
Abstract
Plants are major source for discovery and development of anticancer drugs. Several plant-based anticancer drugs are currently in clinical use. Fagonia indica is a plant of medicinal value in the South Asian countries. Using mass spectrometry and NMR spectroscopy, several compounds were purified from the F. indica extract. We have used one of the purified compounds quinovic acid (QA) and found that QA strongly suppressed the growth and viability of human breast and lung cancer cells. QA did not inhibit growth and viability of non-tumorigenic breast cells. QA mediated its anticancer effects by inducing cell death. QA-induced cell death was associated with biochemical features of apoptosis such as activation of caspases 3 and 8 as well as PARP cleavage. QA also upregulated mRNA and protein levels of death receptor 5 (DR5). Further investigation revealed that QA did not alter DR5 gene promoter activity, but enhanced DR5 mRNA and protein stabilities. DR5 is one of the major components of the extrinsic pathway of apoptosis. Accordingly, Apo2L/TRAIL, the DR5 ligand, potentiated the anticancer effects of QA. Our results indicate that QA mediates its anticancer effects, at least in part, by engaging DR5-depentent pathway to induce apoptosis. Based on our results, we propose that QA in combination with Apo2L/TRAIL can be further investigated as a novel therapeutic approach for breast and lung cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plants from land and aquatic sources have proven to be a rich source for the discovery and development of anticancer drugs. The agents isolated from these natural sources generally fall under a variety of groups including diterpenes, diterpenoquinone, lactonic sesquiterpene, alkaloids, macrocyclic polyethers, proteins, purine-related compounds, peptides, cyclic depsipeptide and similar others [1]. Several important anticancer drugs currently in the clinic have their origins linked to plants. For example (1) vinca alkaloids, isolated from the plant Madagascar periwinkle, (2) paclitaxel, a taxane diterpenoid, isolated from the bark of pacific yew tree Taxus brevifolia Nutt, (3) camptothecin, isolated from the stem of Camptotheca acuminate [2,3,4]. Etoposide and teniposide, also extensively used anticancer drugs, are derivatives of podophyllotoxin that is present in the plant Podophyllum peltatum L., also known as mayapple or American mandrake [5].
The incidence of cancer has been increasing worldwide and thus, newer and more efficacious therapeutics are needed to better manage human malignancies. In this context, we have investigated the potential of plant Fagonia indica (Zygophyllaceae) as a source of newer cancer therapeutics. F. indica is utilized by the practitioners of traditional medicine to treat various aliments in parts of Pakistan and India. F. indica has a rich source of secondary metabolites such as phenolics, flavanoids, alkaloids, triterpenoids, coumarins and tannins [6, 7]. In Pakistan, parts of the plants are also used by local healers to treat malignancies. However, the scientific basis for the use of this plant in traditional medicine has not been systematically investigated. There is some scientific evidence to implicate anticancer potential attributable to this plant. For example, it has been reported that the aqueous extract of F. indica (reported as Fagonia cretica) induced growth arrest and apoptosis in human breast cancer cells in culture [8]. In another study, steroidal saponin glycoside was isolated from F. indica that was found to induce apoptosis or necrosis in MDA-MB-468, MCF-7 breast cancer cells and Caco-2 colon cancer cells [9].
Previously, bioactivity-guided isolation was performed on the crude extract of F. indica (reported as Fagonia cretica) to isolate ursane-type pentacyclic triterpenoid quinovic acid (QA) and its derivatives. Experimental evidence indicated that QA and its derivatives harbored anti-diabetic properties [10, 11]. For example, in one study, these agents were found to exhibit dipeptidyl peptidase-4 (DPP-4) inhibitory activities [11] and in another study, these agents activated Takeda G-protein-coupled receptor 5 (TGR5) and stimulated glucagon-like peptide (GLP-1) secretion [10].
In view of the medicinal value of QA, we sought to also investigate its potential as an anticancer therapeutic. Here, we report that QA strongly suppressed the growth and viability of various human cancer cell lines including breast and lung cancer cells. QA did not inhibit growth and viability of non-tumorigenic cells. QA mediated its anticancer effects by inducing cell death coupled with upregulation of death receptor 5 (DR5). Apo2L/TRAIL, the DR5 ligand, also potentiated the anticancer effects of QA. Our results indicate that QA mediates its anticancer effects, at least in part, by engaging DR5-depentent pathway to induce apoptosis. Thus, QA in combination with Apo2L/TRAIL can be further investigated to develop as a novel therapeutic approach for breast and lung cancer.
Materials and methods
Cell lines and cell culture
The cell lines used in this study include, A549 and H1299 human lung cancer cells (from NIH), MCF-7 and MDA-MB-231 human breast cancer cells (from NIH). Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gemini Bio-Products Inc., West Sacramento, CA) was used for culture. Non-tumorigenic MCF-10A human breast epithelial cells were grown in mammary epithelial cell growth medium with supplements provided in the SingleQuots kit (Lonza). MCF-10A were purchased from American Type Culture Collection (ATCC).
