
Abstract
Tumor cells increase glucose metabolism through glycolysis and pentose phosphate pathways to meet the bioenergetic and biosynthetic demands of rapid cell proliferation. The family of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB1-4) are key regulators of glucose metabolism via their synthesis of fructose-2,6-bisphosphate (F2,6BP), a potent activator of glycolysis. Previous studies have reported the co-expression of PFKFB isozymes, as well as the mRNA splice variants of particular PFKFB isozymes, suggesting non-redundant functions. Majority of the evidence demonstrating a requirement for PFKFB activity in increased glycolysis and oncogenic properties in tumor cells comes from studies on PFKFB3 and PFKFB4 isozymes. In this study, we show that the PFKFB2 isozyme is expressed in tumor cell lines of various origin, overexpressed and localizes to the nucleus in pancreatic adenocarcinoma, relative to normal pancreatic tissue. We then demonstrate the differential intracellular localization of two PFKFB2 mRNA splice variants and that, when ectopically expressed, cytoplasmically localized mRNA splice variant causes a greater increase in F2,6BP which coincides with an increased glucose uptake, as compared with the mRNA splice variant localizing to the nucleus. We then show that PFKFB2 expression is required for steady-state F2,6BP levels, glycolytic activity, and proliferation of pancreatic adenocarcinoma cells. In conclusion, this study may provide a rationale for detailed investigation of PFKFB2’s requirement for the glycolytic and oncogenic phenotype of pancreatic adenocarcinoma cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Reprogramming of glucose metabolism is recognized as a key feature of neoplastic transformation by proto-oncogenes such as KRAS and required for the maintenance of malignant properties [1, 2]. Tumor cells increase glucose uptake and metabolism via glycolysis to meet the bioenergetic and biosynthetic requirements of rapid cell division [3]. Preponderance of evidence suggests that while most tumor cells are capable of extracting sufficient energy through metabolism of alternative nutrients, e.g., glutamine [4], they still require an unhindered glycolytic flux to fuel biosynthetic pathways (e.g., nucleotide synthesis) [5], protect tumor cells from reactive oxygen species (through NADPH production via pentose phosphate pathway) [6], and modify immune cell metabolism [7] and microenvironment via lactate efflux [8, 9].
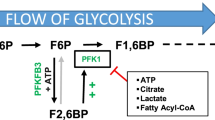
Regulation of the glycolytic activity is accomplished at several steps via multiple mechanisms. The phosphorylation of fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F1,6BP) by the 6-phosphofructo-1-kinase (PFK1) enzyme is an irreversible reaction that operates far from equilibrium and constitutes a critical control point in regulation of the glycolytic flux [10]. As an allosteric enzyme, PFK1 is subject to positive and negative regulation by a variety of glycolytic and non-glycolytic metabolites. A shunt product of glycolysis, fructose-2,6-bisphosphate (F2,6BP), functions as a key activator of glycolysis by antagonizing the allosteric inhibition of PFK1 by adenosine triphosphate (ATP) and citrate while simultaneously increasing PFK1’s affinity for its substrate, F6P [11]. Stimulation of the glycolytic activity by various oncogenic stimuli, including proto-oncogenes, e.g., Ras [12], growth factors, and transforming growth factor β1 [13], has been shown to coincide with increase in intracellular F2,6BP levels, supporting the notion that F2,6BP may play an essential role in the glycolytic phenotype of tumor cells. Intracellular F2,6BP levels are set by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) enzyme family, which have both kinase and phosphatase activities, i.e., they interconvert F6P and F2,6BP and are encoded by four genes, PFKFB1–4 [14, 15]. While in vitro and preclinical studies involving various tumor cell line and mouse xenograft models demonstrate a requirement for PFKFB activity for the glycolytic flux [16, 17] as well as cell proliferation [18], cell survival [19] and tumor growth [20], majority of the data demonstrating a requirement for PFKFB activity comes from studies on the third (PFKFB3) and fourth (PFKFB4) isozymes. Little is known about the expression and requirement of PFKFB2 isozyme in tumor cells.
In the current study, we show that PFKFB2 (i) is overexpressed in pancreatic adenocarcinomas; (ii) localizes to the nucleus; (iii) contributes to F2,6BP synthesis and glycolytic phenotype in pancreatic adenocarcinoma cells; and (iv) is required for proliferation of pancreatic adenocarcinoma cells.
Materials and methods
Cell culture
PANC-1 (ATCC; Cat.#CRL-1469), HeLa (ATCC; Cat.#CCL-2), A375 (ATCC; Cat.#CRL-1619), DU145 (ATCC; Cat.#HTB-81), MCF7 (ATCC; Cat.#HTB-22), and S2VP10 cells (a metastatic subclone of the SUIT-2 pancreatic adenocarcinoma cell line; a gift from Lacey McNally, Wake Forest School of Medicine, Winston-Salem, NC) were cultured in DMEM (Sigma) supplemented with 10% fetal bovine serum (FBS) (Gibco). MIA PaCa-2 (ATCC; Cat.#CRL-1420) cells were cultured in DMEM supplemented with 10% FBS and 2.5% horse serum. A549 (ATCC; Cat.#CCL-185) and BxPC-3 (ATCC; Cat.#CRL-1687) cells were cultured in RPMI-1640 supplemented with 10% FBS. HCT116 (ATCC; Cat.#CCL-247) cells were cultured in McCoy’s 5A media supplemented with %10 FBS. Cells were grown at 37 °C in 5% CO2.
Database analysis
The Oncomine database tool (www.oncomine.com) was used to compare PFKFB2 mRNA expression between normal and tumor samples. The following publicly available datasets were included in analyses: Cervix squamous cell carcinoma (GEO#: GSE9750), melanoma (GEO#: GSE3189), invasive ductal breast carcinoma (GEO#: GSE5764), and pancreatic adenocarcinoma (GEO#: GSE3654). Nucleotide and amino acid sequences, and mRNA splice variants of the human PFKFB2 gene were retrieved from the Ensembl genome browser (www.ensembl.org; PFKFB2: ENSG00000123836) and confirmed in NCBI database (www.ncbi.nlm.nih.gov) (NCBI Gene ID#5208).
Cloning
FLAG-tagged cDNA plasmids of PFKFB2 mRNA splice variant 1 (Origene; Cat.# RC211329) and variant 2 (Origene; Cat.# RC219676) were linearized with BamH1 and Pme1 enzymes and the resulting DNA fragments were then ligated into the BamH1/Pme1-cut pCMV6-A-BSD expression vector with the blasticidine resistance gene (bsr) (Origene; Cat.#PS100022).
siRNA and plasmid transfection
Two separate mammalian non-targeting control (ThermoFisher; Cat.#4390846 and AM4613) and two PFKFB2-specific siRNA molecules (ThermoFisher; Cat.#s10358 and 104811) were used in transfections. Transfection of siRNA molecules into cells was carried out using Lipofectamine RNAiMAX (ThermoFisher) following the manufacturer’s instructions. Cells were 40–50% confluent at the time of transfection. For ectopic expressions, pCMV6-A-BSD expression vector carrying FLAG-tagged PFKFB2 mRNA splice variants was transfected into cells using Lipofectamine 3000 reagent (ThermoFisher) following the manufacturer's instructions. Cells were 70–80% confluent at the time of transfection. For stable transfections, cells were treated with 5 μg/ml blasticidine (Sigma; Cat.#15205) for one week and maintained in 2 μg/ml blasticidine thereafter.
