
Abstract
The long non-coding RNA (lncRNA) prostate cancer-associated ncRNA transcript 1 (PCAT-1) has been shown to promote prostate cancer cell proliferation through c-Myc and is associated with the poor prognosis of CRC patients. In the current study, it was hypothesized that the effect of PCAT-1 on the aggressiveness of CRC cells was dependent on the function of c-Myc. Human CRC cell lines Caco-2 and HT-29 were transfected with specific PCAT-1 shRNAs, and cell migration, invasiveness, and resistance to 5-fluorouracil were measured. To elucidate the role of c-Myc in PCAT-1 function, c-Myc was overexpressed in PCAT-1-silenced CRC cells and the effect of c-Myc overexpression on the aggressiveness of PCAT-1-silenced cells was detected. The results showed that knockdown of PCAT-1 in CRC cells suppressed cell motility and invasiveness, and sensitized the cells to 5-fluorouracil, as evidenced by the reduced viability and induced apoptosis in PCAT-1-silenced cells compared to the parental cells in response to 5-fluorouracil treatment. The expression of c-Myc in PCAT-1-silenced CRC cells was down-regulated, and forced expression of c-Myc partially restored the invasiveness in PCAT-1-silenced cells. In summary, the findings outlined in the current study suggest that PCAT-1 regulates the invasiveness and drug resistance in CRC cells and that PCAT-1 may promote CRC cell invasion by modulating the expression of c-Myc.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the last decade, non-coding RNAs (ncRNAs) have been proposed to be important regulators of cancer-related pathways and promising predicators of cancer prognosis [1,2,3,4]. With the advancement of high-throughput sequencing technology, it is possible for scientists to conduct complicated analyses of the cellular transcriptomes, which leads to the discovery of numerous novel ncRNA species [3, 5]. Among those newly identified classes, long non-coding RNAs (lncRNAs) have received much attention for their fundamental roles in a diversity of molecular processes, including cell differentiation, lineage specificity, and tumorigenesis [6,7,8]. LncRNAs, such as HOTAIR, HULC, and H19, have shown their potential values as therapeutic targets in breast, lung, and liver cancers [9,10,11]. Given the fact that cancer is one of the leading causes of death worldwide, a comprehensive understanding of the function of lncRNAs in oncogenesis and cancer progression will help with the prevention, diagnosis, and treatment of this devastating disease.
Prostate cancer-associated ncRNA transcript 1 (PCAT-1) was firstly identified in prostate cancer (PCa) and implicated in the progression of PCa [12, 13]. Up-regulation of PCAT-1 has been shown to promote the proliferation of PCa cells. In line with the pro-survival role of PCAT-1, knockdown of PCAT-1 effectively reduces the viability of PCa cells, implying a potential of PCAT-1 as a therapeutic target for the treatment of PCa [12, 13]. Moreover, Shi et al. has demonstrated that up-regulation of PCAT-1 correlates with an advanced clinical stage and poor prognosis in esophageal squamous carcinoma [14]. In addition, PCAT-1 is also associated with the poor prognosis in colorectal cancer (CRC) patients [15].
As one of the cancer malignancies, CRC affects more than one million new patients each year with a mortality rate as high as 33% in the developed countries [16, 17]. Despite the progression in the understanding of the molecular mechanisms and the treatment regimens for CRC, the overall survival rate of CRC patients remains barely changed in the recent years [15]. One of the factors leading to the poor prognosis of CRC is the high metastasis potential of CRC cells. Previously, He et al. showed that transcription factor c-Myc could induce the activity of lncRNA CCAT1 and contributed to colon cancer metastasis [18]. c-Myc is an evolutionarily conserved member of the MYC family [19] and one of the most widely studied proto-oncogenes [20]. The MYC family genes are often deregulated in several human neoplasias, including diffuse large B cell lymphoma, multiple myeloma, colon cancer, glioblastoma, melanoma, ovarian cancer, and PCa [21, 22]. Recently, it is found that PCAT-1 promotes cancer progression through c-Myc in PCa [23]. Taken together, the function of PCAT-1 in CRC is likely to be associated with the action of c-Myc as well. Therefore, in the current study, a series of in vitro assays were performed to verify this hypothesis. CRC cell lines with high PCAT-1 expression were selected and transfected with specific shRNAs targeting PCAT-1, followed by assessments of cell migration, invasion, and multidrug resistance (MDR). The role of c-Myc in PCAT-1-regulated invasiveness of CRC cells was investigated by overexpressing c-Myc in PCAT-1 knockdown cells. The findings in the current study support the hypothesis that PCAT-1 promotes the metastatic potential and MDR in CRC cells through c-Myc.