Reagents
The antibodies used in this study are anti-vinculin (Santa Cruz Biotechnology, Dallas, TX) as well as antibodies against cleaved PARP, procaspase 3, procaspase 8 and DR5 (Cell Signaling Technologies, Danvers, MA). The peroxidase-conjugated horse anti-mouse, goat anti-rat and goat anti-rabbit antibodies were obtained from Vector Laboratories (Burlingame, CA, USA). DMEM was purchased from Mediatech Inc. (Manassas, VA, USA) and fetal bovine serum from Gemini Bio-Products Inc. (West Sacramento, CA). For transfections, PolyJet and LipoJet reagents were purchased from SignaGen Laboratories (Rockville, MD, USA). Plasmid preparation and RNA extraction kits were purchased from Qiagen (Hilden, Germany). The quantitative PCR reagents were from Bio-Rad (Hercules, CA, USA). Most of the other chemicals and molecular and cell biology reagents were obtained from Sigma-Aldrich and Thermo Fisher Scientific.
Plant Fagonia indica material collection and identification
The fresh aerial parts of the plant were collected from Mianwali (a city in the Punjab Province of Pakistan). It is locally known as “Dhamasa/Suchi booti”. DNA was extracted from the plant material using previously reported method [12]. Plant identification was carried out by DNA barcoding approach that has been adopted as the standard barcoding method for land plants [13]. Chloroplast region ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit (rbcL) gene was selected for DNA barcoding. rbcL gene was amplified by PCR using forward primer 5′-ATATTTTGGCAGCATTCCGA-3′ and reverse primer 5′-TCACATGTACCTGCAGTAGC-3′ respectively. Rapid PCR Purification System 9700 (Marligen Biosciences, Ijamsville, MD, USA) was employed to purify the PCR product, which was then sequenced via the dideoxy-chain termination method using an ABI Prism 310 Automated DNA Sequencer (PE, Applied Biosystems, Foster City, CA, USA). Sequences were aligned and identified via the BioEdit sequence alignment tool.
The DNA sequence obtained as a result of DNA barcoding on the plant material was analyzed by nucleotide BLAST analyses. The sequence was found to be 96% similar to that of plant F. indica, a member of family Zygophyllaceae. The DNA sequence is as follows:
5′-GTGCATCGCGTCACCTGAGGAGCAGGGGCTGCAGTAGCTGCTGAATCTTC
TACTGGTACATGGACAGCTGTGTGGACAGATGGTCTTACCAGTCTTGATC
GTTACAAAGGACGATGCTACAACCTAGAGGCTGTTCCTGGAGAAGACAAT
CAATACATTGCTTATGTAGCTTACCCTTTGGACCTTTTTGAAGAAGGTTC
TGTTACTAACATGTTGACTTCCATTGTGGGTAACGTATTTGGGTTCAAAG
CCCTACGTGCTCTACGTTTGGAAGATTTACGAATCCCTACTGCGTATACT
AAAACTTTCCAAGGGCCTCCTCACGGTATCCAAGTTGAGAGAGATAAATT
AAATAAGTATGGTCGTCCATTATTGGGCTGTCCTATTAAGCCTAAATTGG
GGCTATCCACAAAGAATTATGGTAGAGCAGTTTATGATGGTCCTCGACGT
GGAGGTGCTATTACCACCGAATATGAGAAAGTCACGGGCCACCTATAATC
TGTATAGAGAGACGTTCTCCTTTGTCATGGCTGCGCATTCTTCGCGCTAC
TAGCTCAGGGGGGAGATCTGGGGGGGGTTAAGTGGGAGTAGTGGCTGAAA
AAAAAGTCTCGGTGTGGAGTTTATTTTCTATTGTGTGTATTTGTGTGGGT
TATTGGTGTTTTTGTTTTAGTTAGTGGTTGGA-3′
Extraction and isolation of quinovic acid (QA) from Fagonia indica
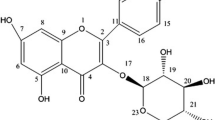
The fresh aerial parts of F. indica yielded 22 kg (dry weight of plant material), which was macerated in methanol–chloroform (1:1) solution to obtain the crude plant extract. It was later suspended in ethyl acetate to obtain an ethyl acetate fraction. Quinovic acid (QA) was isolated from ethyl acetate fraction on a silica gel column through chromatographic separation. Its isolation and identification have been described in detail [11]. Briefly, the identification of QA was achieved via a combination of mass spectrometry and NMR spectroscopy (Bruker AVANCE 400 MHz NMR). Chemical structures were confirmed by comparison of its chemical and spectroscopic properties. The compound was isolated yielding a white amorphous powder having molecular formula as C30H46O5 (m/z 486). 1H and 13C NMR analysis of the compound was performed that revealed its triterpenoid nature with ursane-type characteristics and distinctive signals for the presence of 30 carbons: six methyl groups, nine methylenes, seven methines, eight quaternary carbons along with the presence of two carboxyl groups. Accordingly, the compound was named as (3β)-3-Hydroxyurs-12-ene-27,28-dioic acid or quinovic acid.
Crystal violet staining and MTT assay
Crystal violet staining was performed to determine adherent cell viability according to the protocol described by Dr Gjoerup [14]. Briefly, cells were washed with PBS, and then fixed with 4% paraformaldehyde. Fixed cells were subjected to 0.1% crystal violet staining solution. After the excessive stain was drained, the plates were water-washed and air-dried. The plates were later scanned using a flat-bed scanner (Perfection V550, Epson, Japan) and images were captured as presented in the results section.
