
Abstract
Toll-like receptors (TLRs) are major receptors that mediate the innate immune and inflammatory responses, of which TLR4 has been found most closely related to human atherosclerosis. After ligands are polymerized and activated by TLR, the mitogen-activated protein kinase and nuclear factor-κB (NF-κB) pathways are activated, leading to promotion of NF-κB–regulated transcription of inflammatory factors, thus playing a role in the physiological and pathological processes in atherosclerosis. Oxidized lipoproteins or their components, oxidized lipids, have been confirmed as endogenous TLR receptors. Lysophosphatidic acid (LPA) is an active component of low-density lipoprotein that induces vascular endothelial lesions. However, the mechanism of the TLR4/NF-κB signaling system involved in LPA-induced atherosclerosis has not been fully elucidated. In this study, we investigated the effects of LPA on TLR4 expression, nuclear translocation of NF-κB p65 subunit, and changes in the cytokine tumor necrosis factor α (TNF-α) in human THP-1 cells. LPA upregulated expression of the TLR4 mRNA and protein in THP-1 cells in a dose- and time-dependent manner, induced NF-κB p65 activation synchronously in THP-1 cells, and increased TNF-α secretion. After TLR4 was blocked using TLR4 monoclonal antibody, NF-κB p65 expression and TNF-α secretion were inhibited significantly. These data suggest that LPA can significantly upregulate TLR4 expression and promote NF-κB activation and proinflammatory cytokine secretion in THP-1 cells; it is possible that the TLR4/NF-κB signaling pathway mediates the atherogenic effect of LPA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toll-like receptors (TLRs) are major receptors that mediate the innate immune and inflammatory responses; they were recently found to be closely related to human atherosclerosis. Oxidized lipoproteins or their components, oxidized lipids, have been confirmed as endogenous TLR receptors. The mitogen-activated protein kinase and nuclear factor-κB (NF-κB) pathways are activated following ligand polymerization and activation by TLR, leading to the promotion of NF-κB-regulated inflammatory factor transcription [1–3].
NF-κB is a transcriptional regulatory factor usually bound with the inhibitor of κB (IκB) protein in dimers and is present in the cytoplasm. IκB is phosphorylated when cells are stimulated (by cytokines, hypoxia, virus, bacterial infection). NF-κB is thus activated, localizing from the cytoplasm to the nucleus to activate expression of various target genes. Likewise, TLR-mediated signals induce the abundant expression of genes encoding proinflammatory cytokines, and are thus involved in the inflammatory response and the physiological and pathological processes in atherosclerosis.
To date, TLRs have been found in 12 mammals, of which TLR4 is most closely related to human atherosclerosis [4]. TLR4 is expressed in endothelial cells and macrophages in human coronary atherosclerotic plaques, and oxidized low-density lipoproteins (oxLDL) can induce the upregulation of TLR4 in macrophages [5, 6].
As an active component of LDL, lysophosphatidic acid (LPA) is a low-molecular weight lysophospholipid enriched in platelets and a mildly oxidized LDL that exerts a range of effects in the cardiovascular system that includes modulation of platelet activation, recruitment and activation of inflammatory cells, migration, phenotypic modulation, and proliferation of vascular smooth muscle cells (VSMC) [7, 8]. Thus, its involvement in thrombosis and vascular disease, such as atherosclerosis and vascular remodeling, is critical [9]. However, the role TLR4 plays in LPA-induced atherosclerosis is unclear. In the present study, the effect of LPA on the TLR4/NF-κB signaling pathway in the human monocytic THP-1 cell line was studied to investigate the mechanism of LPA-induced atherosclerosis.
Materials and methods
Reagents
THP-1 cells were from Biochain (Newark, CA, USA); 18:1 LPA (1-oleoyl-sn-glycero-3-phosphate) was purchased from Sigma-Aldrich (St Louis, MO, USA), Purity (TLC) ≥98 %. LPA was dissolved in phosphate-buffered saline (PBS) containing 0.1 % fatty acid-free bovine serum albumin (BSA). Nuclear protein and cytoplasmic protein isolation kits, mouse anti-TLR4 antibody, and rabbit anti–Phospho -NF-κB p65 antibody were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). TLR4 monoclonal antibody (TLR4 mAb, Anti-Human CD284, Clone: HTA125) were from eBioscience (San Diego, CA, USA), SYBR Green PCR Master Mix was from ABI Research (Foster City, CA, USA), and 4′6-diamidino-2-phenylindole (DAPI) was from Invitrogen (Carlsbad, CA, USA).