Cytoplasmic and nuclear fractionation
Nuclear and cytosolic fractions were extracted using the NE-PER Nuclear and Cytoplasmic Extraction Kit (ThermoFisher; Cat.#78833). The extraction procedure was conducted according to the manufacturer's protocol.
Quantitative PCR
RNA isolation and cDNA synthesis were performed using commercial kits (ThermoFisher; Cat.#K0731 and #4387406) following manufacturer’s instructions. Synthesized cDNA was used in PCR reactions to amplify splice variants using variant-specific primers. The primers used were as follows: PFKFB2 variant 1: 5′-GACTGCTGAATCTCTCGGGG-3′ (forward) and 5′-CCCTTCTTGCATGTCCTGGG-3′ (reverse); PFKFB2 variant 2: 5′-CCTCAAAGTGTGGACAAGCC-3′ (forward) and 5′-CAAGGCAACATGAGGGACG-3′ (reverse); and β-actin: 5′-AGAGCTACGAGCTGCCTGAC-3′ (forward) and 5′-AGCACTGTGTTGGCGTACAG-3′ (reverse). Resulting products were separated in 1% agarose gel electrophoresis and imaged by ChemiDocMP imaging system (BioRad, USA). Real-time quantitative (q) PCR was performed with StepOnePlus (ThermoFisher, NY, USA) to analyze total PFKFB2, vimentin and fibronectin mRNA expressions using TaqMan® probes for PFKFB2 (ThermoFisher; Cat.#Hs01015408_m1), Vimentin (ThermoFisher; Cat.#Hs00958111_m1), Fibronectin (ThermoFisher, Cat.#Hs01549976_m1) and β-actin (ThermoFisher; Cat.#Hs01060665_g1). β-actin was used as the internal control for normalization of cDNA.
Western blot
SDS-PAGE and Western blotting were performed following standard protocols. Primary antibodies specific to PFKFB2 (Abcam; Cat.#70175), PFKFB3 (Proteintech; Cat.#13763-1-AP), FLAG (DYKDDDDK) tag (Origene; Cat.#TA50011), Oct1 (Cell Signaling; Cat.#8157), β-actin (Cell Signaling; Cat.#3700), α-tubulin (Cell Signaling; Cat.#2125) and Histone H3 (Cell Signaling; Cat.#4499) proteins and HRP-conjugated goat anti-rabbit (Cell Signaling; Cat.#7074) or anti-mouse (Cell Signaling; Cat.#7076) secondary antibodies were used. Signals were detected using ECL prime (Amersham; Cat.#RPN2232) and bands were visualized in ChemiDocMP (BioRad).
Immunofluorescence and immunohistochemistry analyses
HeLa cells grown in chamber slides were transfected either of PFKFB2 variants and fixated with 4% formalin. An anti-FLAG primary (Origene; Cat.#TA50011) and an Alexa Fluor 488-conjugated goat anti-mouse secondary (Cell Signaling; Cat.#4408) antibodies were used to visualize the subcellular localization of PFKFB2. Cell silhouettes and nuclei were identified by rhodamine-conjugated phalloidin (which stains actin) (Cell Signaling; Cat.#8953) and DAPI (Cell Signaling; Cat.#8961), respectively. Images were acquired on Nikon A1R confocal microscope (NY, USA).
Immunohistochemical staining of the PFKFB2 protein was carried out on paraffin-embedded formalin-fixed samples (Biomax; Cat.#PA241d) using anti-PFKFB2 antibody (Millipore; Cat.#07-1530; 1/400 dilution, overnight incubation) as described elsewhere [21]. Detection of specific signal was achieved using SignalStain® Boost IHC detection reagent (Cell Signaling; Cat.#8114).
Cell proliferation
Cells were lifted with trypsin post-transfections at indicated times and stained with trypan blue for 1 min. Cells excluding trypan blue were counted under an inverted microscope (Accu-Scope, China) using a standard hemocytometer (Neubauer improved) to determine the total number of viable cells.
Fructose-2,6-bisphosphate analysis
Cells were lysed in 100 mM NaOH and 50 mM Tris–acetate buffer and then heated at 80 °C for 5 min. Lysates were neutralized with ice-cold acetic acid and HEPES. F2,6BP levels in supernatants were measured spectrophotometrically following a coupled enzyme method as described elsewhere [22], except that potato tuber pyrophosphate-dependent phosphofructokinase (PPi-PFK) was replaced by the hetero-hexameric PPi-PFK from Citrus sinensis obtained recombinantly to high purity as recently described [23]. F2,6BP levels were normalized to total protein.
Glycolytic activity and glucose uptake assays
Glucose uptake and glycolytic activity were determined as described elsewhere [16]. For glucose uptake, stably transfected cells and transiently transfected cells (48 h post-transfection) were incubated for 30 min in glucose-free media and for additional 15 min after adding 2-[1-14C]deoxyglucose (0.1 μCi/μl; Perkin-Elmer). Cells were then lysed in 500 μl 0.1% SDS and mixed with Microscint 40 scintillation fluid (Perkin-Elmer) and counts were measured on Tri-Carb 2910 liquid scintillation analyzer (Perkin-Elmer). Counts were normalized to protein concentration. For glycolytic activity, cells were incubated in medium supplemented with [5-3H]glucose (1 μCi; Perkin-Elmer) for 60 min. Separation of 3H2O formed by the enolase activity in glycolysis from medium was achieved by an evaporation technique in a sealed system. 3H2O formed was then measured on a liquid scintillation analyzer and compared to [5-3H]glucose and 3H2O standards. Counts were normalized to protein concentration.
Glucose and lactate levels in the media were measured colorimetrically using kits employing enzyme-based assays following the manufacturer’s protocol (Biovision; Cat. #K606-100 and K607-100).
Wound healing assay
Cells were plated into 6-well cell culture plates and allowed to grow for 24 h. When cells reached confluency, a scratch was made across the cell layer using a sterile pipette tip and the zone of scratch was visualized with an inverted microscope at 0,12, 24 and 48 h time points. Images were taken from three different zones per sample analyzed.
Colony formation assay
One thousand cells were seeded in 6-well plates and allowed to grow for one week. Cells were fixated with methanol in − 20 °C for 10 min and stained with 0.2% crystal violet. Colonies were visualized and evaluated both microscopically (10 × magnification) and macroscopically.
Statistical analyses
All experiments were performed three times. Data are expressed as the mean ± s.d. of triplicate measurements of a single experiment. Statistical significance was assessed by the two-tailed t tests and p < 0.05 was considered significant.