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide-based assays for cell growth were done as we have described previously [15]. Briefly, the cells seeded in 12-well plates were treated with vehicle or QA, then at the end of the treatment incubated with MTT 1 mg/ml for approximately 1–2 h. The formazan precipitate resulted after MTT incubation were dissolved into isopropanol containing 0.1 N HCl and 10% triton X-100. The sample absorbance was read using a Bio-Rad Smart-Spec 3000 spectrophotometer at the wavelengths 570 and 690 nm. Quantitative values were obtained after subtracting the absorbance at 690 nm wavelength for each sample, blank 570 nm and blank 690 nm wavelengths from the original A570 value to eliminate background readings respectively.
Western blot analyses
Western blot analyses were performed according to the procedures we have described previously [16, 17]. Proteins of interest (~ 60 μg/sample) were separated via 10% SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked using TBST and 5% milk for 1 h and then incubated with primary antibodies (at 4 °C) overnight on a shaker. Membranes were washed three times with TBST or TBST plus 5% milk and probed with secondary antibodies. Signals were developed following peroxidase reactions using SuperSignal Pico and Femto West chemiluminescent substrate (Thermo Fisher Scientific) and detected by Bio-Rad ChemiDoc XRS+ imaging system. Image Lab v4.1 software (Bio-Rad, Hercules, CA, USA) or ImageJ (NIH) was used to measure the band intensities for quantitative analyses.
Luciferase assays
DR5 luciferase promoter construct pGL2-Full was a kind gift from Dr. WS El-Deiry; cloning of the vector has been described previously [18]. This vector contains ~ 3.6 kb DR5 genomic sequence of which ~ 1.6 kb is upstream of ATG and remaining runs through intron 2 [18]. The promoter regulation analyses were performed using Luciferase Assay System (Promega, Madison, WI, USA) according to the manufacturer’s protocol and also as we have described previously [16]. Briefly, MCF-7 breast cancer cells and A549 lung cancer cells were transiently transfected with the DR5 gene promoter luciferase construct using LipoJet transfection kit (SignaGen Laboratories, Rockville, MD, USA). After 24 h, the media with transfection mix was removed, and fresh media containing either DMSO (control) or indicated doses of QA were added to the cells. After completion of the treatment, the cells were lysed with the lysis buffer and equal amount (~ 20 µg/sample) of protein lysates were mixed with 100 μl of Beetle luciferin substrate solution (Promega, Madison, WI, USA) followed by immediate measurement of luminescence using the luminometer (Lumat LB-9507, Berthold Technologies, Germany).
RNA extraction and quantitative RT-PCR
RNA was extracted using RNeasy Plus Mini kit (Qiagen, Hilden, Germany) per the manufacturer’s instructions. Analyses of mRNA expression were done via two-step quantitative real-time PCR (qRT-PCR) assays using the iScript cDNA Synthesis Kit and iQ SYBR Green Supermix from Bio-Rad (Hercules, CA, USA) according to manufacturer’s protocol. Ct values for DR5 were normalized with respect to the Ct values for GAPDH mRNA within the same sample; fold changes in mRNA expression were ascertained via the ΔΔCt method as reported earlier [19]. The following primer sets were used:
DR5 –
forward: 5′-AAGACCCTTGTGCTCGTTGT-3′;
reverse: 5′-AGGTGGACACAATCCCTCTG-3′;
GAPDH –
forward: 5′-CACCATCTTCCAGGAGCGAG-3′;
reverse: 5′-GCAGGAGGCATTGCTGAT-3′
Statistical analysis
For statistical analyses, Rcmdr 2.5-3 package based on R software (version 3.6.1) was used [20]. Mean values were compared using the two-tailed student’s t test or one-way ANOVA. The p < 0.05 values were considered statistically significant.
Results
QA, (3β)-3-hydroxy-urs-12-ene-27,28-dioic acid, is a ursane-type triterpene; its structure is shown in Fig. 1a. To investigate the anticancer potential of QA, its effect was tested on cancer cell lines representing human breast and lung cancers. QA effect on cell growth and viability was assessed by multiple approaches including phase contrast morphology, DAPI fluorescent (nuclear) staining, MTT assays and crystal violet staining. Our results indicated that QA inhibited proliferation and survival of these cell lines. Figure 1b shows representative results of crystal violet staining performed on A549, a human non-small cell lung cancer (NSCLC) cell line and MCF-7, a human breast cancer cell line. As is shown, QA inhibited cell viability of lung and breast cancer cells in a concentration-dependent manner. Figure 2a shows cellular morphology of A549 cells that were either vehicle-treated or treated with QA for 24 h, and Fig. 2b shows results of MTT assays performed on A549 cells treated with increasing concentrations of QA for 24 h. Lower concentrations of QA also inhibited A549 cell growth when they were treated with QA for 48 h (Fig. 2c) or 72 h (Fig. 2d). QA also inhibited growth and viability of other cancer cell types including MDA-MB-231 human breast cancer cells (Fig. 2e) and H1299 human lung cancer cells (data not shown). QA effect on normal cells (non-tumorigenic cells) was also investigated, and to that end, MCF-10A non-tumorigenic breast cells were used. As is shown (Fig. 2f), QA did not affect the viability of non-tumorigenic MCF-10A cells even at higher concentrations up to 40 µM. Thus, QA affected cell growth and viability of cancer cells, but did not have such effects on non-tumorigenic (normal) cells.

