
Abstract
Modulation of Ca2+ homoeostasis in cardiac myocytes plays a major role in beat-to-beat regulation of heart function. Previous studies suggest that sphingosine-1-phosphate (S1P), a biologically active sphingomyelin metabolite, regulates Ca2+ handling in cardiac myocytes, but the underlying mechanism is unclear. In the present study, we tested the hypothesis that S1P-induced functional alteration of intracellular Ca2+ handling includes the L-type calcium channel current (ICa,L) via a signalling pathway involving P21-activated kinase 1 (Pak1). Our results show that, in rat ventricular myocytes, S1P (100 nM) does not affect the basal activity of ICa,L but is able to partially reverse the effect of the β-adrenergic agonist Isoproterenol (ISO, 100 nM) on ICa,L. S1P (25 nM) also significantly prevents ISO (5 nM)-induced Ca2+ waves and diastolic Ca2+ release in these cells. Our further molecular characterisation demonstrates that Pak1 activity is increased in myocytes treated with S1P (25 nM) compared with those myocytes without treatment of S1P. By immunoprecipitation we demonstrate that Pak1 and protein phosphatase 2A (PP2A) are associated in ventricular tissue indicating their functional interaction. Thus the results indicate that S1P attenuates β-adrenergic stress-induced alteration of intracellular Ca2+ release and L-type Ca2+ channel current at least in part via Pak1–PP2A-mediated signalling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sphingosine-1-phosphate (S1P) is a circulating bioactive sphingolipid and has been identified as an important signalling mediator implicated in the regulation of several cellular processes such as proliferation, migration, contraction, and intracellular calcium mobilisation [1]. S1P has been found to be an intracellular messenger as well as an intercellular messenger [2–4]. The extracellular actions of S1P are mediated through its interaction with the G-protein-coupled receptors. Five structurally related receptors with high affinity for S1P have been so far identified. Evidence suggests that the cardio-pulmonary system has the highest S1P1, S1P2, and S1P3 receptors expression [5, 6], whereas the lymphatic system [7] and the central nervous system [8] express mainly S1P4 and S1P5 receptors, respectively. Animal studies suggest that the administration of S1P may lead to a decrease in cell shortening as well as antagonising isoproterenol (ISO)-induced increases in cAMP and positive inotropy [9]. Interestingly, the S1P1 receptor agonist, SEW2871, was found as efficacious as S1P at antagonising ISO-stimulated contractility, while the administration of the S1P1 + 3 receptor antagonist, VPC23019, blocked the negative inotropic actions of SEW2871, which was observed in S1P3 receptor knockout cardiac myocytes [9]. These findings suggest that the S1P1 receptor may be the primary receptor that mediates the negative inotropic effects of S1P in ventricular myocytes; while the S1P3 receptor may have only a small contribution. These results are consistent with the findings obtained by Levkau group demonstrating that the inhibition of ISO-induced contractility by S1P, may be abolished in cardiac myocytes lacking S1P1 receptor [1].
In summary, the above evidence suggests that S1P may regulate electrophysiological and contractile properties in cardiac myocytes under both basal and β-adrenergic stress conditions, however the underlying signalling mechanisms mediating such regulation largely remain elusive. In the present study, we tested the hypothesis that S1P-induced functional alteration of intracellular Ca2+ handling includes the L-type calcium channel current (ICa,L) via a signalling pathway involving P21-activated kinase 1 (Pak1), a serine–threonine protein kinase regulated by Ras-related small G proteins. Our hypothesis is based on our prior demonstration that S1P may regulate the activation of Pak1 and protein phosphatase 2A (PP2A) [10, 11]. The aims of this study are, therefore two-fold: (i) to determine the functional effect of S1P on ICa,L and Ca2+ transients under basal and β-stimulation states in fresh isolated rat ventricular cells; (ii) to determine the molecular relationship of the S1P receptor, Pak1, and PP2A through co-localisation studies.
Materials and methods
As S1P exerts maximal biological effects at concentrations of 10–100 nm [12], 25 and 100 nM were arbitrarily chosen and used in this study.