Materials and methods
Chemicals and antibodies
Antibody against matrix metalloproteinase-2 (MMP-2) (BA3716) was purchased from Boster (Wuhan, China). Antibodies against c-Myc (D199941) and MMP-9 (D261999) were purchased from Sangon (Shanghai, China). Antibody against β-actin (sc-47778) was purchased from Santa Cruz (USA) Secondary goat anti-rabbit (A0208) and goat anti-mouse (A0216) IgG-HRP antibodies were purchased from Beyotime Biotechnology (Shanghai, China). RNA Purified Total RNA Extraction Kit (RP1201) and super M-MLV reverse transcriptase (RP6502) were purchased from BioTeke (Beijing, China). Total Protein Extraction Kit (WLA019) was purchased from Wanleibio (Shenyang, China). 5-fluorouracil (5-Fu) (V900394) and MTT (M-2128) were purchased from Sigma (USA). Matrigel was obtained from BD Biosciences (San Jose, CA, USA). Hoechst staining kit (C0003) was purchased from Beyotime Biotechnology. Plasmids pRNA-H1.1 and pcDNA3.1 were purchased from Sangon Biotech.
Patients and collection of CRC specimens
CRC specimens and the corresponding para-carcinoma tissues were collected from 23 CRC patients hospitalized in The First Hospital of China Medical University. The specimens were fixed, prepared in paraffin blocks, and sectioned. All the patients enrolled in the present study met the following criteria: (1) diagnosed with primary CRC; (2) with complete clinicopathological data and follow-up information. The study was approved by the ethics committee at The First Hospital of China Medical University regarding the related screening, inspection, specimen collection, and analyses. All the subjects had signed the informed consent form, and all the procedures were performed in accordance with the Declaration of Helsinki.
Cell cultures
Human CRC cell lines Caco-2 (TCHu146) and HT-29 (TCHu103) were obtained from the Cell Bank of Chinese Academy of Sciences and maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% (v/v) antibiotics mixture at 37 °C in a humidified atmosphere consisting of 5% CO2.
Construction of PCAT-1 shRNA vectors and cell transfection
Two specific shRNAs targeting PCAT-1 (shRNA-1: 5′-GCTCACGCCTGTAATCTCA-3′; shRNA-2: 5′-GAACCTAACTGGACTTTAA-3′) were ligated to the pRNA-H1.1 plasmid to construct pRNA-H1.1-siPCAT-1-1 and pRNA-H1.1-siPCAT-1-2 vectors. A non-targeting version of shRNA was used as negative control. The ORF of c-Myc gene was amplified by PCR and ligated to the pcDNA3.1 + plasmid to construct the c-Myc overexpression vector. For transfection, 1 × 105 cells were plated in six-well plates and transfected with different vectors using Lipofectamine 2000 (1024993, Invitrogen,USA). Cells with stable PCAT-1 knockdown were selected using 500 μg/ml G418.
Determination of IC50 of 5-Fu and MDR test
The IC50 of 5-Fu in CRC cells were determined by exposing the cells to different concentrations of 5-Fu (0, 5, 10, 15, 25, 50, 100 μg/ml) for 48 h. Thereafter, CRC cells with or without PCAT-1 knockdown were subjected to 5-Fu treatment at IC50 (2.43 μg/ml for Caco-2 cells and 7.33 μg/ml for HT-29 cells) (Table S1) for 96 h. The effect of PCAT-1 knockdown on the MDR of CRC cells was assessed by MTT assay as described below.
