–26472. https://doi.org/10.1074/jbc.M604660200
Ohshima N, Kudo T, Yamashita Y, Mariggiò S, Araki M, Honda A, Nagano T, Isaji C, Kato N, Corda D, Izumi T, Yanaka N (2014) New members of the mammalian glycerophosphodiester phosphodiesterase family: GDE4 and GDE7 produce lysophosphatidic acid by lysophospholipase D activity. J Biol Chem 290:4260–4271. https://doi.org/10.1074/jbc.M114.614537
Rahman IA, Tsuboi K, Hussain Z, Yamashita R, Okamoto Y, Uyama T, Yamazaki N, Tanaka T, Tokumura A, Ueda N (2016) Calcium-dependent generation of N-acylethanolamines and lysophosphatidic acids by glycerophosphodiesterase GDE7. Biochim Biophys Acta 1861:1881–1892. https://doi.org/10.1016/j.bbalip.2016.09.008
Zheng B, Berrie CP, Corda D, Farquhar MG (2003) GDE1/MIR16 is a glycerophosphoinositol phosphodiesterase regulated by stimulation of G protein-coupled receptors. Proc Natl Acad Sci USA 100:1745–1750. https://doi.org/10.1073/pnas.0337605100
Tsuboi K, Okamoto Y, Rahman IA, Uyama T, Inoue T, Tokumura A, Ueda N (2015) Glycerophosphodiesterase GDE4 as a novel lysophospholipase D: a possible involvement in bioactive N-acylethanolamine biosynthesis. Biochim Biophys Acta 1851:537–548. https://doi.org/10.1016/j.bbalip.2015.01.002
Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G (2006) A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA 103:13345–13350. https://doi.org/10.1073/pnas.0601832103
Ayakannu T, Taylor AH, Bari M, Mastrangelo N, Maccarrone M, Konje JC (2019) Expression and function of the endocannabinoid modulating enzymes fatty acid amide hydrolase and N-Acylphosphatidylethanolamine-specific phospholipase D in endometrial carcinoma. Front Oncol 9:1363. https://doi.org/10.3389/fonc.2019.01363
Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV (2012) Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun 3:876. https://doi.org/10.1038/ncomms1871
Sun YX, Tsuboi K, Okamoto Y, Tonai T, Murakami M, Kudo I, Ueda N (2004) Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase. Biochem J 380:749–756. https://doi.org/10.1042/BJ20040031
Szymczak-Pajor I, Kleniewska P, Wieczfinska J, Pawliczak R (2020) Wide-range effects of 1,25(OH)2D3 on group 4A phospholipases is related to nuclear factor k-B and phospholipase-A2 activating protein activity in mast cells. Int Arch Allergy Immunol 181:56–70. https://doi.org/10.1159/000503628
Chang PA, Shao HB, Long DX, Sun Q, Wu YJ (2008) Isolation, characterization and molecular 3D model of human GDE4, a novel membrane protein containing glycerophosphodiester phosphodiesterase domain. Mol Membr Biol 25:557–566. https://doi.org/10.1080/09687680802537605
Bachmann AS, Duennebier FF, Mocz G (2006) Genomic organization, characterization, and molecular 3D model of GDE1, a novel mammalian glycerophosphoinositol phosphodiesterase. Gene 371:144–153. https://doi.org/10.1016/j.gene.2005.11.023
Zheng B, Chen D, Farquhar MG (2000) MIR16, a putative membrane glycerophosphodiester phosphodiesterase, interacts with RGS16. Proc Natl Acad Sci USA 97:3999–4004. https://doi.org/10.1073/pnas.97.8.3999
Aoyama C, Horibata Y, Ando H, Mitsuhashi S, Arai M, Sugimoto H (2019) Characterization of glycerophosphodiesterase 4-interacting molecules Gaq/11 and Gb, which mediate cellular lysophospholipase D activity. Biochem J 476:3721–3736. https://doi.org/10.1042/BCJ20190666
