
Abstract
Hyperglycemia in diabetes causes protein glycation that leads to oxidative stress, release of cytokines, and establishment of secondary complications such as neuropathy, retinopathy, and nephropathy. Several other metabolic disorders, stress, and inflammation generate free radicals and oxidative stress. It is essential to study whether oxidative stress independently enhances protein glycation leading to rapid establishment of secondary complications. Oxidative stress was experimentally induced using rotenone and Fenton reagent for in vivo and in vitro studies, respectively. Results showed significant increase in the rate of modification of BSA in the form of fructosamine and protein-bound carbonyls in the presence of fenton reagent. Circular dichroism studies revealed gross structural changes in the reduction of alpha helix structure and decreased protein surface charge was confirmed by zeta potential studies. Use of rotenone demonstrated enhanced AGE formation, ROS generation, and liver and kidney tissue glycation through fluorescence measurement. Similar findings were also observed in cell culture studies. Use of aminoguanidine, a protein glycation inhibitor, demonstrated reduction in these changes; however, a combination of aminoguanidine along with vitamin E demonstrated better amelioration. Thus, oxidative stress accelerates the process of protein glycation causing gross structural changes and tissue glycation in insulin-independent tissues. Use of antioxidants and protein glycation inhibitors in combination are more effective in preventing such changes and could be an effective therapeutic option for preventing establishment of secondary complications of diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protein glycation, oxidative stress, and inflammation have been implicated in causing secondary complications in diabetes, initiating nephropathy, neuropathy, retinopathy, and cardiac disorders. However, the time taken for such changes to occur varies from person to person and depends upon several factors such as diet stress and sedentary lifestyles. While protein glycation stems mainly through hyperglycemia and is reported to cause oxidative stress through gluco-oxidation and protein oxidation, oxidative stress can be an independent component arising due to a number of other reasons. It is important to assess whether this stress exacerbates the process of protein glycation, causing rapid acceleration towards tissue damage in diabetes. Such a study is vital in understanding the role of oxidative stress in diabetes and whether avoidance of such stress can delay progression to secondary complications of diabetes.
Protein glycation is a non-enzymatic reaction between reducing sugars and free amino groups of proteins [1] through Schiff base formation. It proceeds through Amadori rearrangement which leads to the formation of Advanced Glycation End-products—AGEs like carboxy methyl lysine, carboxy ethyl lysine, glyoxal lysine dimer, and methyl glyoxal lysine dimer through several types of reactions, such as rearrangement, dehydration, condensation, fragmentation, oxidation, and cyclization reactions [2, 3]. The products formed cause changes in molecular conformation of proteins, alter enzymatic activity, reduce their degradative capacity, and result in abnormal recognition and clearance by receptors [3,4,5,6].
AGEs are known to initiate signaling cascade to induce the expression of pro-inflammatory mediators and elicit oxidative stress, causing cell damage, which contributes to the diabetic complications [7]. The process of AGEs formation is a complex process involving multiple steps [8]. Several AGEs inhibitors are studied that can interfere in these processes and prevent AGEs formation [9]. Aminoguanidine is a well-known standard glycation inhibitor which inhibits protein glycation by competition, scavenging dicarbonyls, and antioxidant activity by virtue of its guanidino group [10].
It is believed that oxidative stress plays an important role in the development of vascular complications in diabetes. Generation of oxidative stress is a result of overproduction of free radicals like O−, OH, H2O2, and ONOO− in a cellular system (mainly in mitochondria). If cellular antioxidants fail to remove these free radicals, it may lead to radical attack resulting in damage of proteins, lipids, and nucleic acids. Free radical formation in diabetes through non-enzymatic glycation of proteins, glucose oxidation, and increased lipid peroxidation is reported to damage enzymes, cellular machinery, and also increases insulin resistance [11]. Level of ROS elevation may be due to decreased scavenging or the production by the antioxidant enzymes like catalase, superoxide dismutase, and glutathione peroxidase and could be independent of diabetes.
Earlier Fenton reaction has been used to generate oxidative stress in Parkinson’s disease [12]. Rotenone is also a well-characterized, high-affinity specific inhibitor of complex-I of the inner mitochondrial membrane involved in oxidative phosphorylation used for induction of oxidative stress in rat [13]. These agents can be effectively used to induce oxidative stress experimentally. Likewise, different dietary vitamins are used as antioxidant to reduce ROS level. Vitamin E has been more broadly examined in diabetes models and found to prevent diabetes-induced changes [14].