Scratch assay
Cell mobility after PCAT-1 knockdown was detected using scratch assay. After seeding cells in a 24-well plate at a density of 2 × 104 cells/well, reference points were marked to guarantee the same area of image acquisition. The cells were cultured at 37 °C for 2 days to allow the cells to grow into a confluent monolayer. Thereafter, the cell layer was scratched to generate a cell-free straight line, and rinsed with PBS to remove debris at the edges of the scratch. Cell migration towards the midline of the scratch was recorded in reference with the reference points. Three images (at 0, 14, and 24 h, respectively) were captured with a phase-contrast microscope for each well and the gap distances were measured by the ImageJ software (US National Institutes of Health). The migration rate was represented as the percentage of gap closure.
Transwell assay
The invasiveness of CRC cells was measured using the transwell apparatus. Briefly, 2 × 104 cells in 200 μl medium were plated in the upper chamber of a transwell system (BSA-coated porous polycarbonate membrane with a pore size of 8 μm), and each polycarbonate membrane was pre-coated with 40 μl matrigel (1.5 mg/ml) and set at 37 °C for 2 h to allow the formation of a reconstituted basement membrane. CRC cells were allowed to migrate through the porous membrane at 37 °C for 24 h. After completely removing the cells on the upper surface of the membrane, the cells on the lower surface of the membrane were stained with 0.5% (w/v) crystal violet for 5 min. Invasion cell number was determined using the Image-Pro Plus 6.0 software (Nikon) at ×200 magnification.
Reverse-transcription real-time PCR (RT2-PCR)
Total RNA was extracted using an RNA Purified Total RNA Extraction Kit. Super M-MLV reverse transcriptase was used to obtain cDNA templates from total RNA. The mixture of each real-time PCR reaction contained 10 μl SYBR GREEN mastermix, 0.5 μl of each primer [PCAT-1, forward: 5′-ACAGGCTGAGGCAGGAGAAT-3′, reverse: 5′-CTTTGGGAAGTGCTTTGGAG-3′; β-actin (internal reference gene), forward: 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′, reverse: 5′-CTGTCACCTTCACCGTTCCAGTTT-3′], 1 μl cDNA template, and 8 μl Rnase-free H2O. The thermal cycling parameters for the amplification were set as follows: a denaturation step at 95 °C for 10 min, followed by 40 cycles of amplification at 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 30 s, and then the reaction was stopped at 25 °C for 5 min. The relative expression levels of the target gene were calculated by the real-time PCR system (Exicycler 96, BIONEER, South Korea) using the formula of \(2^{{ - \Delta \Delta C_{t} }} .\)
Western blotting
Total protein in CRC cells transfected with NC or shPCAT-1-1 was extracted using the Total Protein Extraction Kit, and the concentration of the protein samples was determined using the BCA method. Forty μg protein from each sample was subjected to 10% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) at 70 V for 2.5 h. Subsequently, the proteins were transferred onto polyvinylidene difluoride (PVDF) membranes, which were then rinsed with TTBS and blocked with skimmed milk solution for 1 h. The membranes were incubated with one of the primary antibodies against c-Myc (1:500), MMP-2 (1:400), MMP-9 (1:500), E-Cadherin (1:500), Snail (1:500), and β-actin (1:1000) (internal reference protein) at 4 °C overnight. Then the membranes were incubated with secondary HRP-conjugated IgG antibodies (1:5000) for 45 min at 37 °C. The blots were developed using the Beyo ECL Plus reagent, and the signals were detected in the Gel Imaging System. The relative expression levels of the targeted proteins were calculated by the Gel-Pro-Analyzer (Media Cybernetics, USA).
Gelatin zymography
Total protein in CRC cells transfected with NC or shPCAT-1-1 was extracted and the concentration was measured using the BCA method. Thirty μg protein was subjected to 10% SDS-PAGE at 70 V for 2.5 h. The gel was soaked in the eluent buffer (2.5% Triton X-100, 50 mM Tris–HCl, 5 mM CaCl2, 1 μM ZnCl2, pH 7.6) twice (40 min each) with constant shaking, and then washed with the wash buffer (50 mM Tris–HCl, 5 mM CaCl2, 1 μM ZnCl2, pH 7.6) twice (20 min each). Afterwards, the gel was incubated in the incubating buffer (50 mM Tris–HCl, 5 mM CaCl2, 1 μM ZnCl2, 0.02% Brij, 0.2 M NaCl) for 40 h before staining with the staining buffer (0.05% Coomassic brilliant blue G-250G-250, 30% Methanol, 10% acetic acid) for 3 h. The gel was imaged using the Gel Imaging System, and the band intensities were analyzed using Gel-Pro-Analyzer.