Cell culture and intervention experiments
THP-1 cells were grown in suspension in RPMI 1640 containing 10 % fetal bovine serum in 5 % CO2 at 37 °C. The medium was replaced every 2–3 days. The cells were passaged when they had grown to 4–6 × 106/ml. Cells were incubated in serum-free medium overnight; seeded in 24-well plates at a density of 106 cells; treated with 0, 0.1, 0.5, 1, 5, and 10 μM LPA; and harvested after 4 h, or treated with 1 μM LPA and harvested after 0, 1, 2, 4, and 8 h. To evaluate the possible contamination of LPA with LPS, the endotoxin content was determined by the chromogenic Limulus amebocyte lysate test, following the manufacturer’s instructions. The endotoxin content in the 100 μM LPA solution was ≤3.45 × 10−3pg/ml, which is far below the concentration to affect THP-1 cells under our assay conditions. The TLR4 mRNA level was determined using fluorescence quantitative reverse transcription-polymerase chain reaction (RT-PCR). Cell proteins were extracted, and the TLR4 protein level was determined using Western blotting. The nucleoproteins were extracted and the expression and translocation of the NF-κB p65 subunit was detected by Western blotting and immunofluorescence, respectively. The secretion of tumor necrosis factor α (TNF-α) into cell culture supernatants was measured by enzyme-linked immunosorbent assay (ELISA).
Fluorescent quantitative RT-PCR
Cellular total RNA was extracted using the one-step TRIzol procedure, and then the reverse transcription reaction was carried out using a Taq RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fermentas, Burlington, Canada) according to its instructions to synthesize the first chain of complementary DNA. Real-time quantitative PCR was carried out using SYBR Green fluorescent dye with β-actin as the internal control. The primer sets for TLR4 and β-actin were as follows: TLR4 forward: 5′-acatcaaatgcccctactca-3′ and reverse: 5′-ctaaaccagccagaccttga-3′; β-actin forward: 5′-cattaaggagaagctgtgct-3′; and reverse: 5′-gttgaaggtagtttcgtgga-3′. The fluorescent quantitative PCR reaction system contained 1 μl cDNA, 12.5 μl 2 × SYBR Green PCR Master mix, and 100 nm each upstream and downstream primers, and was topped up to 25 μL with double-distilled water. The amplification reaction was performed at 95 °C for 5 min, 94 °C for 20 s, 56 °C for 20 s, 72 °C for 20 s, 72 °C for 5 min, and 55 °C for 10 s, for 40 cycles. The melting point curve was analyzed after the end of the amplification, and measurement between 60 and 95 °C was made at increments of 0.5 °C and 5 s each time. The data were automatically collected using an ABI 7500 Real-Time PCR System (Applied Biosystem Research), and the threshold cycle (CT) value was analyzed on a personal computer. The relative level of TLR4 mRNA expression to that of β-actin was derived using ΔCt (ΔCtTLR4 = CtTLR4−Ct β-actin). The data were processed using the 2−ΔΔCt (RQ) method. Three wells were run in parallel three times for each specimen.
Western blot analysis
The cells were harvested to prepare lysates according to a previously described protocol [10]. NF-κB p65 was extracted according to the instructions of a Nuclear Protein Extraction Kit (Beyotime, Jiangsu, China), and the protein concentration was determined using the bicinchoninic acid method. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed to determine the level of TLR4 protein and nucleoprotein NF-κB p65, followed by the Western blot analysis with TLR4- and NF-κB p65-specific antibodies used according to the manufacturer’s instructions. Briefly, the treatment of THP-1 cells with various agents was halted by the addition of trichloroacetic acid, and total cellular proteins were extracted with SDS sample buffer as previously described [10]. An equal amount of protein (5 μg) was resolved by SDS-PAGE and transferred to a nitrocellulose membrane (Pierce, Rockford lL USA). Immunoreactivity was visualized using an enhanced ECL detection kit (Pierce) and exposed to radiographic film. The blots were scanned and quantified with Gelpro32 (Beta 4.02 version for Windows Media Cybernetics, Marlow, UK). When the expression level of nucleoprotein NF-κB p65 was detected, the GAPDH expression level was determined simultaneously.