Although it is well established that glycation of proteins leads to oxidative stress, whether oxidative stress favors rapid protein glycation accelerating the process needs to be established. Therefore, the present study attempts to evaluate the role of oxidative stress in progression of protein glycation in terms of fructosamine, protein-bound carbonyl formation, and tissue AGE formation through in vivo and in vitro studies. Evaluation of their effect on protein structural changes is possible. It also attempts use of antioxidants vis-à-vis protein glycation inhibitor and their relative effect in preventing protein glycation and thereby progression to secondary complications of diabetes.
Materials and methods
Chemicals and reagents
Bovine serum albumin fatty acid free, Aminoguanidine hydrochloride, 2′,7′-dichlorofluorescin diacetate (DCF-DA), Rotenone were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium azide, Nitroblue tetrazolium (NBT), Hydrogen peroxide, Vitamin E (alpha tocopherol), and other chemicals of analytical grades were supplied by Himedia (Mumbai), Huh7 cell line was purchased from National Centre for Cell Science, Pune.
In vitro experimental design
For in vitro glycation studies, BSA-Glucose model was used as published in our previous report to assess the effect of oxidative stress on protein glycation, and Fenton reagent was used to generate oxidative stress [15]. Aminoguanidine was used as a standard protein glycation inhibitor and its inhibitory activity was also checked in the presence of Fenton reagents [10, 12]. The combinatorial effects of Vitamin E (as an antioxidant) and aminoguanidine were checked in the presence of Fenton reagents on progression of the glycation. The reaction sets used in this study were as follows:
In detail, the glycation reaction mixtures contained bovine serum albumin (20 mg/ml), 100 mM d-glucose, Fenton reagent [FeSO4 (10 mM), H2O2 (10 mM) and ascorbic acid (100 mM)] along with Vitamin E (5 mM) and aminoguanidine (2 mM) in 50 mM phosphate buffer (pH 7.4). All reaction mixtures were incubated in the dark for 14 Days at 37 °C. After 14 days of incubation, unbound glucose and small molecules were removed by extensive dialysis against the same buffer. Protein concentration was measured by Bradford’s method and BSA was used as standard.
Measurement of fructosamine
Fructosamine measurement was carried out by Johnson’s method [16] with slight modifications. 0.2 ml of glycated reaction mixture and 0.8 ml of (300 mM) nitro blue tetrazolium (NBT) reagent prepared in (100 mM) sodium carbonate buffer, pH 10.3 was incubated at room temperature for 30 min. and absorbance was measured at 530 nm against blank. The final fructosamine content was expressed as Fructosamine nM/mg protein using an extinction coefficient of 12,640/M/cm for monoformazan [17].
Measurement of protein-bound carbonyls
Non-enzymatic glycation-mediated glyco-oxidative damage of BSA was determined by estimating total protein-bound carbonyls [18]. In brief, protein of each reaction mixture was precipitated with 20% TCA. The precipitates were treated with 2 N HCl containing 10 mM DNPH at room temperature for 1 h followed by centrifugation, each reaction mixture was washed thrice with an ethanol-ethyl acetate (1:1) and the final precipitates were dissolved in 8 M urea (in 2 mM potassium phosphate, pH 2.3). The absorbance was measured at 360 nm and protein-bound carbonyl content was obtained as nM/mg protein using molar extinction coefficient of 22,000/M/cm. Samples processed similarly but without DNPH treatment were used as controls. Protein concentrations of all samples were determined with absorbance at 280 nm.
Circular dichroism studies
All far-UV CD spectra were obtained at room temperature and recorded on a JASCO J-715 (Jasco Inc., Easton, MD, USA) spectropolarimeter using solutions of a protein concentration about 0.2–0.3 mg/ml. All spectra were generated by taking average of three scans and were corrected for the respective blanks. Results are expressed as molar ellipticity, [Θ] (deg cm2/dmol), based on a mean amino acid residue weight (MRW). The molar ellipticity was determined as [Θ] λ = (Θ 100 MRW)/(c l), where, l is the light path length in centimeters, c is the protein concentration in mg/ml, and Θ is the measured ellipticity in degrees at the relevant wavelength. The mean residual ellipticity (MRE) in deg cm2/dmol can be estimated using the formula
where n is the number of residues in the protein, Cp is the molar concentration of the protein, and l is the path length. From the MRE values at 208 nm, we can calculate the α-helical content of free and complexed BSA using the following formula
where MRE208 is to the observed MRE values, 33,000 and 4000 are the MRE values of a pure α-helix and of the β-turn and random coil conformation at 208 nm, respectively [19, 20].