Cell isolation
Male rats (3–4 months of age) were killed by cervical dislocation according to the Home Office Guidance on the operation of the Animals (Scientific Procedures) Act 1986 (H.M.S.O.). The heart was rapidly excised, washed in a modified Tyrode medium containing EGTA and heparin, and mounted on a Langendorff apparatus for retrograde perfusion via the aorta. Perfusion was initially carried out in a modified Tyrode solution containing (mM): NaCl 136, KCl 5.4, NaHCO3 12, Na+ pyruvate 1, NaH2PO4 1, MgCl2 1, EGTA 0.04, glucose 5; gassed with 95 % O2/5 % CO2 to maintain a pH of 7.4. This was replaced after 2 min with a modified Tyrode solution containing 30 mg of collagenase and 10 μL of 0.1 mM CaCl2 (type II, Worthington Biochemical Corp., Lakewood, NJ, USA), but no EGTA. Additional 20 μL aliquots of 0.1 mM CaCl2 were added during perfusion. After this enzymatic digestion of the heart, myocytes were isolated by trituration (mechanical disruption using a flame-smoothed glass pipette) and stored at 4 °C in KB medium containing (mM): KCl 70, MgCl2 5, K+ glutamine 5, taurine 20, EGTA 0.04, succinic acid 5, KH2PO4 20, HEPES 5, glucose 10; pH to 7.2 with KOH.
Electrophysiology recording
Ventricular myocytes were transferred to a small tissue bath on the stage of an inverted microscope on a vibration isolation table and superfused with modified Tyrode containing (mM): NaCl 125, NaHCO3 25, KCl 5.4, NaH2PO4 1.2, MgCl2 1, CaCl2 1.8, glucose 5.5; gassed with 95 % O2/5 % CO2 to maintain a pH of 7.4. Solutions were perfused by gravity and experiments were performed at 36 ± 1 °C. Calcium currents were recorded using the perforated patch clamp technique (Axoclamp-2B amplifier, Axon Instruments, USA). Patch pipettes (4–6 MΩ) contained (mM): KCl 150, NaCl 5, MgCl2 2, K2ATP 2, HEPES 5, pH 7.2 with KOH; perforation using amphotericin (Sigma-Aldrich, 250 µg/mL) occurred within a few minutes after seal formation. Electrophysiology data were analysed using pCLAMP software (Axon Instruments).
Measurements of Ca2+ transients
Fresh isolated rat ventricular myocytes in Krebs–Henseleit solution were treated with 5 µM of Fluo-4AM and incubated at 32 °C for 15 min. After having absorbed the calcium dye, cells were transferred to a Perspex 30 × 15 × 5 mm perfusion chamber on the stage of a Leica (model no. DMIRM) microscope, ×40 magnification. After 5 min of settling the cells onto the bottom plate of the chamber, the cells were superfused with Krebs solution at the rate of 2.5–3 ml/min at 37 °C. The cells were stimulated by field stimulation at ×1.5 threshold of contraction of the cell at frequency of 1 Hz with a pulse width of 2 ms. Calcium fluorescence was imaged by Cairn Fluorescence Imaging system (Cairn Research Ltd, UK). Fluorescence was excited at 488 nm and emitted light at 494/506 nm. Measured fluorescence was normalised to resting levels (F/F0). The transients were recorded and analysed using the Axon pCLMAP™ 10.