Results
PFKFB2 is expressed in tumor cell lines
Given the scarcity of studies on PFKFB2 expression levels in tumor cells, we first conducted a real-time qPCR analysis of PFKFB2 mRNA expression in established tumor cell lines derived from cervix (HeLa), lung (A549), skin (A375), breast (MCF7), colon (HCT116), prostate (DU145), and pancreas (PANC-1) tumors and found that all the tumor lines tested expressed PFKFB2 mRNA to various degrees (Fig. 1a), as well as PFKFB3 and PFKFB4 mRNAs (Fig S1). Western blot analysis confirmed the expression of PFKFB2 at protein levels in the tumor cells (Fig. 1b). In silico analysis of PFKFB2 mRNA expression in publicly available datasets using the Oncomine database (www.oncomine.com) demonstrates that PFKFB2 mRNA expression is upregulated in cervix squamous cell carcinoma (GEO#: GSE9750), melanoma (GEO#: GSE3189), invasive ductal breast carcinoma (GEO#: GSE5764), and pancreatic adenocarcinoma (GEO#: GSE3654) samples relative to normal tissues (Fig. 1c).

PFKFB2 expressed in tumors and cell lines. a Real-time qPCR analysis of PFKFB2 mRNA levels of indicated cell lines. b Western blot analysis of the PFKFB2 protein in indicated cell lines. β-actin was used as loading control. c Oncomine analysis of PFKFB2 mRNA expression in cervix squamous cell carcinoma, melanoma, breast carcinoma, and pancreatic adenocarcinoma. Fold changes represent relative PFKFB2 mRNA levels in tumor tissues relative to normal tissues. d Exon (E) structure of PFKFB2 mRNA splice variant 1 (PFKFB2-V1) and 2 (PFKFB2-V2) (upper left panel); PCR analysis using PFKFB2-V1 and PFKFB2-V2 plasmids as templates to show the specificity of primers that are used to analyze the expression of each variant in cell lines (upper right panel); qPCR analysis of PFKFB2-V1 and PFKFB2-V2 mRNA expressions in indicated cell lines. β-actin mRNA was used as housekeeping gene control (bottom panel)
Tumor cells co-express two PFKFB2 mRNA splice variants
Ensemble search of the PFKFB2 gene (Ensembl#ENSG00000123836) for alternative splice variants reveals that there exists two mRNA variants that encode full-length transcripts containing both the kinase and phosphatase domains, which are denoted as PFKFB2-202 and PFKFB2-201, and, in NCBI database, correspond to transcript variant 1 (NM_006212.2) and 2 (NM_001018053.1) (hereafter, referred to as PFKFB2-V1 and PFKFB2-V2, respectively). Although both variants contain 14 exons, they differ at the inclusion/exclusion of one exon at carboxy (C) terminus (Fig. 1d, upper left panel). Because tumor cells express one dominant mRNA splice variant of PFKFB3 as previously shown by us [18] and others [24], we sought to determine whether expression of either of the PFKFB2 variants also dominates in cell lines examined above. Since the Taqman probe which was used to analyze PFKFB2 mRNA expression in real-time qPCR does not distinguish between PFKFB2-V1 and PFKFB2-V2, we performed reverse transcription (RT) followed by PCR using primers that span the variable C terminus of the PFKFB2 mRNA. The PCR product was then run on 1% agarose. We first confirmed the specificity of the primers using commercially available plasmid vectors encoding PFKFB2-V1 and PFKFB2-V2 cDNAs (Fig. 1d, upper right panel). All cell lines examined express both variants to various degrees, with PFKFB2-V1 expression appears to be higher relative to PFKFB2-V2 (Fig. 1d, bottom panel), though we cannot rule out a difference in amplification efficiencies of the primers used. Nevertheless, results obtained so far suggest that both PFKFB2 are expressed in tumor cells and PFKFB2 mRNA is upregulated in several tumors, warranting efforts directed toward understanding if PFKFB2 serves an essential function in tumor cell metabolism and biology.
PFKFB2 is highly expressed in pancreatic adenocarcinoma cells and PFKFB2-V1 localizes to nucleus
Given the extensive reprogramming of glucose metabolism in pancreatic adenocarcinoma cells, as well as the association of the glycolytic phenotype with the aggressive and chemoresistant phenotype of pancreatic tumors [25, 26], for the remainder of the study, we have chosen pancreatic adenocarcinoma tumor lines as models to examine the expression and function of PFKFB2. We first started out analyzing PFKFB2 mRNA and protein expressions in pancreatic adenocarcinoma cell lines MIA PaCa-2, PANC-1, S2VP10, and BxPC-3 by real-time qPCR and Western blot, respectively. Based on relative threshold cycles (Ct) in real-time qPCR analysis, PFKFB2 mRNA expression is lowest in MIA PaCa-2 cells and highest in PANC-1 cells (Fig. 2a). We found that all examined cell lines express the PFKFB2 protein at levels comparable with, or even higher than, PFKFB3 (Fig. 2b, left panel), a PFKFB isoform that has been shown to be ubiquitously expressed across various types of tumors and cell lines, although we cannot rule out a difference in amplification efficiencies of PFKFB2 and PFKFB3 antibodies used. Consistent with the Western blot data, PFKFB2 was the highest expressed PFKFB isoform at mRNA level in PANC-1 cells (Fig S2). While the immunoreactivity of the antibody with both isoforms was verified by Western blot on MIA PaCa-2 cells that were transiently transfected with either of vectors encoding PFKFB2-V1 and PFKFB2-V2 cDNAs (Fig. 2b, right panel), with the conditions used (i.e., gel size, acrylamide %, antibody, etc.), we were not able to readily distinguish endogenously expressed PFKFB2-V1 and PFKFB2-V2 proteins in these cells, although careful examination of PFKFB2-immunoreactive bands in left and right panels in Fig. 2b, and given that PFKFB2-V2 should run faster compared to PFKFB2-V1 on the gel (three is ~ 3.4 kD difference) suggest that PFKFB2-V1 protein expression may dominate over PFKFKB2-V2 in MIA PaCa-2, PANC-1, and S2VP10 cells, whereas BxPC-3 cells may preferentially express PFKFB2-V2 at protein level. (Note: The exposure time that was used to detect only ectopically expressed PFKFB2 variants on the right panel in Fig. 2b was less than 2 s.) RT-PCR analysis of each isoform using specific primers corroborates this conclusion (Fig S3).