The GAPDH expression was negative which indicated that the nuclear extracts are free of contaminating cytoplasmic protein.
Immunofluorescence staining
Immunofluorescence staining was performed according to a previously described protocol [11]. Briefly, cells were fixed with 4 % paraformaldehyde in phosphate-buffered saline (PBS) containing 10 % fetal calf serum for 20 min at room temperature, followed by permeabilization for 10 min using 0.3 % (w/v) Triton X-100 in PBS. Primary antibody dilutions were Anti-NFkB p65 antibody 1:100 and secondary antibody dilutions were Goat Anti-Rabbit IgG H&L Alexa Fluorj 488 1:200 (Sigma-Aldrich). After incubation with primary antibody overnight at 4 °C, slides were incubated for 1 h with a secondary antibody. Slides with stained cells were mounted in 90 % glycerol with 1 μg/ml DAPI and examined with a fluorescence microscope. Images were captured with a Nikon inverted fluorescent microscope with attached CCD (charge-coupled device) camera at ×400 magnification.
Statistical analysis
Data are presented as mean ± SE; statistical significance was determined using analysis of variance for multiple comparisons. P < 0.05 was considered statistically significant.
Results
Effects of LPA on TLR4 mRNA and protein levels, and NF-κB p65 nuclear translocation
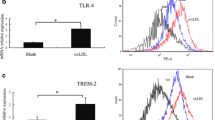
To investigate the effects of LPA on the expression levels of TLR4 mRNA and protein and NF-κB p65 in THP-1 cells, cells were cultured in serum-free medium overnight. Cells were then treated using 0, 0.1, 0.5, 1, 5, and 10 μM LPA for 4 h, or 1 μM LPA for 0, 1, 2, 4, and 8 h. Total RNA was extracted and the level of TLR4 mRNA was determined using fluorescent quantitative RT-PCR. The levels of extracted TLR4 protein and NF-κB p65 nucleoprotein were determined with Western blotting. Nuclear translocation of NF-κB p65 was detected by immunofluorescence. The expression levels of TLR4 mRNA and protein and NF-κB p65 increased in THP-1 cells synchronously with the increased LPA titer, peaking at 1 μM LPA; subsequently, the expression levels of TLR4 mRNA, protein (Fig. 1a), and NF-κB p65 (Fig. 2a) decreased. Following different durations of 1 μM LPA treatment, the expression levels of TLR4 mRNA, protein (Fig. 1b), and NF-κB p65 nucleoprotein (Fig. 2c) in THP-1 cells increased synchronously with time and peaked at 4 h before subsequently declining. At the same time, nuclear translocation of NF-κB p65 was examined with a fluorescence microscope. In unstimulated cells, NF-κB p65 was mainly located in the cytoplasm. In LPA-stimulated cells, NF-κB p65 translocation to the nucleus significantly increased (Fig. 2b, d). This indicated that LPA upregulated the expression of TLR4 mRNA and protein and NF-κB p65 activation in a dose- and time-dependent manner in THP-1 cells.