Immunocytochemistry, imaging with confocal microscopy, western immunoblotting, and co-immunoprecipitation
Experiments were carried out on ventricular myocytes from 7 rats aged 3–4 months. All immunocytochemistry experiments in this study were carried out using a two-step staining method. This method involves an unlabelled primary antibody that reacts with tissue antigen, and a labelled secondary antibody which reacts with the primary antibody. In addition, the following key steps are included in the protocol: (i) Fixation: Paraformaldehyde was used to help formation of chemical cross-links between the cellular proteins; (ii) Triton treatment: after fixation and subsequent wash with PBS, the cell/section was treated with triton, a detergent capable of penetrating liposomal bilayers; (iii) Blocking of non-specific sites: the specimens were treated with a protein solution (bovine serum albumin) prior to staining with antibodies to reduce non-specific antibody binding; (iv) Labelling of antibodies: Once delivered, primary antibody was reconstituted according to the supplier’s suggestions. Stock antibody solution was divided into small volumes and stored at −20 °C until use. Before experimenting, primary antibody was diluted in PBS solution containing 1 % BSA according to supplier’s suggestion or optimised by titration. The antigen–antibody reaction was allowed to take place at 4 °C over night. The secondary antibody was also diluted in 1 % BSA buffer according to the manufacture’s protocol. The incubation was kept in dark box at room temperature for 1.5 h. In order to limit the destruction of fluorescence by light over long time due to the production of molecular oxygen two mounting media that contain anti-fading reagent were used to increase the photostability of the fluorescence. CitiFluor anti-fading medium (Agar Scientific, UK) and Vectorshield H-1000 (Vector Laboratories, Burlingame, CA USA). Various primary antibodies (Abs) (IgGs) were used for Immunocytochemistry: rabbit anti-EDG01 (S1P1) against human EDG-1 (S1P1) residue 241–253 (1:100 dilutions, Cayman Chemical, Tyne & Wear, UK); rabbit anti-EDG-3 (S1P3) against human EDG-3 (S1P3) residue 12–25 (1:100 dilutions, Cayman Chemical); rabbit anti-PAK1 (1:1000 dilutions, Cell Signaling Technology, Hitchin, UK); rabbit anti-phospho-Pak1 (T423) (1:1000 dilutions, Cell Signaling Technology, Hitchin, UK); and rabbit anti-Akt and anti-phospho-Akt Thr308 residue (1:1000 dilutions, Cell Signaling Technology).
Generation of protein samples from tissue, as well as Western immunoblotting, co-immunoprecipitation, and chemifluorescent detection have been described previously [13].
Confocal microscopy and image processing
A Zeiss LSM 5 Pascal Confocal Laser Scanning Microscope (Carl Zeiss Jena, Germany) equipped with HeNe and Argon laser were used in this study. For experiments in which subcellular distribution was the main aim, the microscope and laser setting for each acquisition were adjusted to achieve maximal image quality for any individual cell. For experiments in which fluorescence intensity was used as a measure of protein expression, the imaging protocol was fixed in order to allow quantitative comparisons of relative fluorescence intensity among comparable samples.
Data analysis
All data passed normality and equal variance tests. Analysis was performed using ANOVA, the paired or unpaired Student’s t-test as appropriate (SigmaStat, Systat Software Inc), and significance was accepted at p < 0.05. Data are presented as mean ± SE.
Results
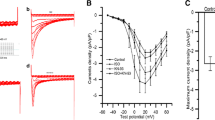
We first determined the effect of S1P (100 nM) on L-type Ca2+ current (ICa,L) in the absence and presence of β-stimulation by perfusion of isoproterenol (ISO, 100 nM). ICa,L was activated by 100 ms step depolarisation pulse from to 0 mV from a holding potential of -40 mV at a rate of 0.2 Hz. Figure 1 shows the effect 100 nM S1P on ICa,L in ventricular myocytes under the basal condition. Basal ICa,L in cells in the conditions of absence and presence of 100 nM S1P and wash-off were statistically indistinguishable, which indicates that 100 nM S1P has no effect on ICa,L in these cells under basal conditions. Figure 2 shows the effect of S1P on ISO stimulation of ICa.L in ventricular myocytes. The amplitude of ICa.L was significantly increased in the presence of 100 nM ISO, the peak current at 0 mV increased by 82 %, from −344 ± 0.07 pA at the basal condition to −625 ± 0.76 pA in the presence of 100 nM ISO (p < 0.05, n = 995 % C.I.). Addition of 100 nM ICa,L of ISO on ICa,L; in 100 nM ISO + 100 nM S1P cells, the peak current at 0 mV increased by 32 %, from −344 ± 0.07 pA to −455 ± 1.80 pA (p < 0.05, n = 9, 95 % C.I.). The difference in response to ISO indicates that ISO-induced increase in ICa.L in cells treated with S1P was significantly attenuated.