PFKFB2 highly expressed in pancreatic adenocarcinoma cells and PFKFB2-V1 localizes to nucleus. a Real-time qPCR analysis of PFKFB2 mRNA levels of indicated cell lines. b Western blot analysis of PFKFB2 and PFKFB3 proteins in indicated cells lines. β-actin was used as loading control (left panel); Western blot analysis of the PFKFB2 protein in MIA PaCa-2 cells transiently transfected with PFKFB2-V1 and PFKFB2-V2 plasmids using an antibody that recognizes both variants (right panel). c, Alignment of carboxy-terminal of PFKFB3, PFKFB2-V1 and PFKFB2-V2 proteins. pat4 nuclear localization signal (NLS) in PFKFB3 and PFKFB2-V1, and pat7 NLS in PFKFB2-V1 are shown in bold and boxed (upper panel); Confocal microscopy analysis of HeLa cells that were transfected with FLAG-PFKFB2-V1 and FLAG-PFKFB2-V2 plasmids using an anti-FLAG primary and Alexa Fluor 488 secondary antibodies. Cell silhouettes and nuclei were visualized by rhodamine-conjugated phalloidin and DAPI, respectively (bottom panel). d Cytoplasmic (CE) and nuclear extracts (NE) were isolated and subjected to Western blot analysis using antibodies to PFKFB2, Oct1 (a nuclear protein), and α-tubulin (a cytoplasmic protein) proteins
The oxidation of glucose to pyruvate followed by reduction to lactate takes place in cytosol necessitating the localization of the main and, presumably, accessory (i.e., regulatory molecules such as PFKFBs) glycolytic machinery to cytosol. However, studies consistently show both inducible and constitutive localization of enzymes involved in glycolysis to various compartments of the cells [27], supporting the hypothesis that they serve alternative functions that may be required to coordinate glucose metabolism and with other functions of the cells, e.g., cell cycle. Lending support to this hypothesis, we had previously demonstrated that the dominant mRNA splice variant of PFKFB3 partially localizes to the nucleus, where it regulates cell cycle [18, 28]. Trafficking of PFKFB3 protein to the nucleus is dictated by a stretch of amino acids at C terminus that constitutes a well-known nuclear localization signal (NLS) known as pat4—K(R/K)X(R/K) (X: K,R,P,V,A) [29]. Alignment of amino acid sequences of PFKFB2-V1 and PFKFB2-V2 with PFKFB3 demonstrate that PFKFB2-V1 carries a nuclear localization motif similar to PFKFB3, as well as a second NLS motif, PVRMRRN, known as pat7 (Fig. 2c, upper panel), suggesting it may also localize to the nucleus. First, to determine if PFKFB2-V1 and PFKFB2-V2 isoforms differ in their intracellular localization pattern, we transfected HeLa cells with a vector encoding FLAG-tagged PFKFB2-V1, PFKFB2-V2, or empty vector, and 48 h later, performed immunofluorescence analysis using an anti-FLAG primary followed by Alexa Fluor 488-conjugated secondary antibodies. Cell silhouettes and nuclei were identified by rhodamine-conjugated phalloidin (which stains actin) and DAPI, respectively. While ectopic PFKFB2-V1 isoform partially localized to the nucleus, PFKFB2-V2 isoform displayed a cytoplasmic pattern (Fig. 2c, bottom panel). To determine whether PFKFB2-V1 isoform is associated with the chromatin, a Western blot analysis was performed on cytoplasmic, nuclear, and chromatin fractions of the cells transfected with the same ectopic constructs. As shown in Fig S4, while Histone H3, a protein associated with chromatin, was enriched in the non-soluble fraction, PFKFB2-V1 was not detected in this fraction of cell extracts, indicating that PFKFB2-V1 is partially found in soluble fraction of the nucleus.
To determine the intracellular localization of the endogenous PFKFB2, we performed a Western blot analysis on cytoplasmic and nuclear extracts obtained from MIA PaCa-2, PANC-1, S2VP10, and BxPC-3 cell lines, and found that PFKFB2 protein expression was distributed in both compartments in all four cell lines (Fig. 2d). We next conducted an immunohistochemistry analysis on a tissue array containing human normal pancreas and pancreatic adenocarcinoma paraffin-embedded tissues, to assess intracellular localization of the endogenously expressed PFKFB2 protein in situ. We noticed that not only was there a higher level of immunostaining in tumor tissues relative to normal tissues, correlating with in silico PFKFB2 mRNA analysis in Fig. 1c, but also the PFKFB2 protein in nuclei of tumor tissues was substantially higher compared with those of normal tissues, indicating a differential intracellular localization of PFKFB2 in normal vs. tumor tissues (Fig. 3 and Fig S5). The specificity of the antibody was verified by an immunofluorescence analysis on PANC-1 cells transfected with either control or PFKFB2-specific siRNA molecules (Fig S6). Functional significance of PFKFB2 expression in the nucleus with regards to pancreatic tumor development and progression, however, remains to be determined.

PFKFB2 overexpressed and localizes to the nucleus in pancreatic adenocarcinoma tissues. Immunohistochemistry analysis of the PFKFB2 protein on a paraffin-embedded pancreatic adenocarcinoma and matched pancreas tissue array. 4 × and 40 × magnifications are shown. Arrows in insets in pancreatic adenocarcinoma samples show the PFKFB2 protein in the nucleus and, in matched pancreas, show exclusion of PFKFB2 from nuclei. H&E, Hematoxylin&Eosin
Transient expression of PFKFB2-V2 increases F2,6BP and glucose uptake in MIA PaCa-2 cells
We chose MIA PaCa-2 as a model to study the effect of each variant on F2,6BP, glucose uptake and glycolytic activity using an ectopic plasmid transfection approach, as, out of four pancreatic cell lines examined, MIA PaCa-2 cells express the PFKFB2 protein at lowest level (Fig. 2b). Previous studies [30, 31] show increased PFKFB2 expression by certain oncogenic stimuli, e.g., AKT, suggesting that activity of the kinase domain of the PFKFB2 protein may dominate over that of the phosphatase domain in tumor cells. Given the cytoplasm is the site of glycolytic activity and PFKFB2-V2 predominantly localizes to the cytoplasm, we therefore predicted that ectopic PFKFB2-V2 expression would result in an increase in F2,6BP, subsequently leading to glucose uptake/glycolytic activity. To test this, MIA PaCa-2 cells were transiently transfected with plasmids encoding FLAG-tagged PFKFB2-V1, PFKFB2-V2, or an empty vector and, 48 h later, increases in PFKFB2 mRNA and protein levels were confirmed by real-time qPCR (Fig. 4a) and Western blot (Fig. 4b) analyses, respectively. As predicted, ectopic PFKFB2-V2 expression resulted in an increase in F2,6BP levels (Fig. 4c, vector: 67 ± 2 pmol/mg protein; PFKFB2-V2: 285 ± 90 pmol/mg protein, p < 0.05) and glucose uptake (Fig. 4d, vector: 1288 ± 14 cpm/mg protein; PFKFB2-V2: 1436 ± 4 cpm/mg protein, p < 0.05) whereas PFKFB2-V1 expression did not affect glucose uptake or glycolysis, even though, it increased intracellular F2,6BP levels (Fig. 4c, PFKFB2-V1:155 ± 46 pmol/mg protein, p < 0.05), albeit, to a lesser degree compared with PFKFB2-V2. Similar results were obtained with PANC-1 cells (Fig S7). However, neither variant changed glycolytic activity in MIA PaCa-2 (Fig. 4e) and PANC-1 cells (Fig S7E).