a Structure of QA. b QA inhibits lung and breast cancer cell growth as a function of dose. b A549 human lung cancer cells and MCF-7 human breast cancer cells were vehicle (DMSO)-treated or treated with QA at the indicated concentrations in µM for 72 and 48 h, respectively. Cells were fixed and stained with crystal violet. The reproducibility of the QA-mediated inhibitory effects on these cells was confirmed via by multiple approaches including crystal violet staining, phase contrast microscopy and MTT assays

QA inhibits growth of cancer cells but not that of non-tumorigenic cells. a Phase contrast photomicrographs show morphologic features of DMSO-treated (control) and QA-treated A549 human lung cancer cells. Cells were treated with the indicated dose of QA for 24 h. b Dose-dependent growth inhibition of A549 human lung cancer cells as determined by MTT assay. Cells were treated with the indicated doses of QA for 24 h. c MTT assay showing growth inhibition of A549 cells with the indicated doses of QA for 48 h. d MTT assay showing growth inhibition of A549 cells with the indicated doses of QA for 72 h. e MTT assay showing growth inhibition of MDA-MB-231 human breast cancer cells with the indicated doses of QA for 24 h. f Growth of non-tumorigenic MCF10A human breast epithelial cells is not inhibited by QA. Non-tumorigenic MCF-10A human breast epithelial cells were treated with increasing concentrations of QA for 24 h. Photomicrographs were captured using a phase contrast microscope
Next, we performed biochemical analyses to confirm that QA indeed induced cell death in cancer cells. Representative results shown in Fig. 3 indicate that QA induced caspases 8 and 3 activation (demonstrated by reduction in procaspase levels) and PARP cleavage (cleaved PARP, a marker of apoptosis) in MDA-MB-231 human breast cancer cells. Biochemical features of QA-induced cell death were also noted in A549 lung cancer cells and MCF-7 breast cancer cells (data not shown). Caspase 8 is an initiator caspase that is activated by death receptors. QA-induced caspase 8 activation suggested that QA appeared to have engaged extrinsic apoptotic pathway. Death receptors are important components of the extrinsic pathway of apoptosis. Therefore, we investigated QA effect on death receptor 5 (DR5) and found that QA indeed, upregulated DR5 protein levels in multiple cancer cell lines including A549 and H1299 human lung cancer cells, and MCF-7 and MDA-231 human breast cancer cells. Representative results for A549 lung cancer cells and MDA-MB-231 breast cancer cells shown in Fig. 4 indicate that QA upregulated DR5 protein levels as a function of time. We also investigated whether QA upregulation of DR5 protein levels was coupled with DR5 mRNA levels. Results for MCF-7 breast cancer cells (Fig. 5) indicate that QA also upregulated DR5 mRNA levels coupled with QA regulation of DR5 protein levels.

Biochemical features of cell death in QA-treated MDA-MB-231 human breast cancer cells. The cells were vehicle-treated or treated with the indicated doses of QA for 24 h. Western blot analyses were performed to detect the indicated molecules. Same blot was probed with the antibodies against the indicated molecules. Presented are representative results for MDA-MB-231 cells; biochemical features of QA-induced cell death were also noted in A549 and MCF-7 human lung and breast cancer cells, respectively

QA upregulates death receptor 5 (DR5) levels. A549 human lung cancer cells and MD-MB-231 human breast cancer cells were treated with 25 µM QA for the indicated periods of time. Western blot analyses were performed to detect DR5 using anti-DR5 antibodies. Same blot for each cell line was sequentially probed for DR5 and vinculin, the latter serving as a loading control. That QA reproducibly upregulated the steady state levels of DR5 was confirmed in multiple separate experiments. Here the effect of QA regulation of DR5 levels as a function of time is shown. The plotted values correspond to a representative experiment for each cell line, which indicate that QA had similar effect on DR5 levels in two different independently treated cell lines. The values are expressed as normalized DR5 band intensities, relative to their respective controls at 4 h. The 24-h values are plotted with respect to their corresponding controls at 24 h

a, b QA upregulates DR5 protein and mRNA levels in MCF-7 human breast cancer cells. Cells were treated with DMSO or QA (25 μM) and harvested at the indicated timepoints and DR5 protein and mRNA levels were determined by Western blotting and quantitative real-time PCR (qRT-PCR), respectively. The plotted values (panel b) represent mean ± SEM of triplicate measurements from each time point. *** p < 0.001. c QA enhances DR5 mRNA stability. MCF-7 human breast cancer cells were pretreated with QA (25 μM) or DMSO as a control for 24 h, and then treated with actinomycin D (4 µg/ml) for up to 4 h without changing the media. The cells were harvested at the indicated timepoints and DR5 mRNA levels were determined by quantitative real-time PCR (qRT-PCR). The plotted values represent mean ± SEM values of triplicate measurements from two independent experiments
We also sought to investigate the mechanism by which QA upregulated DR5 mRNA and protein levels. First, we performed DR5 promoter reporter assays and for that purpose, we used DR5 gene promoter fused to promoterless luciferase reporter (Supplementary Fig. 1A). A549 lung cancer cells were transiently transfected with DR5 gene promoter luciferase construct, and treated with QA for 24 h; control cells were treated with DMSO-vehicle. Our results indicated that QA did not increase DR5 promoter activity in A549 cells (Supplementary Fig. 1B). We have previously shown that thapsigargin (TG) upregulates DR5 mRNA and DR5 protein levels and TG also upregulated DR5 gene promoter activity [21]. Therefore, we used TG as a positive control in these experiments. As is shown (Supplementary Fig. 1B), TG upregulated DR5 gene promoter activity in A549 cells, a finding consistent with our previous report [21], however, induction of DR5 promoter activity was not noted in QA-treated cells. It was possible that QA could increase DR5 gene promoter activity in a different cell line and at an early timepoint. To investigate that possibility, we transiently transfected MCF-7 cells with DR5 promoter luciferase construct and treated them with QA for 4, 8 and 24 h, then performed luciferase assays. Our results indicated that QA also did not upregulate DR5 gene promoter in MCF-7 breast cancer cells. Representative results for 4- and 8-h timepoints are shown in Supplementary Fig. 1C.