LPA upregulated the expression levels of TLR4 mRNA and protein in THP-1 cells. a (Top) TLR4 mRNA levels determined using fluorescent quantitative RT-PCR; (bottom) TLR4 protein levels determined using Western blotting. Cells were incubated with different concentrations of LPA for 4 h. *Compared with the other groups: P < 0.01. b (Top) TLR4 mRNA levels determined using fluorescent quantitative RT-PCR; (bottom) TLR4 protein levels determined using Western blotting. Cells were treated with 1 μM LPA for different durations. *Compared with the other groups: P < 0.01. Values are mean ± SE, n = 3. RQ Relative quotient, GAPDH glyceraldehyde-3-phosphate dehydrogenase

LPA upregulated NF-κB p65 expression and activation in THP-1 cells. a Level of extracted NF-κB p65 determined using western blotting after THP-1 cells had been incubated with different concentrations of LPA for 4 h. *Compared with the other groups: P < 0.01. b Representative immunofluorescence fields showing staining for NF-κB p65 (green) and nucleic acid (blue) in THP-1 cells stimulated with different concentrations of LPA for 4 h (×400). c Level of extracted NF-κB p65 determined using Western blotting after THP-1 cells had been incubated with 1 μM LPA for different durations. *Compared with the other groups: P < 0.01. Values are mean ± SE, n = 3. d Representative immunofluorescence fields showing staining for NF-κB p65 (green) and nucleic acid (blue) in THP-1 cells stimulated with 1 μM LPA for different durations (×400). H2, Histone H2. (Color figure online)
Effect of LPA on levels of secreted TNF-α
The level of TNF-α in the supernatant of THP-1 cells stimulated by 1 μM LPA for 4 h was significantly higher than that in the control group (0 μM LPA), continuously increasing in the groups stimulated with 1–10 μM LPA dose-dependently (Fig. 3a). TNF-α levels in the groups stimulated with 0.1 and 0.5 μM LPA were not significantly different from that of the control group. When THP-1 cells were stimulated using 5 μM LPA over 0, 1, 2, 4, and 8 h, TNF-α levels in the 2, 4, and 8 h groups were significantly increased in contrast to the control group, but there were no significant differences among these groups (Fig. 3b).

The effect of LPA on level of secreted TNF-α in THP-1 cells. TNF-α levels in cell supernatant were determined using ELISA after THP-1 cells were stimulated with a different concentrations of LPA for 4 h, [△Compared with control group (0 μM LPA) and group (0.1 μM LPA) P < 0.01, *Compared with group (0.5 μM LPA) P < 0.05 *Compared with the other groups, P < 0.01); values are mean ± SE, n = 3] or b 5 μM LPA for different durations [*Compared with group (0 h) P < 0.05. *Compared with group (0 h, 1 h) P < 0.01; △Compared with group (8 h) <0.05, ※Compared with group (0 h) P < 0.01; values are mean ± SE, n = 3]
NF-κB p65 activation and TNF-α secretion by TLR4-mediated LPA induction
To investigate the effect of TLR4 on LPA-induced NF-κB p65 activation and TNF-α secretion in THP-1 cells further, TLR4 was blocked with TLR4 mAb. The 1-h TLR4 mAb (5 mg/l) pretreatment of THP-1 cells significantly inhibited NF-κB p65 expression and nuclear translocation (Fig. 4a), and TNF-α secretion (Fig. 4b) in THP-1 cells stimulated by 1 μM LPA.

The effect of TLR4 mAb on LPA-induced NF-κB p65 activation and TNF-α secretion in THP-1 cells. a Level of NF-κB p65 expression determined using Western blotting in THP-1 cells pretreated with 5 mg/l TLR4 mAb for 1 h and then stimulated with 1 μM LPA for 4 h. * P < 0.01 versus with the other groups; △P < 0.05 versus control (LPA alone) *P < 0.05 versus control (Vehicle) values are mean ± SE, n = 3. H2, Histone H2. b Representative immunofluorescence fields showing staining for NF-κB p65 (green) and nucleic acid (blue) in THP-1 cells following 1-h pretreatment with 5 mg/l TLR4 mAb and 4-h stimulation with 1 μM LPA (×400). c The level of TNF-α secreted in the cell supernatant was determined using ELISA. * P < 0.01 versus control (Vehicle), and control (TLR4 mAb alone) # P < 0.05 versus group (LPA alone). (Color figure online)
Discussion
Oxidized LDL is an endogenous immune response activator and is recognized as an atherosclerosis-inducing factor. Studies have shown that vascular or systemic inflammatory diseases are induced by oxLDL, infection, and heat shock protein (three factors involved in the development of atherosclerosis) in part via the TLR4/NF-κB signaling pathway [12]. A larger amount of LPA is generated during mild LDL oxidation and functions by binding to the LDL surface. LPA is an active component of LDL. LDL bound by LPA can promote platelet activation and stimulate the formation of stress fibers and gaps in vascular endothelial cells. However, the mechanism of the TLR4/NF-κB signaling system involved in LPA-induced atherosclerosis is unclear. The present study revealed that LPA could significantly upregulate TLR4 expression and promote NF-κB activation in THP-1 cells. The role of LPA in inducing atherosclerosis might be partially mediated by the TLR4/NF-κB signaling pathway.