Transient expression of PFKFB2-V2 increases F2,6BP and glucose uptake in MIA PaCa-2 cells. MIA PaCa-2 cells were transfected with either FLAG-tagged PFKFB2-V1 or PFKFB2-V2 plasmids, or an empty plasmid (vector) as control and grown for 48 h. a Real-time qPCR analysis of the PFKFB2 mRNA. b Western blot analysis of ectopically expressed PFKFB2-V1 and PFKFB2-V2 using an anti-FLAG antibody. β-actin was analyzed as loading control. c F2,6BP levels. d 2-[1-14C]deoxyglucose uptake. e Glycolytic activity as assessed by release of 3H2O from cells fed with [5-3H]glucose. *p < 0.05; **p < 0.01; ***p < 0.001
Transient silencing of PFKFB2 reduces F2,6BP, glucose uptake and glycolytic activity in PANC-1 and BxPC-3 cells
To determine whether PFKFB2 plays a role in determining the steady-state F2,6BP levels and glucose uptake/glycolytic activity, endogenously expressed PFKFB2 was depleted using the RNAi silencing approach. PANC-1 cells, which highly expresses PFKFB2, were transfected with combination of two control or PFKFB2-specific siRNAs using lipofectamine RNAiMAX as transfection reagent. We first validated the effect of used siRNAs in depleting PFKFB2 mRNA and protein levels by real-time qPCR (Fig. 5a) and Western blot (Fig. 5b) analyses, respectively. Targeting of both splice variants with the siRNA molecules used have been confirmed by RT-PCR (Fig S8A). We also confirmed the specificity of the siRNAs used for the PFKFB2 isoform relative to other co-expressed isoforms (PFKFB3 and PFKFB4) using real-time qPCR (Fig S8B). Compared to control siRNA (siNTC), PFKFB2 siRNA (siPF2) markedly reduced F2,6BP levels (Fig. 5c, siNTC: 85.5 ± 30.8 pmol/mg protein; siPF2: 26.0 ± 6.4 pmol/mg protein, p < 0.001), suggesting that steady-state PFKFB2 expression is required for F2,6BP production and the kinase:phosphatase activity ratio of this isoform is higher than “1” in these cells under the tested conditions. We next assessed the effect of PFKFB2 silencing on glucose uptake and glycolytic activity in PANC-1 cells as described above. We found that while PFKFB2 depletion had no significant effect on glucose consumption (Fig. 5d), it markedly reduced the glycolytic activity (Fig. 5e, siNTC: 48.0 ± 3.0 cpm/mg protein; siPF2: 33.3 ± 2.4 cpm/mg protein, p < 0.01). These data suggest that PFKFB2 serves a non-redundant function in setting the pace of glycolytic activity in PANC-1 cells. In BxPC-3 cells, however, drop in F2,6BP levels upon PFKFB2 silencing did not coincide with a decrease in the glycolytic activity as assessed by lactate production (Fig S9). Consistent with the low endogenous PFKFB2 protein expression, PFKFB2 silencing did not reduce F2,6BP levels, lactate production and glucose consumption in MIA PaCa-2 cells (data not shown).

PFKFB2 silencing reduces F2,6BP levels and glycolytic activity in PANC-1 cells. Cells were transiently transfected with PFKFB2 siRNA (siPF2) or non-targeting control siRNA (siNTC) molecules and allowed to grow for 48 h. a Real-time qPCR analysis of the PFKFB2 mRNA. b Western blot analysis of the PFKFB2 protein. β-actin was analyzed as loading control. c F2,6BP levels. d 2-[1-14C]deoxyglucose uptake. e Glycolytic activity as assessed by release of 3H2O from cells fed with [5-3H]glucose. *p < 0.01; **p < 0.001
Stable PFKFB2-V2 expression increases cell proliferation and migration in MIA PaCa-2 cells
Increased F2,6BP, glucose uptake and glycolytic activity have been linked to enhanced cellular proliferation [32] and oncogenic properties such as invasion [33]. Given that PFKFB2 splice variants had different effects on F2,6BP and glucose uptake in MIA PaCa-2 cells, we postulated that each variant would have non-overlapping effects on cell proliferation and oncogenic properties. Toward this end, we stably transfected MIA PaCa-2 cells with expression vectors carrying PFKFB2-V1 or PFKFB2-V2 and blasticidine as selection marker. Real-time qPCR and Western blot analyses confirmed the expected increases in PFKFB2 mRNA and protein (Fig. 6a, b). We confirmed that stable expression of PFKFB2 variants does not significantly change the expression of the co-expressed PFKFB isoforms, PFKFB3 and PFKFB4 (Fig S10). We measured cell proliferation using trypan blue-exclusion under microscope and found that PFKFB2-V2, but not PFKFB2-V1, strongly stimulated proliferation by 96 h (Fig. 6c), consistent with its effect on glucose uptake and F2,6BP production (Fig. 4). We next performed colony formation assays, to determine whether either variant would have any effect on the capacity of MIA PaCa-2 cells to grow as colonies when seeded at low density. While neither variant had any noticeable effect on colony number and size (Fig S11), we noticed that, under microscopic examination, cells expressing PFKFB2-V2 displayed a distinct phenotype: they appeared more mesenchymal and colonies grown exhibited a scattered and spreaded phenotype, and did not pile on top of each other, as was seen with vector and PFKFB2-V1 expressing cells (Fig. 6d). Given the association of mesenchymal phenotype with cell motility [34], we next compared migratory capacities of vector, PFKFB2-V1 and PFKFB2-V2 expressing cells using wound healing assays and found that cells expressing PFKFB2-V2 closed the wounds at markedly higher rate, compared to vector and PFKFB2-V1 expressing cells (Fig. 6e), indicating that PFKFB2-V2 potentiated the migratory capacity of MIA PaCa-2 cells. Of note, compared to vector and PFKFB2-V1 expressing cells, fewer PFKFB2-V2 cells (50%) were plated for wound healing assays, to rule out the higher proliferative capacity on migration. In fact, cell counts were comparable between clones at the end of wound healing assay (data not shown). Consistent with a mesenchymal phenotype, MIA PaCa-2 transfected with ectopic PFKFB2-V2 expressed fibronectin and vimentin at higher levels than PFKFB2-V1 and vector expressing cells (Fig. 6f). Ectopic expression of PFKFB2-V2 in PANC-1 cells, however, did not increase proliferation (data not shown). Taken together, these data suggest that ectopic expression of PFKFB2-V2 increases the proliferation of MIA PaCa-2 cells cultured as monolayers in 2D-culture, confers a distinct phenotype in colony formation, and induces the expression of mesenchymal markers.

Stable expression of PFKFB2-V2 increases the proliferation and migration of MIA Paca-2 cells. MIA PaCa-2 cells were stably transfected with either PFKFB2-V1 or PFKFB2-V2 plasmids, or an empty plasmid (vector) as control and selected in the presence of blasticidine. a Real-time qPCR analysis of the PFKFB2 mRNA. b Western blot analysis of ectopically expressed PFKFB2-V1 and PFKFB2-V2 using an anti-PFKFB2 antibody. β-actin was analyzed as loading control. c Cell counts at 0 (time of seeding), 24, 48, 72 and 92 h. d Microscopic pictures of cell colonies stained with crystal violet after seeding cells at low density. e Wound healing assay. f Real-time qPCR analysis of fibronectin and vimentin mRNA levels. Data are expressed as the mean ± s.d. of three measurements for A, C, F. *p < 0.05 compared to vector
PFKFB2 expression is required for cell proliferation and colony formation
We next aimed to determine whether the steady-state PFKFB2 expression is required for the proliferation and colony formation capacities of pancreatic adenocarcinoma cells. PFKFB2 expression was depleted using the RNAi approach as described above, and live cells were counted 24, 48, 72 and 96 h after transfection. While, initially, PFKFB2 siRNA led to a faster cell growth compared to control siRNA in PANC-1 cells, as assessed by cell numbers at 48 h (Fig. 7a, siNTC: 286,000 ± 28,400; siPF2: 396,000 ± 64,766 at 48 h, p < 0.05), this effect was lost by 72 h and reversed by 96 h of transfection (siNTC: 384,000 ± 80,000; siPF2: 288,000 ± 58,423 at 96 h, p < 0.05). The effect of PFKFB2 silencing on proliferation was more pronounced in BxPC-3 cells compared with PANC-1 cells, with significantly reduced proliferation in PFKFB2-depleted cells at 48, 72 and 96 h, relative to control siRNA transfected cells (Fig S12, p < 0.001). We next conducted colony formation assays to evaluate the effect of PFKFB2 depletion on the growth of PANC-1 as colonies from single cells. We found that PFKFB2 silencing almost completely abrogated the capacity of PANC-1 cells to grow as colonies in 2D-culture (Fig. 7b, c). However, transient PFKFB2 silencing had no effect on migratory and invasive capacities of PANC-1 cells as assessed by wound healing and Boyden chamber transwell invasion assays (data not shown). BxPC-3 cells did not survive the low density seeding in culture; therefore, we were unable to grow BxPC-3 cells as colonies. Consistent with its lack of effect on F2,6BP and the glycolytic phenotype, transient PFKFB2 silencing did not reduce the proliferation of MIA PaCa-2 cells (data not shown).