Zeta potential study
The zeta potential was measured using laser Doppler velocimetry mode of the Zeta Sizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK). In the solution, fixed charge associated with any particle is always surrounded by counter ions. For moving particles in the measurement zone, shift in the frequency of scattered light is proportional to their velocity. Since the zeta potential is pH dependent, the measurements were performed at pH 7.2. Each result was obtained from the average of 20 runs.
In vivo experimental design
The effect of oxidative stress on high glucose dosing was studied on progression of protein glycation.
Model animals
All the in vivo experiments were carried out on the male Wistar rats weighing 190–200 g. All animal experiments were carried out as per the norms of committee for the purpose of control and supervision of experiments on animals (CPCSEA) formed by the Government of India (Registration No. 233/CPCSEA). The animals were allowed for free access of food (Amrut, Pune), water (ad libitum) and maintained under standard laboratory conditions.
Induction of oxidative stress
Oxidative stress was generated by administering 10 μl (0.1% DMSO as a vehicle) of rotenone (5 mM) on the 1st, 4th, 7th, 10th, and 13th day in Wister male rat for a total treatment of 14 days. High glucose dose 10 g per day with rotenone was given to animals, whereas only glucose-fed animals were used as control. Standard glycation inhibitor aminoguanidine was used at 2 mM concentration while vitamin E (5 mM dissolved in 1% DMSO) was used as antioxidant which were administrated orally. After 14 days of treatment, animals were scarified by cervical dislocation and serum fructosamine, SOD, catalase, tissue fluorescence measurements of liver and kidney were carried out. Animals were divided into six different groups as follows:
Effect on in vivo antioxidant status of treated animals
Superoxide dismutase (SOD) activity was measured by the method of [21] Mishra, et al., 1972. In detail, 0.1 ml of homogenized tissue sample was added with 3.5 ml of buffer [(50 mM phosphate buffer, pH 7.4), 0.3 ml (10 mM) EDTA, 1.2 ml (130 mM) methionine and 0.6 ml NBT (150 µM in 100 mM sodium carbonate buffer, pH 10.35)]. A similar reaction mixture was prepared in which tissue sample was replaced by equal volume of distilled water served as blank. Reaction was started by the addition of 0.4 ml (60 μM) riboflavin. Tubes were radiated in front of 18 W fluorescent bulb for 30 min and the reaction was stopped by placing the tubes in dark. The absorbance was read at 560 nm. Unit of SOD is described as the amount of enzyme required for inhibition of 50% oxidation riboflavin.
Catalase (CAT) was determined in serum by Beers and Sizer method [22]. The change in optical density at 240 nm per unit time was taken as a measure of catalase activity. 2.1 ml of Phosphate buffer (100 mM, pH 7.4) was taken into cuvette along with 50 μl of tissue homogenate and 0.5 ml freshly prepared 30 mM H2O2 solution was added. The rate of decomposition of H2O2 was measured immediately by using UV–Visible spectrophotometer.
Measurement of total reactive oxygen species (ROS)
The ROS measurement was carried out using a fluorescent 2′,7′-dichlorofluorescin diacetate (DCF-DA) dye [23]. The 100 mg of each tissue sample was chopped and homogenized and used for incubation with 10 mM DCF-DA dye at 37. Then the samples were incubated with dye DCF-DA for 30 min at 37 °C. After incubation, the homogenate was sonicated for 15 s with three strokes and centrifuged at 3000×g for 5 min at 4 °C and the fluorescence of supernatant was measured using spectrofluorimeter at 488 nm excitation wavelength and 530 nm emission wavelength. In the presence of ROS, DCFH is oxidized to the highly fluorescent 2′,7′-dichlorofluorescein (DCF). The level of DCF fluorescence is the measure of ROS concentration.