Aoyama C, Sugimoto H, Ando H, Yamashita S, Horibata Y, Sugimoto S, Satou M (2011) The heterotrimeric G protein subunits Ga(q) and Gb(1) have lysophospholipase D activity. Biochem J 440:241–250. https://doi.org/10.1042/BJ20110545
Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, Jeffery D, Mortara K, Sampang J, Williams SR, Buggy J, Clark JM (2006) Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem 281:11002–11010. https://doi.org/10.1074/jbc.M600498200
Tsai SJ, Sen U, Zhao L, Greenleaf WB, Dasgupta J, Fiorillo E, Orrú V, Bottini N, Chen XS (2009) Crystal structure of the human lymphoid tyrosine phosphatase catalytic domain: insights into redox regulation. Biochemistry 48:4838–4845. https://doi.org/10.1021/bi900166y
Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Müller S, Knapp S (2009) Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136:352–363. https://doi.org/10.1016/j.cell.2008.11.038
Yang S, Svensson MND, Harder NHO, Hsieh WC, Santelli E, Kiosses WB, Moresco JJ, Yates JR 3rd, King CC, Liu L, Stanford SM, Bottini N (2020) PTPN22 phosphorylation acts as a molecular rheostat for the inhibition of TCR signaling. Sci Signal 13:eaaw8130. https://doi.org/10.1126/scisignal.aaw8130
Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S (2013) Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res 12:260–271. https://doi.org/10.1021/pr300630k
Yu X, Sun JP, He Y, Guo X, Liu S, Zhou B, Hudmon A, Zhang ZY (2007) Structure, inhibitor, and regulatory mechanism of lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci USA 104:19767–19772. https://doi.org/10.1073/pnas.0706233104
An H, Xu H, Zhang M, Zhou J, Feng T, Qian C, Qi R (2005) Cao X (2005) Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood 105:4685–4692. https://doi.org/10.1182/blood-2005-01-0191
Lamkin TD, Walk SF, Liu L, Damen JE, Krystal G, Ravichandran KS (1997) Shc interaction with src homology 2 domain containing inositol phosphatase (SHIP) in vivo requires the shc-phosphotyrosine binding domain and two specific phosphotyrosines on SHIP. J Biol Chem 272:10396–10401. https://doi.org/10.1074/jbc.272.16.10396
Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143:1174–1189. https://doi.org/10.1016/j.cell.2010.12.001
Cao L, Yu K, Banh C, Nguyen V, Ritz A, Raphael BJ, Kawakami Y, Kawakami T, Salomon AR (2007) Quantitative time-resolved phosphoproteomic analysis of mast cell signaling. J Immunol 179:5864–5876. https://doi.org/10.4049/jimmunol.179.9.5864
Chiang KP, Gerber AL, Sipe JC, Cravatt BF (2004) Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Hum Mol Genet 13:2113–2119. https://doi.org/10.1093/hmg/ddh216
Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF (2006) A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem 281:36569–36578. https://doi.org/10.1074/jbc.M606646200
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87. https://doi.org/10.1038/384083a0
Giang DK, Cravatt BF (1997) Molecular characterization of human and mouse fatty acid amide hydrolases. Proc Natl Acad Sci USA 94:2238–2242. https://doi.org/10.1073/pnas.94.6.2238
Maccarrone M, Bari M, Di Rienzo M, Finazzi-Agrò A, Rossi A (2003) Progesterone activates fatty acid amide hydrolase (FAAH) promoter in human T lymphocytes through the transcription factor Ikaros. Evidence for a synergistic effect of leptin. J Biol Chem 278:32726–32732. https://doi.org/10.1074/jbc.M302123200