MTT assay
MDR of CRC cells after PCAT-1 knockdown was assessed by MTT assay. Cells (3 × 103/well) were cultured in 96-well plates for 96 h. Every 24 h, three randomly wells in each group were selected and added with 5 mg/ml MTT. After another 4-h incubation at 37 °C, the supernatant was aspirated and 200 μl DMSO was added into each well. Cell viability, represented by OD490 value, was measured using a Microplate Reader (ELX-800, BIOTEK, USA).
Hoechst staining
The morphological changes in the nuclei of CRC cells 48 h after 5-Fu treatment were detected using a Hoechst staining kit according to the manufacturer’s instructions, and the results were observed under a fluorescence microscope at a magnification of ×400.
Flow cytometry
The effect of PCAT-1 knockdown on the apoptotic process in CRC cells was measured using a PI/Annexin V-FITC Apoptosis Detection Kit (JingMei Biotech, Beijing, China) and analyzed by a FACScan flow cytometer (Accuri C6, BD, USA). The total apoptotic rate was the sum of the late apoptotic rate (UR, upper right quadrant-advanced apoptosis) and the early apoptotic rate (LR, lower right quadrant-prophase apoptosis).
Statistical analysis
The data were expressed as mean ± SD and each assay was performed with at least three replicates. One-way ANOVA and post hoc Duncan test were performed with a significant level of 0.05 (two-tailed P value). All the statistical analyses and graph plotting were conducted on SPSS version 19.0 (IBM, Armonk, NY, USA).
Results
The expression of PCAT-1 was up-regulated in clinical CRC specimens
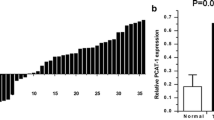
Twenty-three pairs of CRC specimens and the corresponding para-carcinoma tissues were collected to examine the expression of PCAT-1 in CRC. Based on the RT2-PCR result, it was confirmed that the expression of PCAT-1 was up-regulated in CRC tissues when compared with the adjacent non-tumorous tissues (Fig. 1), and the difference was statistically significant (P < 0.05).

The expression of PCAT-1 was up-regulated in clinical CRC specimens. “*,” significantly different from para-carcinoma tissues, P < 0.05
Knockdown of PCAT-1 inhibited the migration and invasion abilities in CRC cells
As shown in Fig. 2, the expression of PCAT-1 was stably silenced by the specific shRNAs in Caco-2 and HT-29 cells. The effect of PCAT-1 knockdown on the migration and invasion of CRC cells was assessed by scratch and transwell assays, respectively. The scratch assay showed that PCAT-1 knockdown delayed the process of scratch wound closure as compared with the parental cells for both cell lines (Fig. 3a, b), representing markedly suppressed motility in PCAT-1 knockdown cells. In addition, the number of PCAT-1-silenced cells passing through the matrigel-coated membrane was lower compared with that in the other two groups, and the differences were statistically significant (P < 0.05), indicating that the invading ability in both cell lines was reduced upon PCAT-1 knockdown (Fig. 4a, b). Further, the expression levels and the activities of the enzymes related to cell invasion were measured by western blotting and gelatin zymography. The results of both assays showed reduced expressions and activities of MMP-2 and MMP-9 in CRC cells with PCAT-1 knockdown (Figs. 5, 6). For the proteins associated with the epithelial-to-mesenchymal transition (EMT) process in cancer cells, the expression of E-cadherin was up-regulated by PCAT-1 knockdown, while the expression of Snail was down-regulated (Fig. 5), representing an inhibited EMT process upon PCAT-1 knockdown. Taken together, these results demonstrated a key role of PCAT-1 in regulating the invasiveness of CRC cells.