Effect on tissue fluorescence
Tissue AGE content was determined by the method described by Nakagawa [24]. Various tissues like heart, kidney, lungs, liver, and muscle were processed for tissue AGEs content measurement. The tissues were rinsed thoroughly in PBS to remove residual blood clots. Tissues were minced and delipidated by shaking gently and incubated with mixture of chloroform and methanol (2:1 v/v) overnight. The delipidated tissue was homogenized in 0.1 N NaOH, followed by centrifugation at 8000×g for 15 min at 4 °C. The amount of total and specific AGE in the supernatant was measured by using spectrofluorometer (Cary Eclipse Fluorescence Spectrophotometer, USA) at an excitation/emission wavelength of 370/440 nm and 335/385 nm. The fluorescence intensities of the samples were measured and expressed as arbitrary units AU/mg of protein.
Flow cytometry
Experimental design, sample preparation, and staining protocol
Huh7 cell line was used for the analysis of oxidative stress induced by rotenone using 2,7, Dichlorofluorescein Diacetate (DCF-DA) dye [25]. DCF has excitation and emission spectra at 388/530 nm, respectively. 0.1 Million Huh7 cells were seeded in each well of 12-well plate. The cells were studied in the presence of hyperglycemic and oxidative condition in the presence and absence of anti-glycating and anti-oxidating agents. The treatment given to the cells was as follows: (a) Untreated cells, (b) Glucose (30 mM), (c) Rotenone (200 µM), (d) Glucose (30 mM) + Rotenone (200 µM), (e) Glucose (30 mM) + AMG (300 µM), (f) Glucose (30 mM) + Vitamin E (300 µM), (g) Glucose (30 mM) + Rotenone (200 µM) + AMG (300 µM), (h) Glucose (30 mM) + Rotenone (200 µM) + AMG (300 µM) + Vitamin E (300 µM), in the essential media and incubated for 24 h in CO2 incubator at 37 °C. Cells were washed with PBS and transferred to 1.5 ml vial. After two washes with PBS, cell pellet was incubated with DCF in dark for 30 min at room temperature. Then cells were suspended in 200 µl PBS and subjected for flow cytometry analysis on Attune NxT Flow cytometer (Thermo Scientific, USA). After the cells were acquired on Attune NxT Flow cytometer, the obtained data were analyzed using the FlowJo software (version 8.8.7) Tree star, USA. The data analysis includes specific gating strategy for selection of cells positive for DCF fluorescent dye showing oxidative stress.
Statistical analysis
All results were expressed as Mean ± SD for n = 6 (six rats in each group) for in vivo and in vitro studies. The mean difference among the two groups is calculated using students T test, and p value < 0.05 was considered as statistically significant. All the cell culture studies were performed in triplicates (n = 3).
Results
In vitro studies
Effect of oxidative stress (Fenton reaction) on fructosamine and protein-bound carbonyl measurements
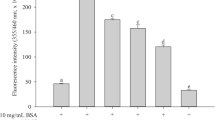
Fructosamine measurement is used as a marker for early glycation. Glycation of BSA in the presence of glucose produces higher fructosamine, while the same is reduced in the presence of aminoguanidine, a known inhibitor of protein glycation [26, 27] (Fig. 1a). Oxidative stress leads to higher glycation which can be seen through a 33.57% increase in fructosamine in the presence of Fenton reagent and 22.50% increase in protein-bound carbonyls (Fig. 1b).

Effect of oxidative stress (Fenton reaction) on early glycation end-product formation. Fructosamine (a), PCO (b), and changes in secondary structure composition (c) of protein in glycation of BSA.The data were expressed as molar ellipticity (deg cm2/dmol). The spectrum given here is an averaged spectrum of three experimental samples
Circular dichroism
The circular dichroism measurements were effectively used to observe structural changes in protein owing to glycation (Fig. 1c). Glycation of BSA reduces helical structure by 18.3% and increases the β-sheet structure by 9.5%. However, it is very surprising to note that in the presence of oxidative stress, the alpha helix is reduced to as low as 2.8% with a strong increase in the beta pleated structure (Table 1).
Zeta potential study
Zeta potential was studied to observe the change in protein surface charge caused by the changes in protein structure. Oxidative stress demonstrates higher charge difference than protein glycation. Again as compared to antioxidant, protein glycation inhibitor seems to have better outcomes (Table 2).