Dainese E, De Fabritiis G, Sabatucci A, Oddi S, Angelucci CB, Di Pancrazio C, Giorgino T, Stanley N, Del Carlo M, Cravatt BF, Maccarrone M (2014) Membrane lipids are key modulators of the endocannabinoid-hydrolase FAAH. Biochem J 457:463–472. https://doi.org/10.1042/BJ20130960
Sirrs S, van Karnebeek CD, Peng X, Shyr C, Tarailo-Graovac M, Mandal R, Testa D, Dubin D, Carbonetti G, Glynn SE, Sayson B, Robinson WP, Han B, Wishart D, Ross CJ, Wasserman WW, Hurwitz TA, Sinclair G, Kaczocha M (2015) Defects in fatty acid amide hydrolase 2 in a male with neurologic and psychiatric symptoms. Orphanet J Rare Dis 10:38. https://doi.org/10.1186/s13023-015-0248-3
Kaczocha M, Glaser ST, Chae J, Brown DA, Deutsch DG (2010) Lipid droplets are novel sites of N-acylethanolamine inactivation by fatty acid amide hydrolase-2. J Biol Chem 285:2796–2806. https://doi.org/10.1074/jbc.M109.058461
Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N (2005) Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem 280:11082–11092. https://doi.org/10.1074/jbc.M413473200
West JM, Zvonok N, Whitten KM, Wood JT, Makriyannis A (2012) Mass spectrometric characterization of human N-acylethanolamine-hydrolyzing acid amidase. J Proteome Res 11:972–981. https://doi.org/10.1021/pr200735a
Zhao LY, Tsuboi K, Okamoto Y, Nagahata S, Ueda N (2007) Proteolytic activation and glycosylation of N-acylethanolamine-hydrolyzing acid amidase, a lysosomal enzyme involved in the endocannabinoid metabolism. Biochim Biophys Acta 1771:1397–1405. https://doi.org/10.1016/j.bbalip.2007.10.002
Yu M, Ives D, Ramesha CS (1997) Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem 272:21181–21186. https://doi.org/10.1074/jbc.272.34.21181
Snider NT, Sikora MJ, Sridar C, Feuerstein TJ, Rae JM, Hollenberg PF (2008) The endocannabinoid anandamide is a substrate for the human polymorphic cytochrome P450 2D6. J Pharmacol Exp Ther 327:538–545. https://doi.org/10.1124/jpet.108.141796
Walker VJ, Griffin AP, Hammar DK, Hollenberg PF (2016) Metabolism of anandamide by human cytochrome P450 2J2 in the reconstituted system and human intestinal microsomes. J Pharmacol Exp Ther 357:537–544. https://doi.org/10.1124/jpet.116.232553
Snider NT, Kornilov AM, Kent UM, Hollenberg PF (2007) Anandamide metabolism by human liver and kidney microsomal cytochrome p450 enzymes to form hydroxyeicosatetraenoic and epoxyeicosatrienoic acid ethanolamides. J Pharmacol Exp Ther 321:590–597. https://doi.org/10.1124/jpet.107.119321
Sridar C, Snider NT, Hollenberg PF (2011) Anandamide oxidation by wild-type and polymorphically expressed CYP2B6 and CYP2D6. Drug Metab Dispos 39:782–788. https://doi.org/10.1124/dmd.110.036707
Hampson AJ, Hill WA, Zan-Phillips M, Makriyannis A, Leung E, Eglen RM, Bornheim LM (1995) Anandamide hydroxylation by brain lipoxygenase: metabolite structures and potencies at the cannabinoid receptor. Biochim Biophys Acta 1259:173–179. https://doi.org/10.1016/0005-2760(95)00157-8
Sakura Y, Tsuboi K, Uyama T, Zhang X, Taoka R, Sugimoto M, Kakehi Y, Ueda N (2016) A quantitative study on splice variants of N-acylethanolamine acid amidase in human prostate cancer cells and other cells. Biochim Biophys Acta 1861:1951–1958. https://doi.org/10.1016/j.bbalip.2016.09.018