These results suggested that QA-mediated DR5 mRNA upregulation did not correlate with concomitant regulation of DR5 promotor activity. Therefore, we further investigated whether QA altered DR5 mRNA stability and thus, upregulated DR5 mRNA via post-transcriptional mechanism(s). To that end, MCF7 cells were treated with QA for ~ 24 h to induce DR5 mRNA expression; the control cells were treated with DMSO-vehicle; cells were then treated with actinomycin D to block gene transcription and harvested at different timepoints thereafter. Our results in Fig. 5c indicate that QA-treated cells exhibited DR5 mRNA half-life of ~ 4 h, whereas vehicle-treated cells had DR5 mRNA half-life of ~ 2 h. These results indicate that QA upregulated DR5 levels, at least in part, by increasing DR5 mRNA stability.
Because QA also enhanced DR5 protein levels, we investigated QA effect on DR5 protein stability. To that end, MCF-7 and A549 cells were treated with QA for ~ 24 h; the control cells were treated with DMSO; cells were then treated with cycloheximide to stop protein synthesis and harvested at different timepoints. Representative results in Fig. 6 indicate that QA-treated cells exhibited increased DR5 protein half-life compared to the vehicle-treated cells. These results indicate that QA upregulates DR5 protein levels also by increasing DR5 protein stability.

QA enhances DR5 protein stability. MCF-7 human breast cancer cells were pretreated with DMSO or QA (25 μM) for 24 h. Then, the medium was replaced with the medium containing 25 µg/ml cycloheximide (CHX). Cells were harvested at the indicated timepoints and the protein levels of DR5 and vinculin were analyzed by Western blotting. a Images of representative experiment are shown. Similar results were obtained in four independent experiments. b The plotted values of relative normalized DR5 band intensities as mean ± SEM from four independent experiments. c The values showing QA enhances DR5 protein stability in A549 human lung cancer cells
TRAIL is the natural ligand for DR5 [22]. Given that QA upregulated DR5, next we investigated the biological significance of QA-mediated DR5 upregulation. We reasoned that because QA upregulates DR5 levels, QA-mediated cell death should be potentiated by concomitant treatment with TRAIL. Accordingly, we investigated the effects of QA in combination with TRAIL and for that purpose, MCF-7 and A549 cells were treated with QA and TRAIL either agent alone and in combination,control cells were treated with vehicle. Figure 7 shows morphological features of A549 lung cancer cells and MCF-7 breast cancer cells, and as is shown, TRAIL potentiated the anticancer effects of QA in both the lung and breast cancer cells.

TRAIL potentiates QA-mediated effects on breast and lung cancer cells. MCF-7 human breast cancer cells were treated with vehicle (DMSO), QA (15 µM), TRAIL (50 ng/ml) and the combination at these concentrations for 48 h. A549 human lung cancer cells were treated with vehicle (DMSO), QA (20 µM), TRAIL (50 ng/ml) and the combination at these concentrations for 24 h. Photomicrographs were captured using a phase contrast microscope. TRAIL potentiation of QA effects was also confirmed by MTT assays
Discussion
Cancer remains a major cause of morbidity and mortality and thus, newer and better therapeutic approaches are highly desirable. Biomedical research in the past decades has led to the development of a number of FDA-approved anticancer drugs; molecular targets of which have also been identified. These targets include kinases, cell surface proteins such as growth factor receptors and cell adhesion molecules, growth factors such as vascular endothelial growth factor (VEGF), poly ADP ribose polymerase (PARP), proteasomes and components of apoptotic machinery [23,24,25].