In vivo studies
Effect of protein glycation and rotenone on tissue oxidative stress
The reactive oxygen species were measured in tissue homogenate of liver and kidney. It is shown in Fig. 2 that the increased glycation can lead to 39.09% increase in ROS formation in the liver. In the presence of rotenone, it increases 2.3 times by 134%. ROS production is seen to be much higher in liver compared to kidney.

Measurement of reactive oxygen species in liver and kidney using DCFD dye
Effect of oxidative stress and protein glycation on SOD and Catalase activity
The activity of SOD and catalase in the presence of rotenone is found to increase by almost 3.5 to 4 times as shown in Table 3. It is to be observed that aminoguanidine has lesser effect on SOD activity but in the presence of Vitamin E in the liver there is reduction in activity; however, SOD activity continues to be high in the case of kidney tissue. Catalase activity on the other hand reduces considerably in the presence of aminoguanidine and Vitamin E.
Tissue fluorescence
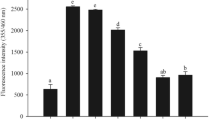
The tissue fluorescence was measured after 14 days of treatment in experimental animals. It can be observed that liver and kidney showed higher fluorescence in the presence of rotenone. Both vitamin E as well as aminoguanidine are effective in lowering tissue fluorescence; however, in combination, they can reduce the tissue fluorescence to near normal. This is also true for both general and specific fluorescence (Fig. 3).

Effect of rotenone-induced oxidative stress in the presence of high glucose dose on tissue fluorescence Specific AGEs (a) and Total AGEs (glycation inhibitors and probiotics can ameliorate the changes caused by high fructose feed)
Flow cytometry
Evaluation of ROS production is performed by flow cytometry method by using Huh-7 human liver cell line and fluorescent DCFD dye, which specifically binds to the ROS. The results showed that the 10% of the cells treated with high glucose showed total ROS generation compared to untreated cells, whereas additional treatment with strong anti-glycating agent AMG showed significant decrease in the number of cells (1.56%) with ROS generation (Fig. 4). Further oxidative stress induced by treating the cells with Rotenone also showed increased number of cells (11.5%) with total ROS generation and are further increased in the presence of high glucose (12.1%). Whereas cells treated with AMG in the presence of high glucose and rotenone showed a significant decrease in the number of cells (6.75%) showing ROS generation. Further cells were treated with AMG in the presence of high glucose and in the presence of rotenone and vitamin E (known anti-oxidative agent) and found that the number of cells showing ROS generation decreased significantly (2.28%). Hence, the use of anti-glycating agents like AMG has significant impact on reducing the induction of oxidative stress due to higher glucose levels and equally important finding is the use of Vit E as strong antioxidant to curb the adverse effects of increased oxidative stress in high glucose conditions.

Effect of rotenone-induced oxidative stress on generation of ROS formation in Huh 7 cell line at hyperglycemic condition measured by using a flow cytometry
Discussion
The role of protein glycation in initiating secondary complications such as neuropathy, nephropathy, and retinopathy is well established [28,29,30]. Glycation of proteins leads to structural and functional changes in proteins [31, 32]. Protein glycation is known to enhance oxidative stress through auto oxidation of sugars as well as amino acids in the proteins [33, 34]. It is also reported that the formation of Advanced Glycation End-Products (AGEs) leads to their binding to the cellular receptors-RAGEs and initiates cytokine signaling leading to progressive immunological response and tissue damage [35]. Though progression of this pathway leading to tissue damage is well established, the time taken is likely to vary from months to years depending upon the concentration of AGEs generated. However, the process could be accelerated by oxidative stress and immunological activation in a feed-forward manner. It is important to understand the individual contribution and interplay between these three processes.
Oxidative stress and inflammation are often seen associated with Alzheimer’s disease [36, 37]; however, Plucinska, et al., reported that almost 80% of Alzheimer’s patients develop some form of diabetes [38]. It is also suggested that rather than people with diabetes developing Alzheimer as is often believed, it is the other way round. The most likely cause can be attributed to oxidative stress that is predominant in Alzheimer and is perhaps helping in the progression of protein glycation. The present study intends to demonstrate that oxidative stress can lead to rapid acceleration of protein glycation thereby reducing the time of manifestation of tissue damage. It further delves into use of inhibitors of oxidative stress and protein glycation inhibitors to observe their relative inhibition and to assess the benefit of combinatorial action.