Gorelik A, Gebai A, Illes K, Piomelli D, Nagar B (2018) Molecular mechanism of activation of the immunoregulatory amidase NAAA. Proc Natl Acad Sci USA 115:E10032–E10040. https://doi.org/10.1073/pnas.1811759115
Wang J, Zhao LY, Uyama T, Tsuboi K, Tonai T, Ueda N (2008) Amino acid residues crucial in pH regulation and proteolytic activation of N-acylethanolamine-hydrolyzing acid amidase. Biochim Biophys Acta 1781:710–717. https://doi.org/10.1016/j.bbalip.2008.08.004
Tai T, Tsuboi K, Uyama T, Masuda K, Cravatt BF, Houchi H, Ueda N (2012) Endogenous molecules stimulating N-acylethanolamine-hydrolyzing acid amidase (NAAA). ACS Chem Neurosci 3:379–385. https://doi.org/10.1021/cn300007s
Pavlopoulos S, Pelekoudas DN, Benchama O, Rawlins CM, Agar JN, West JM, Malamas M, Zvonok N, Makriyannis A (2018) Secretion, isotopic labeling and deglycosylation of N-acylethanolamine acid amidase for biophysical studies. Protein Exp Purif 145:108–117. https://doi.org/10.1016/j.pep.2017.12.005
Biringer RG (2020) The enzymology of the human prostanoid pathway. Mol Biol Rep 47:4569–4586. https://doi.org/10.1007/s11033-020-05526-z
Boutaud O, Montine TJ, Chang L, Klein WL, Oates JA (2006) PGH2-derived levuglandin adducts increase the neurotoxicity of amyloid beta1-42. J Neurochem 96:917–923. https://doi.org/10.1111/j.1471-4159.2005.03586.x
Salomon RG, Miller DB, Zagorski MG, Coughlin DJ (1984) Solvent-induced fragmentation of prostaglandin endoperoxides. New aldehyde products from PGH2 and a novel intramolecular 1,2-hydride shift during endoperoxide fragmentation in aqueous solution. J Am Chem Soc 106:6049–6060
Urquhart P, Wang J, Woodward DF, Nicolaou A (2015) Identification of prostamides, fatty acyl ethanolamines, and their biosynthetic precursors in rabbit cornea. J Lipid Res 56:1419–1433. https://doi.org/10.1194/jlr.M055772
Woodward DF, Liang Y, Krauss AH (2008) Prostamides (prostaglandin-ethanolamides) and their pharmacology. Br J Pharmacol 153:410–419. https://doi.org/10.1038/sj.bjp.0707434
Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, Jakobsson PJ, Marnett LJ (2002) Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem 277:44877–44885. https://doi.org/10.1074/jbc.M206788200
Woodward DF, Wang JW, Poloso NJ (2013) Recent progress in prostaglandin F2a ethanolamide (prostamide F2a) research and therapeutics. Pharmacol Rev 65:1135–1147. https://doi.org/10.1124/pr.112.007088
Nemeth JF, Hochgesang GP Jr, Marnett LJ, Caprioli RM (2001) Characterization of the glycosylation sites in cyclooxygenase-2 using mass spectrometry. Biochemistry 40:3109–3116. https://doi.org/10.1021/bi002313c
Alexanian A, Miller B, Chesnik M, Mirza S, Sorokin A (2014) Post-translational regulation of COX2 activity by FYN in prostate cancer cells. Oncotarget 5:4232–4243. https://doi.org/10.18632/oncotarget.1983
Kim SF, Huri DA, Snyder SH (2005) Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 310:1966–1970. https://doi.org/10.1126/science.1119407
Cuendet M, Mesecar AD, DeWitt DL, Pezzuto JM (2006) An ELISA method to measure inhibition of the COX enzymes. Nat Protoc 1:1915–1921. https://doi.org/10.1038/nprot.2006.308