Plants are rich source of natural products of medicinal value. Many plants-derived anticancer drugs such as paclitaxel, camptothecin, etoposide, vinblastine and the like have proven effective in the clinic. Here, we have investigated the potential of QA, isolated from plant F. indica, as an anticancer therapeutic. Our results indicate that QA strongly suppressed the growth and viability of breast and lung cancer cells. QA was specific towards cancer cells as it did not inhibit growth and viability of non-tumorigenic cells. QA-mediated anticancer effects were associated with activation of cell death associated with caspases 8 and 3 activation, cleavage of PARP and upregulation of death receptor 5 (DR5). Apo2L/TRAIL, the DR5 ligand, also potentiated the anticancer effects of QA. From mechanistic standpoint, our results indicate that QA mediates its anticancer effects, partly, by engaging DR5-depentent pathway to induce cell death.
QA upregulated both mRNA and protein levels of DR5. Because QA also upregulated DR5 mRNA levels, its effect on DR5 gene promoter activity was investigated and the results indicated that QA did not induce DR5 gene promoter activity when tested in breast and lung cancer cells. However, QA treatment was associated with increased DR5 mRNA stability suggesting that QA-mediated increase in DR5 mRNA levels could occur, at least in part, via its effect on DR5 mRNA stability. We noted that QA effect on DR5 mRNA stability was evident until 4 h following actinomycin treatment; the effect after 4 h and until 6 h following actinomycin D treatment was not informative. Given that QA effect on steady state DR5 mRNA levels was more pronounced compared to the extent of its effect on DR5 mRNA stability, it is possible that QA may upregulate DR5 mRNA levels also via other mechanisms.
It is also possible that QA may increase DR5 mRNA production via epigenetic modification of the DR5 gene. Epigenetic changes, such as DNA methylation, histone modification and microRNA (miRNA) expression, could affect DR5 gene expression following treatment with QA. It is also possible that, QA may enhance DR5 gene transcription via response elements residing further upstream of the DR5 gene promoter region utilized in our study. Interestingly, QA also enhanced DR5 protein stability indicating that QA regulates DR5 levels via multifaceted mechanisms.
Lung cancer is the most common cancer is the world. In 2018, a total of 2,093,876 new cases of lung cancer were diagnosed for both men and women. With about 1.8 million deaths in 2018, lung cancer remains as one of the leading causes of deaths globally [26, 27]. In the US, 228,150 cases of lung cancer were expected to be diagnosed and 142,670 deaths occurring due to lung cancer for both genders in 2019. Thus, lung cancer was expected to account for 23–24% of cancer-related deaths in both men and women in 2019 [28]. Clearly, lung cancer remains a major cause of morbidity and mortality.
In the past decades, identification of driver mutations/alterations has led to the use of targeting agents, but this therapeutic strategy is useful for relatively smaller subset of patients. A large number of patients without targetable abnormalities cannot benefit from the targeted therapies. In the recent years, immunotherapies such as monoclonal antibodies against PD-1 or PD-L1 have been approved for the treatment of NSCLC [29, 30]. Although a promising approach, immunotherapies involving checkpoint inhibitors as monotherapy yield about 25–30% 5-year overall survival rate [31]. There is also the problem of relapse after initial response [30, 31]. Clearly, discovery and development of newer agents that can be used alone or in combination with existing therapeutics are highly desirable to manage lung cancer.
Breast cancer in women is the most common cancer in the world. In 2018, a total of 2,088,849 new cases of breast cancer were diagnosed, a figure that reflects 24.2% of all cancers diagnosed in women worldwide [27]. Breast cancer is also one of the common causes of cancer-related deaths in women globally. In the US on the other hand, 268,600 cases of breast cancer were expected to be diagnosed in 2019, a figure that corresponds to 30% of all cancers diagnosed in women [28]. In the US, although mortality due to breast cancer has declined since 1989, management of metastatic breast cancer remains challenging [28]. Thus, there is an urgent need to also develop novel therapeutic approaches for this debilitating and deadly disease.
In context to morbidity and mortality associated with lung and breast cancers, our results that QA strongly suppressed the growth and viability of breast and lung cancer cells are relevant and highlight its potential as a novel anticancer agent. QA is a ursane-type triterpene. Because of their hydrophobic nature, the water solubility of triterpenes is expected to be low. However, triterpenes are dietary constituents present in fruits and vegetables, and the health benefits of fruits and vegetables-derived triterpene are well-known. Given that oral delivery is generally desirable mode of drug administration, the oral absorption and bioavailability of QA can be investigated in the future and improved via various approaches involving nanocarriers. Furthermore, because QA upregulated DR5, Apo2L/TRAIL, the natural ligand for DR5, potentiated the effects of QA in breast and lung cancer cells. Apo2L/TRAIL (named as dulanermin), an important anticancer agent with selective toxicity towards cancer cells, is currently in clinical trials [32]. Recently, a phase III study reported the effectiveness and safety of Apo2L/TRAIL in combination with vinorelbine and cisplatin in advanced NSCLC patients [33]. It was found that Apo2L/TRAIL when combined with chemotherapy significantly improved progression-free survival and overall response rate [33]. Clearly, Apo2L/TRAIL can also be regarded as a promising agent particularly for use in the combination therapy. Based on our results, which indicate that the combination of QA and Apo2L/TRAIL exhibits stronger cancer cell killing effect than either agent alone, we propose that QA in combination with Apo2L/TRAIL can be developed as a novel therapeutic approach for breast and lung cancer.