The Fenton reaction is a suitable method for generating in vitro oxidative stress through generation of hydroxyl anion [39]. While use of Vitamin E is seen to reduce the fructosamine content, aminoguanidine is a better inhibitor. Together they are more effective in reducing both the fructosamine as well as protein carbonyl content. It is to be noted that aminoguanidine in the absence of fenton reagent is able to generate fructosamine on par with normal control. Hence, it is vital to realize that oxidative stress does lead to enhanced protein glycation and further establishment of secondary complications.
Oxidative stress is also observed to induce strong alpha–beta transitions in protein structure. It should be noted that use of antioxidants is not as effective as protein glycation inhibitor in preventing the alpha–beta transitions. In combination with protein glycation inhibitor they are able to totally prevent the adverse changes in protein structure. The same is also observed in normalizing the surface charge of the protein as measured by its zeta potential.
Although demonstration of the role of oxidative stress in enhancing protein glycation in the in vitro studies is clear, it is equally important to assess whether the same is true in the in vivo condition. Rotenone is a known inhibitor of the mitochondrial electron transport chain and known to induce oxidative stress in the in vivo system. Superoxide dismutase and catalase activity are found to increase by almost fourfold. While catalase activity is fairly normalized through treatment of Vitamin E and aminoguanidine, SOD activity continues remaining fairly high. We have observed that aminoguanidine is able to reduce the level of SOD in the absence of induced oxidative stress [20, 40]. It can be observed from Fig. 3 that rotenone is able to induce a 39.52% increase in AGE formation in comparison with glucose control as well 47.37% as specific AGEs formation. Although aminoguanidine is able to reduce total AGEs by 22.25% and specific AGEs by 25% (with glucose + rotenone control), there is need of an antioxidant to reduce it to near normal levels. Especially in the kidney it can be observed that antioxidant action is more important. The same picture is also reflected in cell culture studies. While both glycation and oxidative stress causes cell apoptosis, the process is enhanced in the presence of both rotenone and glucose. Both aminoguanidine and Vitamin E are able to control cell apoptosis on glycation, but in the presence of rotenone a combination of both is needed to control cell damage.
Conclusion
The present study clearly demonstrates through in vitro, in vivo, and cell culture studies that oxidative stress independently can enhance the progression of protein glycation thereby accelerating the process of tissue damage. Further, though vitamin E, an anti-oxidant is not able to independently ameliorate the changes nor is aminoguanidine a protein glycation inhibitor able to do so, a combination of both is more effective in preventing tissue damage. Although a person suffers from diabetes the time taken to progress to secondary complications depends on the number of factors including maintenance of a good glycemic control. More important, it is likely that oxidative stress may be a crucial factor to precipitate secondary complications such as nephropathy, neuropathy, and retinopathy as observed in the present study. Hence, the study is a pointer to evaluate the therapy options for diabetes to also include an antioxidant therapy along with glucose lowering drugs to have better management and prevent progression to secondary complications.