L-type Ca2+ current (ICa,L) was recorded from ventricular cells in the presence of 100 nM S1P for 2 min. Current was recorded by 100 ms step depolarisation pulse from −40 to 0 mV from holding potential of −40 mV (left). There is no statically significant difference in the amplitude of ICa,L, in control and presence of 100 nM S1P, and wash-off conditions in these cells (n = 8). Data are presented as mean ± SEM. Asterisk indicates p > 0.05

Representative current traces demonstrating the effects of 100 nM S1P on the response of L-type Ca2+ current to 100 nM ISO. 100 ms steps depolarisations were applied from −40 to 0 mV from holding potential of −40 mV (n = 9). Addition of 100 nM S1P significantly blunts the effect of ISO on ICa,L. Data are presented as mean ± SEM. Asterisk indicates p < 0.05
We subsequently studied the effect of S1P (25 nM) on field-stimulated Ca2+ transients under i) baseline, ii) S1P, iii) S1P + isoprenaline (ISO), and iv) ISO conditions, as illustrated in Fig. 3. Transients were recorded for 60 s after five minutes of perfusion under a specific condition. As shown in Fig. 3, administration of S1P significantly prevents ISO (5 nM)-induced spontaneous Ca2+ waves and diastolic Ca2+ release in isolated rat ventricular myocytes under physiological conditions.

a Ca2+ transients in field-stimulated cardiac myocytes under (i) baseline, (ii) sphingosine-1-phosphate (S1P), (iii) S1P and isoprenaline (ISO), (iv) ISO conditions. Administration of S1P shows significant reduction in β-adrenergic stress conditions induced by ISO. b Effect of S1P (25 nM), ISO (5 nM), and S1P + ISO on spontaneous cytosolic calcium signals during regular field-stimulated pacing. Quantification was based on the total number of irregular calcium transients divided by the total number of calcium transients measured in 60 s. Calcium-dependent fluorescence (Fluo 4-AM) transients were recorded from isolated rat ventricular myocytes. Transients were illuminated and recorded at 490 nm. Measurements were recorded for 60 s after 5 mins of solvent perfusion. Data are presented as mean ± SEM. Asterisk indicates p < 0.05
Our previous studies demonstrated the link between S1P1 receptor signalling, activation of Pak1 and regulation of Pak1 on LTCC and SR Ca2+ release [14, 15]. Thus, we hypothesised that S1P regulates the LTCC and Ca2+ transients through Pak1 signalling [14]. We examined the effect of S1P on the activity of Pak1. Immunocytochemistry and confocal microscopy were used to detect phosphorylation of Pak1 protein in ventricular myocytes. The specificity of the primary antibody was examined by antigenic peptide. As illustrated in Fig. 4, labelling of rat ventricular myocytes with non-phospho-specific Pak1 antibody (indicating total protein, as opposed to ‘active’ kinase), in the absence of S1P, shows a striated pattern and some nuclear labelling (Fig. 4a). In the presence of 25 nM S1P, labelling with the same antibody shows more intense nuclear staining (Fig. 4b). Immunofluorescence signal intensity measurements for non-phospho-specific Pak1 were higher in ventricular myocytes treated with S1P compared to controls (Fig. 4c). We also employed Western blot analysis to determine Pak1 activation with an anti-phospho Thr 423 antibody (Thr-423 phosphorylation being a requirement for activation of Pak1). As shown in Fig. 5, there was a significant (16 %) increase in the levels of phospho-Pak1 in myocytes treated with 25 nM S1P compared with non-treated myocytes.

Effect of S1P on non-phospho-specific Pak1 expression. a Confocal images of isolated non-treated rat ventricular myocytes labelled for non-phospho-specific Pak1. (a) Labelling of a representative rat ventricular myocyte under control conditions with the non-phospho-specific Pak1 antibody (Biocompare) shows a striated pattern and nuclear labelling. (b) Labelling of a representative rat ventricular myocyte under control conditions, following omission of the primary antibody, shows no discernable pattern of labelling. b Confocal images of isolated, S1P-treated rat ventricular myocytes labelled for non-phospho-specific Pak1. (a) Labelling of a representative rat ventricular myocyte treated with S1P shows more diffuse pattern and more intense nuclear labelling. (b) Labelling of a representative rat ventricular myocyte after exposure to S1P, following omission of the primary antibody, shows no discernable pattern of labelling. c Density of labelling of non-phospho-specific Pak1 in control and treated ventricular myocytes. Immunofluorescence signal intensity measurements for non-phospho-specific Pak1 were higher in ventricular myocytes treated with S1P compared to the control. Scale bars, 20 μm. *p < 0.05, n = 18

a Representative Western blot to show Phopho-Pak1 and Phopho-Akt in isolated rat cardiac myocytes treated with 25 nM of S1P. Ventricular cardiac myocytes were exposed for 15 min. b Quantification of Pak1 and Phospho-Pak1 (Thr 423) proteins in these cells
Our previous studies indicated that Pak1 regulates LTCC through PP2A. We therefore examined the effect of S1P on Pak1–PP2A activity. As illustrated in Fig. 6, double labelling of rat ventricular myocytes with Pak1 and PP2A antibodies shows co-localisation of Pak1 with PP2A when cells were treated with S1P. We further performed immunoprecipitation (IP) to study the relationship between Pak1 and PP2A. IP was run first and then the proteins were resolved by SDS-PAGE and stained with Coomassie blue. As shown in Fig. 7, a 37 kD band (PP2Ac) is the major protein co-precipitated with Pak1 and PP2A-B antibodies. These experiments suggest that S1P may blunt the β-adrenergic stimulation via Pak1–PP2A interaction.