Atherosclerosis is a chronic inflammatory disease. The immune response-associated inflammatory process is present throughout the atherosclerotic process. The inflammation and immune responses are critical in the occurrence and development, and formation, evolution, and rupture of unstable plaque in atherosclerosis [13, 14]. The innate immune mechanism is an important participant in promoting atherosclerotic inflammation. Two major risk factors, in atherosclerosis, hyperlipidemia, and inflammatory lesions, are interlinked through the immune response [5]. TLRs are a family of pattern recognition receptors for the molecular patterns associated with immune cell surface recognition of pathogens, playing a key role in the signal transduction of cell activation, and acting as a bridge between natural and acquired immunity [15, 16].
An increasing amount of evidence supports the important effect of the TLR signaling pathway in the initiation and development of atherosclerosis [6]. Many in vivo experiments on atherosclerosis mouse models have provided compelling verification that TLR2 and TLR4 in the TLR signaling pathway are mainly involved in the pathophysiological process in atherosclerosis [17]. TLR2 and TLR4 are expressed in macrophages, dendritic cells, endothelial cells, and VSMC, which are involved in atherosclerosis. TLR2 is also expressed in some T lymphocytes [17–19]. In an atherosclerosis mouse model, with deleted LDL receptors (LDLR-/-) and apolipoprotein E (ApoE-/-), the expression levels of TLR2 and TLR4 were significantly higher in atherosclerotic plaques [6, 20]. Edfeldt et al. found that TLR4 expression was significantly increased in atherosclerotic plaques, and confirmed that a series of atherosclerosis-linked cytokines were synthesized and subsequently released via the recognition function of TLR4 [6]. Comparing a normal mouse model with an atherosclerosis mouse model, Michelsen et al. and Mullick et al. found that the atherosclerotic lesion area in TLR4-deleted or TLR2-missing and ApoE-/- mice was reduced by 55 % [16, 21]; the atherosclerotic lesion area of the aortic arch in ApoE-/- and TLR4-deleted mice was reduced by 25 %, and the volume of lipid core in the aortic sinuses was reduced in the ApoE-/- and TLR4-deleted mice. The macrophages invading the atherosclerotic lesions were reduced by 65 % [21]. Hollestelle et al. [22] showed that TLR4 was a key receptor for arterial remodeling; it promoted plaque formation and outer arterial wall remodeling when exposed to lipopolysaccharide in ApoE Leiden transgenic mice with atherosclerosis in a femoral artery cuff model, but no outer arterial wall remodeling occurred in TLR4-deleted mice.
The present study found that LPA could upregulate the expression levels of TLR4 mRNA and protein in THP-1 cells in a dose- and time-dependent manner. At the same time, LPA could activate NF-κB and promote NF-κB p65 nuclear translocation and expression with the same change trend as TLR4 expression. LPA could also promote secretion of the inflammatory factor TNF-α. The expression and nuclear translocation of NF-κB p65 and the level of TNF-α declined significantly after the TLR4 signal transduction pathway was blocked by TLR4 antibody. Mononuclear macrophages are one of the major inflammatory cells that are involved in the atherosclerotic inflammatory response (Fig. 5). TNF-α from macrophages can promote atherosclerosis and cause the apoptosis of VSMC. LPA can promote endothelial injury and increase cell adhesion and endothelin. This study demonstrated that it could upregulate TLR4 expression in monocytes as a main initiator in the vascular wall inflammatory response. TLR4 can promote the subsequent activation of NF-κB and upregulation of the expression of proinflammatory cytokines such as TNF-α after binding with ligands, which are involved in the atherosclerosis process [23].

TLR4/NF-κB signaling pathways involved in LPA-induced atherosclerosis suggested by the present results. LPA could upregulate TLR4 expression in monocytes, which has probably been involved with GPCR signaling, promotes NF-κB activation, and facilitates the secretion of the inflammatory cytokines, such as TNF-α in THP-1 cells, involved in the atherosclerotic inflammatory response
Conclusions
We have demonstrated that LPA significantly upregulates TLR4 expression, promotes NF-κB activation, and facilitates secretion of the inflammatory cytokine TNF-α in THP-1 cells. TLR4 and inflammatory pathways were activated through LPA stimulation..