Abbreviations
- BSA:
-
Bovine serum albumin
- Fent:
-
Fenton reagent
- Rot:
-
Rotenone
- AMG:
-
Aminoguanidine
- Vit E:
-
Vitamin E
- DCF-DA:
-
2′,7′-Dichlorofluorescin diacetate
- HuH-7:
-
Hepatocellular carcinoma cells
- SOD:
-
Superoxide dismutase
- CAT:
-
Catalase
- PCO:
-
Protein-bound carbonyls
- AGEs:
-
Advanced Glycation End-Products
- TCA:
-
Tricarboxylic acid
- CD:
-
Circular dichroism
References
Turk Z, Ljubic S, Turk N, Benko B (2001) Detection of autoantibodies against advanced glycation end products and AGE-immune complexes in serum of patients with diabetes mellitus. Clin Chim Acta 303:105–115. https://doi.org/10.1016/S0009-8981(00)00389-2
Takeuchi M, Kikuchi S, Sasaki N et al (2004) Involvement of advanced glycation end-products (AGEs) in Alzheimer’s disease. Curr Alzheimer Res 1:39–46. https://doi.org/10.2174/1567205043480582
Vistoli G, De Maddis D, Cipak A et al (2013) Advanced glycoxidation and lipoxidation end products (AGES and ALES): an overview of their mechanisms of formation. Free Radic Res 47:3–27. https://doi.org/10.3109/10715762.2013.815348
Brownlee M (1994) Glycation products and the pathogenesis of diabetic complications. Diabetes 43:836–841. https://doi.org/10.2337/diab.43.6.836
Watkins NG, Thorpe SR, Baynes JW (1985) Glycation of amino groups in protein. Studies on the specificity of modification of RNase by glucose. J Biol Chem 260:10629–10636
Vitek MP, Bhattacharya K, Glendening JM et al (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA 91:4766–4770. https://doi.org/10.1073/pnas.91.11.4766
Schmidt MA, Stern DM (2000) RAGE: a new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol Metab 11:368–375. https://doi.org/10.1016/S1043-2760(00)00311-8
Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344:109–116. https://doi.org/10.1042/bj3440109
Rahbar S, Figarola JL (2003) Novel inhibitors of advanced glycation end products. Arch Biochem Biophys 419:63–79. https://doi.org/10.1016/j.abb.2003.08.009
Brownlee M, Vlassara H, Kooney A et al (1986) Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science 232:1629–1632. https://doi.org/10.1126/science.3487117
Maritim AC, Sanders RA, Watkins JB (2003) Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol 17:24–38. https://doi.org/10.1002/jbt.10058
Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, Liu B et al (2009) Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer’s disease. Eur J Neurosci 29:1335–1347. https://doi.org/10.1111/j.1460-9568.2009.06701.x
Omar ME, Abdel-Salam Yasser AK et al (2014) Effect of a single intrastriatal rotenone injection on oxidative stress and neurodegeneration in the rat brain. Comp Clin Pathol 23:1457–1467. https://doi.org/10.1007/s00580-013-1807-4
Li D, Devaraj S, Fuller C, Bucala R, Jialal I (1996) Effect of tocopherol on LDL oxidation and glycation: in vitro and in vivo studies. J Lipid Res 37:1978–1986
Jagdale AD, Bavkar LN et al (2016) Strong inhibition of the polyol pathway diverts glucose flux to protein glycation leading to rapid establishment of secondary complications in diabetes mellitus. J Diabetes Complicat 30:398–405
Johnson RN, Metcalf PA, Baker JR (1983) Fructosamine: a new approach to the estimation of serum glycosylprotein: an index of diabetic control. Clin Chim Acta 127:87–95. https://doi.org/10.1016/0009-8981(83)90078-5
Ansari NA, Ali R (2011) Physicochemical analysis of poly-l-lysine: an insight into the changes induced in lysine residues of proteins on modification with glucose. IUBMB Life 63:26–29. https://doi.org/10.1002/iub.410
Nakamura A, Goto S (1996) Analysis of protein carbonyls with 2, 4-dinitrophenyl hydrazine and its antibodies by immunoblot in two-dimensional gel electrophoresis. J Biochem 119:768–774. https://doi.org/10.1093/oxfordjournals.jbchem.a021306
Kelly SM, Price NC (2000) The use of circular dichroism in the investigation of protein structure and function. Curr Protein Pept Sci 1:349–384. https://doi.org/10.2174/1389203003381315
Joglekar M, Bavkar L, Sistla S, Arvindekar A (2017) Effective inhibition of protein glycation by combinatorial usage of limonene and aminoguanidine through differential and synergistic mechanisms. Int J Biol Macromol 99:563–569. https://doi.org/10.1016/j.ijbiomac.2017.02.104
Misra HP, Fridovich I (1972) The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 247:3170–3175
Beers RF, Sizer IW (1952) Spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem 195:133–140