PFKFB2 silencing reduces the colony formation capacity of PANC-1 cells. a Cells were transiently transfected with PFKFB2 siRNA (siPF2) or non-targeting control siRNA (siNTC) molecules and 0 (time of transfection), 24, 48, 72, and 92 h later, live cells were counted. b Macroscopic pictures of cell colonies stained with crystal violet after seeding cells at low density 48 h post-transfection. c Image J analysis of colony counts. *p < 0.05; **p < 0.001
Discussion
Despite the relatively high number of studies on the expression and functional relevance of PFKFB3 and PFKFB4 isoforms of the PFKFB family in tumor cells, examination of PFKFB2 function in tumor cell metabolism and biology has not been initiated until recently. Our report, to our knowledge, is the first study to show that (1) the two mRNA splice variants of PFKFB2 (PFKFB2-V1 and PFKFB2-V2) that encode functional proteins with intact kinase and phosphatase domains are expressed in multiple tumor cell lines; (2) PFKFB2-V1 is partially expressed in the soluble fraction of the nucleus; (3) the PFKFB2 protein is overexpressed and predominantly localizes to the nucleus in the pancreatic adenocarcinoma tissue relative to adjacent normal pancreatic tissue; and (4) PFKFB2 expression is required for the steady-state F2,6BP concentration, glycolytic activity, and the growth of PANC-1 cells as colonies in 2D-culture.
Studies consistently demonstrate the co-expression of PFKFB isoforms, PFKFB3 and PFKFB4 in particular [35, 36], in tumor cells, indicating non-redundant functions. Our current report demonstrates that tumor cell lines of various origin, including cervix, lung, breast, and pancreas, express PFKFB2 both at mRNA and protein levels. Combined with results from recent studies demonstrating the expression of the PFKFB2 protein in osteosarcoma [37], gastric carcinoma [38], and melanoma [39] cells, these data indicate that PFKFB2 is widely expressed across multiple tumor cell lines of various origin. Majority of the cell lines examined in this study and other studies are also known to express PFKFB3 and/or PFKFB4 [13, 40, 41], supporting the hypothesis that each PFKFB isozyme may serve a unique and non-redundant function in tumor cells. Lending support to this hypothesis, we [18] and others [28, 42] showed that the variant of PFKFB3 expressed in the nucleus is involved in regulation of the cell cycle and proliferation. Evidence suggests that PFKFB3 shuttles between the cytosol and nucleus, depending on the particular needs of a cell in a given context. For example, DNA damage promotes the acetylation of PFKFB3 at Lys472, leading to its cytoplasmic retention and subsequent glycolytic activation that is required for protection against apoptosis induced by DNA damage [43]. Whether PFKFB2 also shuttles in and out of the nucleus in response to various stimuli is currently unknown. Nevertheless, in human pancreatic adenocarcinoma tissues, the PFKFB2 protein appears to mainly localize to the nucleus, whereas it is largely cytoplasmic in the adjacent normal pancreatic tissue, suggesting a role for PFKFB2 in the nucleus for malignancy. Whether absolute levels of PFKFB2 protein expression or its activity is required for pancreatic tumorigenesis has not been evaluated in this study.
PFKFB family of enzymes have distinct kinase:phosphatase (K:P) activity ratios and PFKFB2 was shown to display nearly equal kinase and phosphatase activities [44]. PFKFB3 is presumed to be the main contributor to intracellular F2,6BP, the product of the kinase function, due to its extremely high relative kinase activity (K:P = ~ 760). Contrasting with these notions, we find that transient PFKFB2 depletion caused a marked reduction in F2,6BP levels in PANC-1 (Fig. 5c) and BxPC-3 cells (Fig S4C), both of which also highly express PFKFB3 [13, 45]. This finding suggests that the kinase function of the PFKFB2 isozyme dominates in these cells and rivals that of PFKFB3. This result is in agreement with studies demonstrating the increased kinase activity of PFKFB2 by oncogenic signals such as AKT [31] and p90 ribosomal S6 kinase (RSK) downstream MAPK [39] through post-translational modifications. Considering that the published K:P levels are based on in vitro measurements using bacterial recombinant proteins and the fact that post-translational modifications in eucaryotic cells can drastically change enzymatic activity, we suggest that relative importance of a particular isozyme in dictating F2,6BP levels and whether the lack of a particular isozyme can be compensated by another in a given cell type or tumor should be determined by genetic approaches.
We found that ectopically expressed of PFKFB2-V2 increased F2,6BP and glucose uptake more potently than PFKFB2-V1 in the pancreatic adenocarcinoma cell lines MiaPaCa-2 and PANC-1. The predominant localization of PFKFB2-V2 to the cytosol—the site of glycolytic activity where PFKFB2-V2 may have better access to its substrate, F6P—as opposed to the nuclear localization of PFKFB2-V1, may have led to the observed differences. Consistent with this notion, we had previously showed that cytoplasmically re-routed PFKFB3 increased F2,6BP and glycolytic activity more potently than the nuclear PFKFB3 [18]. However, we cannot rule out a potential difference in intrinsic K:P activity of each PFKFB2 splice variant. In the current study, increase in F2,6BP by ectopic PFKFB2-V2 coincided with an increase in glucose uptake but not with glycolytic activity, suggesting that additional factors may be required to further stimulate glycolytic activity in these glycolytically active cells. Depletion of the endogenous PFKFB2 protein in PANC-1 cells, however, reduced both glucose uptake and glycolytic activity, suggesting that the steady-state PFKFB2 expression is required to sustain glycolytic activity. The PFKFB4 isozyme, which is solely expressed in the cytosol [18], has been shown to increase glucose utilization via the pentose phosphate pathway [46], which runs parallel to glycolysis and essential to synthesis of nucleotides required for cell proliferation. Although our observations suggesting PFKFB2 expression affects glucose uptake, glycolysis as well as cell proliferation may provide a rationale for detailed interrogation of PFKFB2 with respect to its potential effect on glycolysis and pentose phosphate pathways, future studies should incorporate metabolic flux studies by NMR and MS using labeled glucose analogs (e.g., uniformly labeled glucose ([U-13C6]-glucose) and [1,2-13C2]-glucose) that can distinguish glycolytic and pentose phosphate pathway activities, in order to reach a firm conclusion on the requirement of PFKFB2 on the regulation of glucose metabolism via respective pathways.