Abbreviations
- TLRs:
-
Toll-like receptors
- NF-κB:
-
Nuclear factor-κB
- LPA:
-
Lysophosphatidic acid
- TNF-α:
-
Tumor necrosis factor α
References
Raveendran VV, Tan X, Sweeney ME, Levant B, Slusser J, Stechschulte DJ, Dileepan KN (2011) Lipopolysaccharide induces H1 receptor expression and enhances histamine responsiveness in human coronary artery endothelial cells. Immunology 13:578–588
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Dunzendorfer S, Lee HK, Soldau K, Tobias PS (2004) Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of LBP and sCD14 in mediating LPS responses. FASEB J 18:1117–1119
Balogh S, Kiss I, Csaszar A (2009) Toll-like receptors: link between “danger” ligands and plaque instability. Curr Drug Targets 10:513–518
Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu XP, Rajavashisth TB, Yano J, Kaul S, Arditi M (2001) Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 104:3103–3108
Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ (2002) Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation 105:1158–1161
Clària J (2006) Regulation of cell proliferation and apoptosis by bioactive lipid mediators. Recent Pat Anticancer Drug Discov 1:369–382
Tigyi G (2001) Physiological responses to lysophosphatidic acid and related Glycerol-phospholipids. Prostaglandins Other Lipid Mediat 64:47–62
Siess W, Tigyi G (2004) Thrombogenic and atherogenic activities of lysophosphatidic acid. J Cell Biochem 92:1086–1094
Ai S, Kuzuya M, Koike T, Asai T, Kanda S, Maeda K, Shibata T, Iguchi A (2001) Rho–Rho kinase is involved in smooth muscle cell migration through myosin light chain phosphorylation-dependent and -independent pathways. Atherosclerosis 155:321–327
Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, Wagstaff KM, Dunstone MA, Sloan J, Whisstock JC, Kaper JB, Robins-Browne RM, Jans DA, Frankel G, Phillips AD, Coulson BS, Hartland EL (2010) The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog 6:e1000898
Banchereau J, Steinman RM (1998) Dendritic cells and control of immunity. Nature 392:245–252
Libby P (2002) Inflammation in atherosclerosis. Nature 420:868–874
Hansson GK, Libby P, Schonbeck U, Yan ZQ (2002) Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res 91:281–291
Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R (2001) Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2:947–950
Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U.S.A. 101:10679–10684
Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K (2000) Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol 164:3471–3475
Ueta M, Nochi T, Jang MH, Park EJ, Igarashi O, Hino A, Kawasaki S, Shikina T, Hiroi T, Kinoshita S, Kiyono H (2004) Intracellularly expressed TLR2s and TLR4s contribution to an immunosilent environment at the ocular mucosal epithelium. J Immunol. 173:3337–3347
Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW (2002) Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 298:1025–1029
Mullick AE, Soldau K, Kiosses WB, Bell TA 3rd, Tobias PS, Curtiss LK (2008) Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J Exp Med 205:373–383
Mullick AE, Tobias PS, Curtiss LK (2005) Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest 115:3149–3156
Hollestelle SC, De Vries MR, Van Keulen JK, Schoneveld AH, Vink A, Strijder CF, Van Middelaar BJ, Pasterkamp G, Quax PH, De Kleijn DP (2004) Toll-like receptor 4 is involved in outward arterial remodeling. Circulation 109:393–398
Knuefermann P, Nemoto S, Baumgarten G, Misra A, Sivasubramanian N, Carabello BA, Vallejo JG (2002) Cardiac inflammation and innate immunity in septic shock:is there a role for Toll-like receptors? Chest 121:1329–1336
Acknowledgments
This work was supported by a Grant from the Nature Science Foundation of Fujian province, China (No.2010D010).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Bo Yang, Zhibin Zhou, Xiaohao Li and Jianping Niu contributed to the work equally and should be regarded as co-first authors.
Rights and permissions
About this article

Cite this article
Yang, B., Zhou, Z., Li, X. et al. The effect of lysophosphatidic acid on Toll-like receptor 4 expression and the nuclear factor-κB signaling pathway in THP-1 cells. Mol Cell Biochem 422, 41–49 (2016). https://doi.org/10.1007/s11010-016-2804-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2804-0