References
Lichota A, Gwozdzinski K (2018) Anticancer activity of natural compounds from plant and marine environment. Int J Mol Sci 19:3533. https://doi.org/10.3390/ijms19113533
Amin A, Gali-Muhtasib H, Ocker M, Schneider-Stock R (2009) Overview of major classes of plant-derived anticancer drugs. Int J Biomed Sci 5:1–11
Cragg GM, Grothaus PG, Newman DJ (2009) Impact of natural products on developing new anti-cancer agents. Chem Rev 109:3012–3043. https://doi.org/10.1021/cr900019j
Cragg GM, Newman DJ (2004) A tale of two tumor targets: topoisomerase I and tubulin. The Wall and Wani contribution to cancer chemotherapy. J Nat Prod 67:232–244. https://doi.org/10.1021/np030420c
Lau W, Sattely ES (2015) Six enzymes from mayapple that complete the biosynthetic pathway to the etoposide aglycone. Science 349:1224–1228. https://doi.org/10.1126/science.aac7202
Shaker KH, Bernhardt M, Elgamal MH, Seifert K (1999) Triterpenoid saponins from Fagonia indica. Phytochemistry 51:1049–1053. https://doi.org/10.1016/s0031-9422(98)00750-x
Ullah N, Nadhman A, Siddiq S, Mehwish S, Islam A, Jafri L, Hamayun M (2016) Plants as antileishmanial agents: current scenario. Phytother Res 30:1905–1925. https://doi.org/10.1002/ptr.5710
Lam M, Carmichael AR, Griffiths HR (2012) An aqueous extract of Fagonia cretica induces DNA damage, cell cycle arrest and apoptosis in breast cancer cells via FOXO3a and p53 expression. PLoS ONE 7:e40152. https://doi.org/10.1371/journal.pone.0040152
Waheed A, Barker J, Barton SJ, Owen CP, Ahmed S, Carew MA (2012) A novel steroidal saponin glycoside from Fagonia indica induces cell-selective apoptosis or necrosis in cancer cells. Eur J Pharm Sci 47:464–473. https://doi.org/10.1016/j.ejps.2012.07.004
Jafri L, Saleem S, Calderwood D, Gillespie A, Mirza B, Green BD (2016) Naturally-occurring TGR5 agonists modulating glucagon-like peptide-1 biosynthesis and secretion. Peptides 78:51–58. https://doi.org/10.1016/j.peptides.2016.01.015
Saleem S, Jafri L, ul Haq I, Chang LC, Calderwood D, Green BD, Mirza B (2014) Plants Fagonia cretica L. and Hedera nepalensis K. Koch contain natural compounds with potent dipeptidyl peptidase-4 (DPP-4) inhibitory activity. J Ethnopharmacol 156:26–32. https://doi.org/10.1016/j.jep.2014.08.017
Ahmed I, Islam M, Arshad W, Mannan A, Ahmad W, Mirza B (2009) High-quality plant DNA extraction for PCR: an easy approach. J Appl Genet 50:105–107. https://doi.org/10.1007/BF03195661
Group CPW (2009) A DNA barcode for land plants. Proc Natl Acad Sci USA 106:12794–12797. https://doi.org/10.1073/pnas.0905845106
Gjoerup O (2018) Growth curve (crystal violet staining and quantification protocol). https://labs.mmg.pitt.edu/gjoerup/growth%20curve%20cryst%20violet.doc. Accessed 15 Dec 2018
An J, Shi J, He Q, Lui K, Liu Y, Huang Y, Sheikh MS (2012) CHCM1/CHCHD6, novel mitochondrial protein linked to regulation of mitofilin and mitochondrial cristae morphology. J Biol Chem 287:7411–7426. https://doi.org/10.1074/jbc.M111.277103
Luo X, Huang Y, Sheikh MS (2003) Cloning and characterization of a novel gene PDRG that is differentially regulated by p53 and ultraviolet radiation. Oncogene 22:7247–7257. https://doi.org/10.1038/sj.onc.1207010
Shi J, He Q, An J, Sun H, Huang Y, Sheikh MS (2009) Sulindac sulfide differentially induces apoptosis in Smac-proficient and -deficient human colon cancer cells. Mol Cell Pharmacol 1:92–97. https://doi.org/10.4255/mcpharmacol.09.11
Takimoto R, El-Deiry WS (2000) Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19:1735–1743. https://doi.org/10.1038/sj.onc.1203489
Lui K, An J, Montalbano J, Shi J, Corcoran C, He Q, Sun H, Sheikh MS, Huang Y (2013) Negative regulation of p53 by Ras superfamily protein RBEL1A. J Cell Sci 126:2436–2445. https://doi.org/10.1242/jcs.118117
Fox J (2005) Getting started with the R commander: a basic-statistics graphical user interface to R. J Stat Softw 14:1–42
He Q, Lee DI, Rong R, Yu M, Luo X, Klein M, El-Deiry WS, Huang Y, Hussain A, Sheikh MS (2002) Endoplasmic reticulum calcium pool depletion-induced apoptosis is coupled with activation of the death receptor 5 pathway. Oncogene 21:2623–2633. https://doi.org/10.1038/sj.onc.1205345
Huang Y, Sheikh MS (2007) TRAIL death receptors and cancer therapeutics. Toxicol Appl Pharmacol 224:284–289. https://doi.org/10.1016/j.taap.2006.12.007
Falzone L, Salomone S, Libra M (2018) Evolution of cancer pharmacological treatments at the turn of the third millennium. Front Pharmacol 9:1300. https://doi.org/10.3389/fphar.2018.01300