PCAT-1 shRNAs suppressed the expression of PCAT-1 in Caco-2 and HT-29 cells. a, b Quantitative analysis of RT2-PCR measurement of PCAT-1 expression in Caco-2 and HT-29 cells. Parental, non-transfected Caco-2 and HT-29 cells; NC, CRC cells transfected with non-targeting shRNA; shPCAT-1-1, CRC cells transfected with PCAT-1 shRNA-1; shPCAT-1-2, CRC cells transfected with PCAT-1 shRNA-2. “*,” significantly different from parental cells, P < 0.05. “#,” significantly different from NC cells, P < 0.05

Knockdown of PCAT-1 inhibited the motility of Caco-2 and HT-29 cells. a, b Representative images and quantitative analysis of scratch assays in Caco-2 and HT-29 cells. The process of scratch wound closure was delayed in PCAT-1 knockdown cells. Parental, non-transfected Caco-2 and HT-29 cells; NC, CRC cells transfected with non-targeting shRNA; shPCAT-1-1, CRC cells transfected with PCAT-1 shRNA-1; shPCAT-1-2, CRC cells transfected with PCAT-1 shRNA-2. “*,” significantly different from parental cells, P < 0.05. “#,” significantly different from NC cells, P < 0.05

Knockdown of PCAT-1 reduced the invasiveness in Caco-2 and HT-29 cells. a, b Representative images and quantitative analysis of transwell assays in Caco-2 and HT-29 cells. The numbers of cell invading through the matrigel-coated membrane were lower in the cells transfected with PCAT-1 shRNAs. Parental, non-transfected Caco-2 and HT-29 cells; NC, CRC cells transfected with non-targeting shRNA; shPCAT-1-1, CRC cells transfected with PCAT-1 shRNA-1; shPCAT-1-2, CRC cells transfected with PCAT-1 shRNA-2. “*,” significantly different from parental cells, P < 0.05. “#,” significantly different from NC cells, P < 0.05. Magnification, ×200

Knockdown of PCAT-1 down-regulated the expression of EMT-related proteins in Caco-2 and HT-29 cells. a, b Representative images and quantitative analysis of western blotting for MMP-2, MMP-9, E-Cadherin, and Snail in Caco-2 and HT-29 cells. Parental, non-transfected Caco-2 and HT-29 cells; NC, CRC cells transfected with non-targeting shRNA; shPCAT-1-1, CRC cells transfected with PCAT-1 shRNA-1; shPCAT-1-2, CRC cells transfected with PCAT-1 shRNA-2. “*,” significantly different from parental cells, P < 0.05. “#,” significantly different from NC cells, P < 0.05

Knockdown of PCAT-1 decreased the activities of MMP-2 and MMP-9 in Caco-2 and HT-29 cells. a, b Representative images and quantitative analysis of gelatin zymography for MMP-2 and MMP-9 activities in Caco-2 and HT-29 cells. Parental, non-transfected Caco-2 and HT-29 cells; NC CRC cells transfected with non-targeting shRNA; shPCAT-1-1, CRC cells transfected with PCAT-1 shRNA-1; “*,” significantly different from parental cells, P < 0.05. “#,” significantly different from NC cells, P < 0.05
Knockdown of PCAT-1 increased the susceptibility of CRC cells to 5-Fu-induced apoptosis
The role of PCAT-1 in MDR in CRC cells was studied by treating Caco-2 and HT-29 cells with 5-Fu. For both cell lines after 5-Fu treatment, the OD490 value in PCAT-1 knockdown cells were significantly lower than that in the parental cells (Fig. 7a, b). Additionally, 5-Fu treatment led to significant increases in the early and late apoptotic rates in PCAT-1-silenced cells as compared with the parental and NC-transfected cells (Fig. 7c, d). Consistently, Hoechst staining showed that the nuclei in the Parental and NC cells were regularly shaped and stained in dark blue, while the nuclei in PCAT-1-silenced cells were fragmented and stained by the Hoechst dye in bright blue (Fig. 7e, f).