Effect of S1P on Pak1–PP2A expression. a Confocal images of isolated rat ventricular myocytes without S1P show co-localisation of Pak1 with PP2A. b Confocal images of isolated S1P-treated rat ventricular myocytes show co-localisation of Pak1 with PP2A. In both a and b, (a) Pak1 antibody labelling (from Spring Bioscience), (b) PP2A antibody (from Exalpha Biologicals) labelling, (c) Co-localisation of Pak1 and PP2A. c Higher magnification of Ac & Bc (square). d Density of labelling of Pak1–PP2A in control and S1P-treated ventricular myocytes. Immunofluorescence signal intensity measurements for Pak1–PP2A were higher in ventricular myocytes treated with S1P compared to the control. Scale bars, 20 μm. *p < 0.05, n = 24

Immunoprecipitation (IP) in rat ventricular muscle homogenate. Lane 1, Pak1 antibody. Lane 2, PP2A B alpha antibody. Coomassie blue staining
Discussion
The data presented here are the first to demonstrate that S1P-induced functional alteration of intracellular Ca2+ handling may include the ICa,L via a signalling pathway involving Pak1–PP2A interaction. Our findings extend, therefore earlier reports providing further evidence for the protective effect of S1P-induced Pak1–PP2A signalling pathway, by counterbalancing the potential deleterious beta-adrenergic overstimulation that commonly occur in the development and progression of diseased states including but not limited to heart failure and arrhythmias.
Major conclusions
In rat cardiac myocytes, S1P does not affect the basal activity of L-type Ca2+ channels but partially reverses the effect of the β-adrenergic agonist ISO. This effect is likely to be mediated by the Pak1–PP2A signalling pathway via S1P1 receptors, although other mechanisms cannot be excluded.
Expression of S1P receptors in cardiac tissues
In our previous study, we showed that S1P1, S1P2, and S1P3 transcripts and proteins are expressed in rat cardiac tissues and myocytes and that the S1P1 receptor is the dominant isoform in this tissue [16]. Mazurais and colleagues investigated the distribution pattern of S1P1, S1P2, and S1P3 receptors within the human heart [17]. Although all three receptors had a similar pattern of distribution across the myocardium, the authors found that the expression of S1P1 was higher than that of S1P2 and S1P3 receptors. These findings were also confirmed by the study conducted by Himmel and colleagues [18].
Functional effect of S1P on L-type calcium current in rat cardiac myocytes
We have demonstrated that, in rat cardiac myocytes, S1P does not affect the basal activity of L-type Ca2+ channels but partially reverses the effect of the β-adrenergic agonist ISO on this channel. Our data also showed S1P significantly prevents ISO-induced Ca2+ waves and diastolic Ca2+ release in isolated rat ventricular myocytes under physiological conditions.