FDA NR (2019) FDA approves new type of therapy to treat advanced urothelial cancer. Mol Cell Pharmacol 11:10–11
Roy A, Wang QJ (2017) Protein kinase D: a potential therapeutic target in prostate cancer. Mol Cell Pharmacol 9:1–4
Jemal A, Torre L, Soerjomataram I, Bray F (2019) The cancer atlas, 3rd edn. American Cancer Society, Atlanta, GA, USA
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. https://doi.org/10.3322/caac.21492
Siegel RL, Miller KD, Jemal A (2019) Cancer statistics. CA Cancer J Clin 69:7–34. https://doi.org/10.3322/caac.21551
FDA NR (2018) FDA expands approval of imfinzi to reduce the risk of non-small cell lung cancer progressing. Mol Cell Pharmacol 10:1–2
Lilenbaum RC (2019) Overview of the initial treatment of advanced non-small cell lung cancer. In: West H, Vora SR (eds) UpToDate. Wolters Kluwer, Waltham, MA, USA. https://www.uptodate.com. Accessed 23 Oct 2019
Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn MJ, Eder JP, Balmanoukian AS, Aggarwal C, Horn L, Patnaik A, Gubens M, Ramalingam SS, Felip E, Goldman JW, Scalzo C, Jensen E, Kush DA, Hui R (2019) Five-year overall survival for patients with advanced nonsmall-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J Clin Oncol 37:2518–2527. https://doi.org/10.1200/JCO.19.00934
ClinicalTrial.gov (2020) https://clinicaltrials.gov/. Accessed 25 Jan 2020
Ouyang X, Shi M, Jie F, Bai Y, Shen P, Yu Z, Wang X, Huang C, Tao M, Wang Z, Xie C, Wu Q, Shu Y, Han B, Zhang F, Zhang Y, Hu C, Ma X, Liang Y, Wang A, Lu B, Shi Y, Chen J, Zhuang Z, Wang J, Huang J, Wang C, Bai C, Zhou X, Li Q, Chen F, Yu H, Feng J (2018) Phase III study of dulanermin (recombinant human tumor necrosis factor-related apoptosis-inducing ligand/Apo2 ligand) combined with vinorelbine and cisplatin in patients with advanced non-small-cell lung cancer. Investig New Drugs 36:315–322. https://doi.org/10.1007/s10637-017-0536-y
Acknowledgements
We thank Dr. Wafik el-Deiry (Brown University) for providing DR5 promoter-luciferase construct. Asma Umer Khayam was supported by a fellowship from Higher Education Commission, Islamabad, Pakistan. This work was supported, in part, by the Carol M. Baldwin Breast Cancer Research Fund and Michael E. Connolly Endowment for Lung Cancer Research, Upstate Cancer Center grant to Dr. M. Saeed Sheikh.
Author information
Authors and Affiliations
Contributions
AUK: Conceptualization; Investigation; Methodology; Validation; Writing—original draft; Writing—review & editing. HP: Investigation; Methodology; Validation; Writing—review & editing. NAF: Methodology; Validation; Writing—review & editing. AEFM: Methodology; Validation; Writing—review & editing. ED: Methodology; Validation; Writing—review & editing. BM: Resources; Supervision; Writing—review & editing. YH: Resources; Supervision; Writing—review & editing. MSS: Conceptualization; Investigation; Funding acquisition; Project administration; Resources; Supervision; Writing—original draft; Writing—review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts to declare in relation to the work described in this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11010_2020_3841_MOESM1_ESM.tif
Supplementary file1 Supplementary Figure 1 QA does not regulate DR5 gene promoter. (A) Schematic of DR5 gene promoter luciferase construct used in this study. (B) QA effect on DR5 promoter activity in A549 lung cancer cells. QA does not upregulate DR5 promoter. In the same experiments, cells were separately treated with thapsigargin (TG), used as a positive control that is known to upregulate DR5 promoter activity. As expected, TG upregulates DR5 promoter activity. QA or TG treatment was for 24 hours. (C) QA does not upregulate DR5 promoter activity in MCF-7 human breast cancer cells. Cells were transiently transfected with DR5 promoter luciferase construct. After ~24 hours, cells were treated with QA or vehicle control for the indicated periods of time. Relative luciferase activity was calculated and plotted as fold induction. The values are presented as mean ± SEM of three independent measurements. (TIF 69 kb)
Rights and permissions
About this article

Cite this article
Khayam, A.U., Patel, H., Faiola, N.A. et al. Quinovic acid purified from medicinal plant Fagonia indica mediates anticancer effects via death receptor 5. Mol Cell Biochem 474, 159–169 (2020). https://doi.org/10.1007/s11010-020-03841-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03841-4