Knockdown of PCAT-1 sensitized Caco-2 and HT-29 cells to 5-Fu. a, c, e Cell viability of PCAT-1-silenced Caco-2 cells was reduced and cell apoptosis in PCAT-1-silenced Caco-2 cells was induced by 5-Fu treatment to a greater extent when compared with parental and NC-transfected Caco-2 cells. b, d, f Cell viability of PCAT-1-silenced HT-29 cells was further reduced and cell apoptosis in PCAT-1-silenced HT-29 cells was further induced by 5-Fu as compared with parental and NC-transfected HT-29 cells. Parental, non-transfected Caco-2 and HT-29 cells; NC CRC cells transfected with non-targeting shRNA; shPCAT-1-1, CRC cells transfected with PCAT-1 shRNA-1; shPCAT-1-2, CRC cells transfected with PCAT-1 shRNA-2. “*,” significantly different from parental cells, P < 0.05. “#,” significantly different from NC cells, P < 0.05. Magnification, ×400
PCAT-1 promotes CRC cell invasion through c-Myc
As shown in Fig. 8a, b, knockdown of PCAT-1 resulted in a dramatic suppression on the production of c-Myc in both Caco-2 and HT-29 cells. To elucidate the role of c-Myc in the function of PCAT-1 in CRC cells, c-Myc was overexpressed in PCAT-1-silenced CRC cells. It was found that transfection of c-Myc expression vector significantly up-regulated the levels of c-Myc in both CRC cell lines (Fig. 8c, d) (P < 0.05). Even with PCAT-1 knockdown, the levels of c-Myc were still significantly higher than that in the respective parental cells (P < 0.05). Results of transwell assays showed that overexpression of c-Myc in CRC cells was associated with an increased invading ability in CRC cells, and it also partially restored the invasiveness in PCAT-1-silenced CRC cells (Fig. 8f) (P < 0.05). Taken together, down-regulation of c-Myc expression by PCAT-1 knockdown impaired the invasiveness of CRC cells, and overexpression of c-Myc reversed the effect of PCAT-1 knockdown. Thus, it was inferred that PCAT-1 may promote CRC cell invasion in a c-Myc-dependent manner.

PCAT-1 promotes invasion of CRC cells through c-Myc. a, c, e The expression of c-Myc was down-regulated by PCAT-1 knockdown in Caco-2 cells and overexpression of c-Myc counteracted the suppression of PCAT-1 knockdown on cell invasiveness. b, d, f The expression of c-Myc was down-regulated by PCAT-1 knockdown in HT-29 cells and overexpression of c-Myc counteracted the suppression of PCAT-1 knockdown on cell invasiveness. Parental + vector, Caco-2 and HT-29 cells transfected with the pcDNA3.1 + vector. shPCAT-1-1 + vector, PCAT-1 knockdown cells transfected with the pcDNA3.1 + vector. Parental + c-Myc, CRC cells transfected with the c-Myc expression vector. shPCAT-1-1 + c-Myc group, PCAT-1 knockdown cells transfected with the c-Myc expression vector. “*,” significantly different from parental cells, P < 0.05. “#,” significantly different from NC cells, P < 0.05. “$,” significantly different from Parental + vector group, P < 0.05. “&,” significantly different from shPCAT-1 + vector group, P < 0.05. Magnification, ×400
Discussion
CRC is a major type of malignancy, and the poor prognosis of CRC is largely attributed to its high metastasis potential [24]. In the recent years, with the development of sequencing techniques, gene expression profiling studies have revealed lots of deregulated lncRNAs in CRC [25, 26]. For example, HOTAIR is a powerful predictor of metastasis and poor prognosis in colon cancer because of its regulatory role in the EMT process [27]. Moreover, CCAT2 contributes to metastatic progression and chromosomal instability in CRC by targeting TCF7L2 and its downstream MYC and WNT signaling [28]. Following these studies, the current study, for the first time, explored the mechanism through which PCAT-1 was correlated with the poor prognosis of CRC patients [15]. It was found that PCAT-1 was highly expressed in clinical CRC samples, and knockdown of this lncRNA suppressed the viability and motility of CRC cells while sensitized cancer cells to 5-Fu treatment. Moreover, our study also showed that knockdown of PCAT-1 resulted in down-regulation of c-Myc in both CRC cell lines. Overexpression of c-Myc partially reversed the inhibitory effect of PCAT-1 knockdown on the invasiveness of CRC cells. These findings suggest that c-Myc, as a downstream target of PCAT-1, mediates the regulatory function of PCAT-1 in CRC cell invasion.