While transient PFKFB2 silencing in PANC-1 cells led to a modest decrease in proliferation, it almost completely abrogated the colony growth upon seeding cells at low density in 2D-culture. In BxPC-3 cells, the effect of transient PFKFB2 silencing on cell proliferation was robust. Taken together, these data suggest that PFKFB2 is required pancreatic adenocarcinoma cell proliferation and add to the recent literature demonstrating the requirement of PFKFB2 for the proliferation of melanoma cells [39] and gastric carcinoma cells [38]. Relative importance of endogenously expressed splice variants in tumor glucose metabolism and oncogenic properties such as proliferation is currently unknown and has not been pursued in the current study. Given the co-expression of the nuclear (PFKFB2-V1) and cytoplasmic (PFKFB2-V2) variants in the examined cell lines, an intriguing possibility is that PFKFB2-V2 may function to increase/maintain glycolytic activity in the cytoplasm, whereas PFKFB2-V1 may serve a seemingly non-metabolic function in the nucleus, such as regulation of the cell cycle, that may be essential to integration of glucose metabolism with malignant properties under nutrient-replete conditions. Dissecting the role of each variant in tumor cell biology will require specifically silencing one variant at a time and/or restoring each variant in PFKFB2-depleted cells.
Although this study is far from providing a mechanistical details as to the nature of PFKFB2’s role in tumor cell biology, we postulate that while, owing to the cytoplasmic localization, PFKFB2-V2 is a key contributor to the steady-state levels of F2,6BP in the cytoplasm where it may play in the reprogramming of glucose metabolism and that, by localizing to the nucleus, PFKFB2-V1 may function to coordinate glucose metabolism with proliferation. Given the extensive metabolic alterations and unique metabolic addictions adopted by pancreatic adenocarcinoma [25], comprehensive metabolomics and transcriptional effects of PFKFB2 inhibition via genetic and pharmacological approaches may reveal vulnerabilities that can be exploited for the development of therapeutics against pancreatic adenocarcinoma.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Abbreviations
- F6P:
-
Fructose-6-phosphate
- F1,6BP:
-
Fructose-1,6-bisphosphate
- F2,6BP:
-
Fructose-2,6-bisphosphate
- PFK1:
-
6-Phosphofructo-1-kinase
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- PFKFB:
-
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase
- PPi-PFK:
-
Pyrophosphate-dependent phosphofructokinase
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149:656–670. https://doi.org/10.1016/j.cell.2012.01.058
DeBerardinis RJ, Chandel NS (2016) Fundamentals of cancer metabolism. Sci Adv 2:e1600200. https://doi.org/10.1126/sciadv.1600200
Chiaradonna F, Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Stephanopoulos G, Alberghina L (2011) Oncogenic K-ras decouples glucose and glutamine metabolism to support cancer cell growth. FEBS J 278:199–199
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. https://doi.org/10.1126/science.1160809
Sharma PK, Bhardwaj R, Dwarakanath BS, Varshney R (2010) Metabolic oxidative stress induced by a combination of 2-DG and 6-AN enhances radiation damage selectively in malignant cells via non-coordinated expression of antioxidant enzymes. Cancer Lett 295:154–166. https://doi.org/10.1016/j.canlet.2010.02.021
Kim J (2018) Regulation of immune cell functions by metabolic reprogramming. J Immunol Res 2018:8605471. https://doi.org/10.1155/2018/8605471
Li X, Yu X, Dai D, Song X, Xu W (2016) The altered glucose metabolism in tumor and a tumor acidic microenvironment associated with extracellular matrix metalloproteinase inducer and monocarboxylate transporters. Oncotarget 7:23141–23155. https://doi.org/10.18632/oncotarget.8153
Pertega-Gomes N, Baltazar F (2014) Lactate transporters in the context of prostate cancer metabolism: what do we know? Int J Mol Sci 15:18333–18348. https://doi.org/10.3390/ijms151018333
Tanner LB, Goglia AG, Wei MH, Sehgal T, Parsons LR, Park JO, White E, Toettcher JE, Rabinowitz JD (2018) Four key steps control glycolytic flux in mammalian cells. Cell Syst 7(49–62):e8. https://doi.org/10.1016/j.cels.2018.06.003
Bartrons R, Simon-Molas H, Rodriguez-Garcia A, Castano E, Navarro-Sabate A, Manzano A, Martinez-Outschoorn UE (2018) Fructose 2,6-bisphosphate in cancer cell metabolism. Front Oncol 8:331. https://doi.org/10.3389/Fonc.2018.00331
Telang S, Yalcin A, Clem AL, Bucala R, Lane AN, Eaton JW, Chesney J (2006) Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene 25:7225–7234. https://doi.org/10.1038/sj.onc.1209709
Yalcin A, Solakoglu TH, Ozcan SC, Guzel S, Peker S, Celikler S, Balaban BD, Sevinc E, Gurpinar Y, Chesney JA (2017) 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor beta1-enhanced invasion of Panc1 cells in vitro. Biochem Biophys Res Commun 484:687–693. https://doi.org/10.1016/j.bbrc.2017.01.178
Chesney J (2006) 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell glycolysis. Curr Opin Clin Nutr Metab Care 9:535–539. https://doi.org/10.1097/01.mco.0000241661.15514.fb
Okar DA, Manzano A, Navarro-Sabate A, Riera L, Bartrons R, Lange AJ (2001) PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem Sci 26:30–35
Imbert-Fernandez Y, Clem BF, O'Neal J, Kerr DA, Spaulding R, Lanceta L, Clem AL, Telang S, Chesney J (2014) Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J Biol Chem. https://doi.org/10.1074/jbc.M113.529990
De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154:651–663. https://doi.org/10.1016/j.cell.2013.06.037
Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, Brock E, Siow D, Wattenberg B, Telang S, Chesney J (2009) Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem 284:24223–24232. https://doi.org/10.1074/jbc.M109.016816
Klarer AC, O'Neal J, Imbert-Fernandez Y, Clem A, Ellis SR, Clark J, Clem B, Chesney J, Telang S (2014) Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab 2:2. https://doi.org/10.1186/2049-3002-2-2
Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Lane A, Trent JO, Chesney J (2008) Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther 7:110–120. https://doi.org/10.1158/1535-7163.MCT-07-0482
Helm J, Coppola D, Ganapathy V, Lloyd M, Centeno BA, Chen DT, Malafa MP, Park JY (2012) SLC5A8 nuclear translocation and loss of expression are associated with poor outcome in pancreatic ductal adenocarcinoma. Pancreas 41:904–909. https://doi.org/10.1097/MPA.0b013e31823f429f
Van Schaftingen E, Lederer B, Bartrons R, Hers HG (1982) A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur J Biochem 129:191–195. https://doi.org/10.1111/j.1432-1033.1982.tb07039.x