Ristimäki A, Garfinkel S, Wessendorf J, Maciag T, Hla T (1994) Induction of cyclooxygenase-2 by Interleukin-la. Evidence for post-transcriptional regulation. J Biol Chem 269:11769–11775
Parfenova H, Balabanova L, Leffler CW (1998) Posttranslational regulation of cyclooxygenase by tyrosine phosphorylation in cerebral endothelial cells. Am J Physiol 274:C72–C81. https://doi.org/10.1152/ajpcell.1998.274.1.C72
Akerman S, Romero-Reyes M, Holland PR (2017) Current and novel insights into the neurophysiology of migraine and its implications for therapeutics. Pharmacol Ther 172:151–170. https://doi.org/10.1016/j.pharmthera.2016.12.005
Snider NT, Nast JA, Tesmer LA, Hollenberg PF (2009) A cytochrome P450-derived epoxygenated metabolite of anandamide is a potent cannabinoid receptor 2-selective agonist. Mol Pharmacol 75:965–972. https://doi.org/10.1124/mol.108.053439
Kozak KR, Marnett LJ (2002) Oxidative metabolism of endocannabinoids. Prostaglandins Leukot Essent Fatty Acids 66:211–220. https://doi.org/10.1054/plef.2001.0359
Edgemond WS, Hillard CJ, Falck JR, Kearn CS, Campbell WB (1998) Human platelets and polymorphonuclear leukocytes synthesize oxygenated derivatives of arachidonylethanolamide (anandamide): their affinities for cannabinoid receptors and pathways of inactivation. Mol Pharmacol 54:180–188. https://doi.org/10.1124/mol.54.1.180
Biringer RG (2018) The Enzymes of the human eicosanoid pathway. Res Rep Med Sci 2:106
van der Stelt M, van Kuik JA, Bari M, van Zadelhoff G, Leeflang BR, Veldink GA, Finazzi-Agrò A, Vliegenthart JF, Maccarrone M (2002) Oxygenated metabolites of anandamide and 2-arachidonoylglycerol: conformational analysis and interaction with cannabinoid receptors, membrane transporter, and fatty acid amide hydrolase. J Med Chem 45:3709–3720. https://doi.org/10.1021/jm020818q
Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL (2011) Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res 50:115–131. https://doi.org/10.1016/j.plipres.2010.10.005
Natarajan R, Rosdahl J, Gonzales N, Bai W (1997) Regulation of 12-lipoxygenase by cytokines in vascular smooth muscle cells. Hypertension 30:873–879. https://doi.org/10.1161/01.hyp.30.4.873
Forsell PK, Brunnström A, Johannesson M, Claesson HE (2012) Metabolism of anandamide into eoxamides by 15-lipoxygenase-1 and glutathione transferases. Lipids 47:781–791. https://doi.org/10.1007/s11745-012-3684-z
Arita M, Ohira T, Sun YP, Elangovan S, Chiang N, Serhan CN (2007) Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol 178:3912–3917. https://doi.org/10.4049/jimmunol.178.6.3912
Chen X, Zhang J, Chen C (2011) Endocannabinoid 2-arachidonoylglycerol protects neurons against a-amyloid insults. Neuroscience 178:159–168. https://doi.org/10.1016/j.neuroscience.2011.01.024
Kuhn H, Gehring T, Schröter A, Heydeck D (2016) Cytokine-dependent expression regulation of ALOX15. J Cytokine Biol 1:106. https://doi.org/10.4172/2576-3881.1000106
Maccarrone M, Dainese E, Oddi S (2010) Intracellular trafficking of anandamide: new concepts for signaling. Trends Biochem Sci 35:601–608. https://doi.org/10.1016/j.tibs.2010.05.008
Haj-Dahmane S, Shen RY, Elmes MW, Studholme K, Kanjiya MP, Bogdan D, Thanos PK, Miyauchi JT, Tsirka SE, Deutsch DG, Kaczocha M (2018) Fatty-acid-binding protein 5 controls retrograde endocannabinoid signaling at central glutamate synapses. Proc Natl Acad Sci USA 115:3482–3487. https://doi.org/10.1073/pnas.1721339115