PCAT-1 gene located on chromosome 8q24 and ~725 kb upstream of the c-Myc gene, in which somatic gain in copy number has been observed in 18% colorectal adenomas and 34% of total CRCs [15]. Ge et al. found that aberrant expression of PCAT-1 in CRC was independent of copy number variation, which was consistent with the characteristics observed in PCa [12]. Regarding the mechanism underlying PCAT-1′s function in CRC, it has been revealed that the molecule, as a target of polycomb repressive complex 2 (PRC2) [12], promotes cancer cell proliferation and aggressiveness [12]. Moreover, Prensner and colleagues have demonstrated that PCAT-1 stabilizes c-Myc in PCa cells by interfering with miR-34a and modulating the 3′UTR activity of MYC [23]. However, the potential interaction between PCAT-1 and c-Myc in CRC cells is unclear, and it was the major objective of the current study.
c-Myc is a well-studied regulator of cell proliferation and is ubiquitously dys-regulated in multiple cancer types [18, 23, 29, 30]. Overexpression of c-Myc is associated with wide-ranging consequences in oncogenesis [31]. It is convinced that up to one-third of all human genes is regulated by c-Myc in certain context [32]. In tumorigenesis, c-Myc is post-transcriptionally regulated by miRs, especially miR-34a [33, 34]. Consistent with previous studies on PCa, our results showed that knockdown of PCAT-1 in CRC cells was associated with down-regulation of c-Myc production. Overexpression of c-Myc in PCAT-1 knockdown cells counteracted the inhibitory effect of PCAT-1 knockdown on the invasiveness of CRC cells. Our findings evidently showed a regulatory role of PCAT-1 on the expression of c-Myc and a central role of c-Myc in mediating PCAT-1-promoted aggressiveness of CRC cells. As mentioned above, Prensner and colleagues showed that PCAT-1 regulated c-Myc expression through the post-transcriptional activity of the MYC 3′UTR in PCa cells, and they also characterized the MYC-targeting microRNA which was regulated by PCAT-1, i.e., miR-34a. However, the authors failed to locate the direct binding site for miR-34a on the PCAT-1 transcript, and thus they speculated that the regulatory action of PCAT-1 on miR-34a was indirect [23]. Although the effect of PCAT-1 knockdown on the level of miR-34a was not detected in the current study, we also hypothesize based on the study by Prensner et al. that PCAT-1 regulates c-Myc expression in CRC cells in a miR-34a-dependent manner. This hypothesis would be validated in our future studies. Moreover, the overexpression of PCAT-1 in CRC may also represent an adaptive cellular mechanism to accelerated proliferation and the development of MDR. As validated by our results, knockdown of PCAT-1 suppressed cell growth and reduced the resistance to 5-Fu in both CRC cell lines. Therefore, the pro-tumor function of PCAT-1 in CRC may be multi-pronged in addition to c-Myc-dependent actions.
PCAT-1 may be a potential candidate of biomarker for CRC given its substantial overexpression in CRC and its functional roles in CRC progression, aggressiveness, and MDR. Although our study only demonstrated the roles of PCAT-1 in the aggressiveness of CRC cells in vitro, the results of previous and current studies suggest a possible direction for the clinical translation of PCAT-1 in the diagnosis and treatment of CRC. For better management of CRC, more work is needed to reveal the molecular mechanisms underlying the functions of PCAT-1 in CRC development and progression.