Muchut RJ, Piattoni CV, Margarit E, Tripodi KEJ, Podestá FE, Iglesias AA (2019) Heterologous expression and kinetic characterization of the α, β and αβ blend of the PPi-dependent phosphofructokinase from Citrus sinensis. Plant Sci 280:348–354. https://doi.org/10.1016/j.plantsci.2018.12.012
Zscharnack K, Kessler R, Bleichert F, Warnke JP, Eschrich K (2009) The PFKFB3 splice variant UBI2K4 is downregulated in high-grade astrocytomas and impedes the growth of U87 glioblastoma cells. Neuropathol Appl Neurobiol 35:566–578. https://doi.org/10.1111/j.1365-2990.2009.01027.x
Blum R, Kloog Y (2014) Metabolism addiction in pancreatic cancer. Cell Death Dis 5:e1065. https://doi.org/10.1038/cddis.2014.38
Wang F, Liu H, Hu L, Liu Y, Duan Y, Cui R, Tian W (2018) The Warburg effect in human pancreatic cancer cells triggers cachexia in athymic mice carrying the cancer cells. BMC Cancer 18:360–360. https://doi.org/10.1186/s12885-018-4271-3
Boukouris AE, Zervopoulos SD, Michelakis ED (2016) Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription. Trends Biochem Sci 41:712–730. https://doi.org/10.1016/j.tibs.2016.05.013
Tudzarova S, Colombo SL, Stoeber K, Carcamo S, Williams GH, Moncada S (2011) Two ubiquitin ligases, APC/C-Cdh1 and SKP1-CUL1-F (SCF)-beta-TrCP, sequentially regulate glycolysis during the cell cycle. Proc Natl Acad Sci USA 108:5278–5283. https://doi.org/10.1073/pnas.1102247108
Kosugi S, Hasebe M, Matsumura N, Takashima H, Miyamoto-Sato E, Tomita M, Yanagawa H (2009) Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J Biol Chem 284:478–485. https://doi.org/10.1074/jbc.M807017200
Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ, Kim JW, Park SW, Kim KS (2011) Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J 433:225–233. https://doi.org/10.1042/BJ20101104
Novellasdemunt L, Tato I, Navarro-Sabate A, Ruiz-Meana M, Mendez-Lucas A, Perales JC, Garcia-Dorado D, Ventura F, Bartrons R, Rosa JL (2013) Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) isoenzyme by amino acids. J Biol Chem 288:10640–10651. https://doi.org/10.1074/jbc.M113.455998
Cordero-Espinoza L, Hagen T (2013) Increased concentrations of fructose 2,6-bisphosphate contribute to the Warburg effect in phosphatase and tensin homolog (PTEN)-deficient cells. J Biol Chem 288:36020–36028. https://doi.org/10.1074/jbc.M113.510289
Du JY, Wang LF, Wang Q, Yu LD (2015) miR-26b inhibits proliferation, migration, invasion and apoptosis induction via the downregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 driven glycolysis in osteosarcoma cells. Oncol Rep 33:1890–1898. https://doi.org/10.3892/or.2015.3797
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP (2009) Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 15:416–428. https://doi.org/10.1016/j.ccr.2009.03.016
Minchenko OH, Opentanova IL, Ogura T, Minchenko DO, Komisarenko SV, Caro J, Esumi H (2005) Expression and hypoxia-responsiveness of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 in mammary gland malignant cell lines. Acta Biochim Pol 52:881–888
Houddane A, Bultot L, Novellasdemunt L, Johanns M, Gueuning MA, Vertommen D, Coulie PG, Bartrons R, Hue L, Rider MH (2017) Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell Signal 34:23–37. https://doi.org/10.1016/j.cellsig.2017.02.019
Zhao SJ, Shen YF, Li Q, He YJ, Zhang YK, Hu LP, Jiang YQ, Xu NW, Wang YJ, Li J, Wang YH, Liu F, Zhang R, Yin GY, Tang JH, Zhou D, Zhang ZG (2018) SLIT2/ROBO1 axis contributes to the Warburg effect in osteosarcoma through activation of SRC/ERK/c-MYC/PFKFB2 pathway. Cell Death Dis 9:390. https://doi.org/10.1038/s41419-018-0419-y
Liu H, Chen K, Wang L, Zeng X, Huang Z, Li M, Dong P, Chen X (2019) miR-613 inhibits Warburg effect in gastric cancer by targeting PFKFB2. Biochem Biophys Res Commun 515:37–43. https://doi.org/10.1016/j.bbrc.2019.05.001
Houles T, Gravel SP, Lavoie G, Shin S, Savall M, Meant A, Grondin B, Gaboury L, Yoon SO, St-Pierre J, Roux PP (2018) RSK regulates PFK-2 activity to promote metabolic rewiring in melanoma. Cancer Res 78:2191–2204. https://doi.org/10.1158/0008-5472.CAN-17-2215
Minchenko OH, Ochiai A, Opentanova IL, Ogura T, Minchenko DO, Caro J, Komisarenko SV, Esumi H (2005) Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie 87:1005–1010. https://doi.org/10.1016/j.biochi.2005.04.007
Minchenko OH, Tsuchihara K, Minchenko DO, Bikfalvi A, Esumi H (2014) Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol 20:13705–13717. https://doi.org/10.3748/wjg.v20.i38.13705
Jia W, Zhao X, Zhao L, Yan H, Li J, Yang H, Huang G, Liu J (2018) Non-canonical roles of PFKFB3 in regulation of cell cycle through binding to CDK4. Oncogene 37:1685–1698. https://doi.org/10.1038/s41388-017-0072-4
Li F-L, Liu J-P, Bao R-X, Yan G, Feng X, Xu Y-P, Sun Y-P, Yan W, Ling Z-Q, Xiong Y, Guan K-L, Yuan H-X (2018) Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat Commun 9(1):508. https://doi.org/10.1038/s41467-018-02950-5
Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L (2004) 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J 381:561–579. https://doi.org/10.1042/BJ20040752
Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA 2nd, Klarer AC, Redman R, Miller DM, Trent JO, Telang S, Chesney J (2013) Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 12:1461–1470. https://doi.org/10.1158/1535-7163.MCT-13-0097
Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, Zamboni N, Schulze A (2012) Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov 2:328–343. https://doi.org/10.1158/2159-8290.CD-11-0234
Acknowledgements
This study was supported by the Scientific and Technological Research Council of Turkey (TUBITAK#114Z496). SCO was the recipient of a predoctoral training grant from TUBITAK (#2214).
Author information
Authors and Affiliations
Contributions
SCO and AY designed the experiments; SCO, AS, THA, SG, YG and ALC carried out cell culture, gene expression and protein analyses; SCO and AY conducted immunofluorescence analyses; AA and SG performed immunohistochemistry analyses; RJM and AAI isolated and tested PPi-PFK activity; SCO, THA and AY performed F2,6BP analyses; SCO and YIF carried out glucose uptake and glycolytic activity analyses; JAC revised the manuscript critically; AY and SCO wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ozcan, S.C., Sarioglu, A., Altunok, T.H. et al. PFKFB2 regulates glycolysis and proliferation in pancreatic cancer cells. Mol Cell Biochem 470, 115–129 (2020). https://doi.org/10.1007/s11010-020-03751-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03751-5