De Maio A, Vazquez D (2013) Extracellular heat shock proteins: a new location, a new function. Shock 40:239–246. https://doi.org/10.1097/SHK.0b013e3182a185ab
Kaczocha M, Lin Q, Nelson LD, McKinney MK, Cravatt BF, London E, Deutsch DG (2012) Anandamide externally added to lipid vesicles containing trapped fatty acid amide hydrolase (FAAH) is readily hydrolyzed in a sterol-modulated fashion. ACS Chem Neurosci 3:364–368. https://doi.org/10.1021/cn300001w
Yáñez-Mó M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, Colás E, Cordeiro-da Silva A, Fais S, Falcon-Perez JM, Ghobrial IM, Giebel B, Gimona M, Graner M, Gursel I, Gursel M, Heegaard NH, Hendrix A, Kierulf P, Kokubun K, Kosanovic M, Kralj-Iglic V, Krämer-Albers EM, Laitinen S, Lässer C, Lener T, Ligeti E, Linē A, Lipps G, Llorente A, Lötvall J, Manček-Keber M, Marcilla A, Mittelbrunn M, Nazarenko I, Nolte-’t Hoen EN, Nyman TA, O’Driscoll L, Olivan M, Oliveira C, Pállinger É, Del Portillo HA, Reventós J, Rigau M, Rohde E, Sammar M, Sánchez-Madrid F, Santarém N, Schallmoser K, Ostenfeld MS, Stoorvogel W, Stukelj R, Van der Grein SG, Vasconcelos MH, Wauben MH, De Wever O (2015) Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4:27066. https://doi.org/10.3402/jev.v4.27066
Gabrielli M, Battista N, Riganti L, Prada I, Antonucci F, Cantone L, Matteoli M, Maccarrone M, Verderio C (2015) Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep 16:213–220. https://doi.org/10.15252/embr.201439668
Bojesen IN, Hansen HS (2005) Membrane transport of anandamide through resealed human red blood cell membranes. J Lipid Res 46:1652–1659. https://doi.org/10.1194/jlr.M400498-JLR200
Fowler CJ (2013) Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J 280:1895–1904. https://doi.org/10.1111/febs.12212
Di Pasquale E, Chahinian H, Sanchez P, Fantini J (2009) The insertion and transport of anandamide in synthetic lipid membranes are both cholesterol-dependent. PLoS ONE 4(3):e4989. https://doi.org/10.1371/journal.pone.0004989
Kaczocha M, Vivieca S, Sun J, Glaser ST, Deutsch DG (2012) Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem 287:3415–34124. https://doi.org/10.1074/jbc.M111.304907
Chicca A, Marazzi J, Nicolussi S, Gertsch J (2012) Evidence for bidirectional endocannabinoid transport across cell membranes. J Biol Chem 287:34660–34682. https://doi.org/10.1074/jbc.M112.373241
McFarland MJ, Porter AC, Rakhshan FR, Rawat DS, Gibbs RA, Barker EL (2004) A role for caveolae/lipid rafts in the uptake and recycling of the endogenous cannabinoid anandamide. J Biol Chem 279:41991–41997. https://doi.org/10.1074/jbc.M407250200
Oddi S, Fezza F, Pasquariello N, De Simone C, Rapino C, Dainese E, Finazzi-Agrò A, Maccarrone M (2008) Evidence for the intracellular accumulation of anandamide in adiposomes. Cell Mol Life Sci 65:840–850. https://doi.org/10.1007/s00018-008-7494-7
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author states that he has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Biringer, R.G. The rise and fall of anandamide: processes that control synthesis, degradation, and storage. Mol Cell Biochem 476, 2753–2775 (2021). https://doi.org/10.1007/s11010-021-04121-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-021-04121-5