References
Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB (2010) Annotating non-coding regions of the genome. Nat Rev Genet 11(8):559–571
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12(12):861–874
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F (2012) Landscape of transcription in human cells. Nature 489(7414):101–108
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM (2013) Gene regulation by the act of long non-coding RNA transcription. BMC Biol 11(1):59
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22(9):1775
Huarte M, Rinn JL (2010) Large non-coding RNAs: missing links in cancer? Hum Mol Genet 19(R2):R152–R161
Gibb EA, Brown CJ, Wan LL (2011) The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10(1):38
Prensner JR, Chinnaiyan AM (2011) The emergence of lncRNAs in cancer biology. Cancer Discov 1(5):391–407
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough H, Helms JA, Farnham PJ, Segal E (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129(7):1311–1323
Gabory A, Jammes H, Dandolo L (2010) The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays 32(6):473–480
Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M (2007) Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132(1):330–342
Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD (2011) Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 29(8):742–749
Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani I, Grasso C, Kominsky HD (2011) Transcriptome sequencing identifies PCAT-1, a novel lincRNA implicated in prostate cancer progression. Nat Biotechnol 29(8):742–749
Shi WH, Wu QQ, Li SQ, Yang TX, Liu ZH, Tong YS, Tuo L, Wang S, Cao XF (2015) Upregulation of the long noncoding RNA PCAT-1 correlates with advanced clinical stage and poor prognosis in esophageal squamous carcinoma. Tumor Biol 36(4):2501–2507
Ge X, Chen Y, Liao X, Liu D, Li F, Ruan H, Jia W (2013) Overexpression of long noncoding RNA PCAT-1 is a novel biomarker of poor prognosis in patients with colorectal cancer. Med Oncol 30(2):588
Ottaiano A, Franco R, Aiello TA, Liguori G, Tatangelo F, Delrio P, Nasti G, Barletta E, Facchini G, Daniele B (2006) Overexpression of both CXC chemokine receptor 4 and vascular endothelial growth factor proteins predicts early distant relapse in stage II-III colorectal cancer patients. Clin Cancer Res 12(9):2795
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66(1):7
He X, Tan X, Wang X, Jin H, Liu L, Ma L, Yu H, Fan Z (2014) C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumor Biol 35(12):12181–12188
Vennstrom B, Sheiness D, Zabielski J, Bishop JM (1982) Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol 42(3):773–779
Dallafavera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM (1982) Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA 79(24):7824–7827
Nesbit CE, Tersak JM, Prochownik EV (1999) MYC oncogenes and human neoplastic disease. Oncogene 18(19):3004–3016
Vita M, Henriksson M (2006) The Myc oncoprotein as a therapeutic target for human cancer. Seminars in cancer biology. Academic Press, San Diego
Prensner JR, Chen W, Han S, Iyer MK, Cao Q, Kothari V, Evans JR, Knudsen KE, Paulsen MT, Ljungman M (2014) The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia 16(11):900–908
Siegel R, Naishadham MMD, Jemal DVMPA (2012) Cancer statistics, 2012. CA Cancer J Clin 62(1):10–29
Chen H, Xu J, Hong J, Tang R, Zhang X, Fang JY (2014) Long noncoding RNA profiles identify five distinct molecular subtypes of colorectal cancer with clinical relevance. Mol Oncol 8(8):1393–1403
Zhu Y, Bian X, Dingwei YE, Yao X, Zhang S, Dai BO, Zhang H, Shen Y (2014) Long noncoding RNA expression signatures of bladder cancer revealed by microarray. Oncol Lett 7(4):1197
Wu ZH, Wang XL, Tang HM, Jiang T, Chen J, Lu S, Qiu GQ, Peng ZH, Yan DW (2014) Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncol Rep 32(1):395–402
Ling H, Spizzo R, Atlasi Y, Nicoloso M, Shimizu M, Redis RS, Nishida N, Gafà R, Song J, Guo Z (2013) CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res 23(9):1446
Sikora K, Chan S, Evan G, Gabra H, Markham N, Stewart J, Watson J (1987) c-myc oncogene expression in colorectal cancer. Cancer 59(7):1289
Albihn A, Johnsen JI, Henriksson MA (2010) MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res 107:163–224
Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8(12):976–990
Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA (2010) c-Myc regulates transcriptional pause release. Cell 141(3):432
Yamamura S, Saini S, Majid S, Hirata H, Ueno K, Deng G, Dahiya R (2012) MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 7(1):81–97
Yamamura S, Saini S, Majid S, Hirata H, Ueno K, Chang I, Tanaka Y, Gupta A, Dahiya R (2012) MicroRNA-34a suppresses malignant transformation by targeting c-Myc transcriptional complexes in human renal cell carcinoma. Carcinogenesis 33(2):294–300
Acknowledgements
This study was supported by a grant from Science and Technology Plan Project of Shenyang City (No. F12-277-1-19).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors disclose no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Qiao, L., Liu, X., Tang, Y. et al. Knockdown of long non-coding RNA prostate cancer-associated ncRNA transcript 1 inhibits multidrug resistance and c-Myc-dependent aggressiveness in colorectal cancer Caco-2 and HT-29 cells. Mol Cell Biochem 441, 99–108 (2018). https://doi.org/10.1007/s11010-017-3177-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3177-8