Yeligar SM, Harris FL, Hart CM, Brown LAS (2012) Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol 188:3648–3657. https://doi.org/10.4049/jimmunol.1101278
Nakagawa T, Yokozawa T, Terasawa K, Shu S, Juneja LR (2002) Protective activity of green tea against free radical and glucose-mediated protein damage. J Agric Food Chem 50:2418–2422. https://doi.org/10.1021/jf011339n
Saengkhae C, Loetchutinat C, Garnier-Suillerot A (2003) Kinetic analysis of fluorescein and dihydrofluorescein effluxes in tumour cells expressing the multidrug resistance protein. MRP1. Biochem Pharmacol 65:969–977. https://doi.org/10.1016/S0006-2952(02)01662-3
Edelstein D, Brownlee M (1992) Mechanistic studies of advanced glycation end product inhibition by aminoguanidine. Diabetes 41:26–29. https://doi.org/10.2337/diab.41.1.26
Thornalley PJ (2003) Use of aminoguanidine (pimagedine) to prevent the formation of advanced glycation end products. Arch Biochem Biophys 419:31–40. https://doi.org/10.1016/j.abb.2003.08.013
Goh SY, Cooper ME (2008) The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab 93:1143–1152. https://doi.org/10.1210/jc.2007-1817
Meerwaldt R, Links T, Zeebregts C, Tio T, Hillebrands J, Smit A (2008) The clinical relevance of assessing advanced glycation end products accumulation in diabetes. Cardiovasc Diabetol 7:29. https://doi.org/10.1186/1475-2840-7-29
Vlassara H, Uribarri J (2014) Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Curr Diab Rep 14:453. https://doi.org/10.1007/s11892-013-0453-1
Awasthi S, Saraswathi NT (2016) Non-enzymatic glycation mediated structure-function changes in proteins: case of serum albumin. Rsc Adv 6:90739–90753. https://doi.org/10.1039/C6RA08283A
Greifenhagen U, Frolov A, Blüher M, Hoffmann R (2016) Site-specific analysis of advanced glycation end products in plasma proteins of type 2 diabetes mellitus patients. Anal Bioanal Chem 408:5557–5566. https://doi.org/10.1007/s00216-016-9651-4
Miyata T, Kurokawa K, De Strihou CVY (2000) Advanced glycation and lipoxidation end products: role of reactive carbonyl compounds generated during carbohydrate and lipid metabolism. J Am Soc Nephrol 11:1744–1752
Hunt JV, Wolff SP (1991) Oxidative glycation and free radical production: a causal mechanism of diabetic complications. Free Radic Res Commun 1:115–123. https://doi.org/10.3109/10715769109145775
Aronson D, Rayfield EJ (2008) How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol 2:89. https://doi.org/10.1186/1475-2840-1-1
Verdile G, Keane KN, Cruzat VF et al (2015) Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediat Inflamm 2:89. https://doi.org/10.1155/2015/105828
Namioka N, Hanyu H, Hirose D et al (2017) Oxidative stress and inflammation are associated with physical frailty in patients with Alzheimer’s disease. Geriatr Geriatr Gerontol Int 17:913–918. https://doi.org/10.1111/ggi.12804
Plucińska K, Dekeryte R, Koss D et al (2016) Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia 59:1513–1523. https://doi.org/10.1007/s00125-016-3960-1
Kanti Das T, Wati MR, Fatima-Shad K (2015) Oxidative stress gated by Fenton and Haber Weiss reactions and its association with Alzheimer’s disease. Arch Neurosci 2:e20078. https://doi.org/10.1007/s00125-016-3960-1
Patil RS, Jagdale AD, Nalawade ML et al (2018) Glycation inhibitors and probiotics can ameliorate the changes caused by high fructose feed. Int J Pharm Pharm Sci 10:28–32. https://doi.org/10.22159/ijpps.2018v10i7.26870
Acknowledgements
Laxman Naghnath Bavkar acknowledges the University Grant Commission (UGC), New Delhi, India for a fellowship under the Special Assistance Program for Basic Scientific Research. Rahul Shivaji Patil acknowledges the Department of Science and Technology (DST), New Delhi, India for DST-INSPIRE fellowship. Chemicals and research work were supported by RUSA, Government of Maharashtra funded to Prof. (Mrs.) A. U. Arvindekar. The authors are thankful to the Department of Biochemistry, Shivaji University, Kolhapur for providing work place and laboratory.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bavkar, L.N., Patil, R.S., Rooge, S.B. et al. Acceleration of protein glycation by oxidative stress and comparative role of antioxidant and protein glycation inhibitor. Mol Cell Biochem 459, 61–71 (2019). https://doi.org/10.1007/s11010-019-03550-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-019-03550-7