Numerous studies have indicated that S1P can alter intracellular ion concentrations in the heart by regulating the activity of various ion channels. Although the influence of S1P on calcium signalling in cardiomyocytes has not well understood, evidence suggests that S1P can modulate intracellular Ca2+ levels in myocytes [19]. It has been reported that ceramide, a vital precursor molecule in S1P synthesis, can decrease ICa,L but increase free intracellular Ca2+ concentrations in cardiomyocytes from adult [20, 21] and neonatal rats [19]. The observed S1P-induced alterations in free intracellular Ca2+ concentration could result from alterations in intracellular Ca2+ handling and/or from alterations in the influx of extracellular Ca2+. In rabbit isolated sinoatrial node cells, S1P did not affect the basal activity of L-type Ca2+ channels but reversed the effects of the β-adrenergic agonist isoprenaline [22]. This is in well line with our results: in rat cardiomyocytes, S1P does not affect the basal activity of L-type Ca2+ channels but reversed the effect of the β-adrenergic agonist ISO on this channel.
Co-localisation between Pak1 and PP2A
The final objective of this study was to explore some aspects of the signalling pathways of S1P effects in adult rat ventricular myocytes. Our data show co-localisation of Pak1 and PP2A in S1P-treated cells. We have also shown that S1P may downregulate ryanodine receptors as well as upregulating L-type Ca2+ channels, which may lead to a neutral effect on calcium handling. We show a significant increase in the levels of phospho-Pak1, by 16 %, in myocytes treated with 25 nM S1P for 15 min compared with non-treated myocytes. Our IP experiments demonstrated that a 37 kD band (PP2Ac) is the major protein co-precipitated with Pak1 and PP2A-B antibodies.
Binding of S1P to its receptors activates different signalling pathways via heterotrimeric G proteins. Fundamental differences in signalling through S1P receptors relate primarily to variations in G-protein coupling. S1P1 couples exclusively via Gi, whereas S1P2 and S1P3 have been shown to couple to either Gi, Gq or G12/13 [23]. The activation of the same second messenger by two receptors does not necessarily imply the same outcome. Although there may be some redundancy in the system, but the receptors do not have the same “power” in activating a particular second messenger. For example S1P1 null lacks support for the blood vessels, which eventually rupture. This receptor is vital for survival and cannot be compensated for by other S1P receptors even if all these receptors are linked to Gi [24]. While S1P1 seems to be the strongest modulator of adenylate cyclase and possibly Rac, S1P2 drives Rho activation and S1P3 is a stronger inducer of the PLC/IP3/Ca2+ pathway [25–27]. Our prior studies showed S1P1 to be the main S1P receptor isoform in rat ventricular myocytes. Therefore, the effects of S1P seem likely to be linked mainly to Gi-mediated reduction in cAMP production/accumulation in these cells. It follows that S1P may antagonise ISO-mediated activation of adenylyl cyclase through activation of Gi, resulting in the inhibition of cAMP accumulation and prevention of cAMP-mediated calcium increase. Our work suggests that Pak1–PP2A-mediated dephosphorylation of ICaL also contributes to the observed effects of S1P, though it remains to be determined whether other mechanisms and/or proteins contribute to these observed effects and signalling pathways.
Paks are a highly conserved family of serine/threonine protein kinases [28].
Nearly all eukaryote genomes contain one or more Pak genes [29]. Pak family members regulate cellular processes including but not limited to proliferation and survival. They also play important roles in cytoskeletal rearrangement during cell migration. Two subfamilies of Pak1 exist in mammals: group A (PAK1, 2, and 3) can be activated by small GTPases such as Rac-GTP or Cdc42-GTP binding [28]. Group B (PAK4, 5, and 6) can interact with Cdc42-GTP but are not activated by this binding. In addition, PAK1, 2, and 3 can be activated by Rac/Cdc42-independent mechanisms such as by caspase-mediated cleavage, membrane recruitment via adaptor proteins, and sphingolipids [28]. Guo et al. [30] demonstrated that an activated form of Rac1 GTPase—but not the related Rho or Cdc42 GTPases—stimulates transmembrane but not soluble guanylyl cyclase (GC) activity in transfected cells. Guo et al. also showed that PAK directly increases the activity of purified GC. This new Rac/PAK/GC/cGMP pathway postulated by the authors could provide a general mechanism for various types of signalling receptors to increase the cellular concentrations of cGMP. Since cGMP regulates physiological processes by activating protein kinases, gating specific ion channels and modulating cellular cyclic nucleotide concentrations through phosphodiesterases [31], this brings up another potential mechanism underlying S1P effects.
In summary, as illustrated in Fig. 8 [32], these S1P/Pak1 effects on Ca2+ homoeostasis complement previously established actions upon PP2A and the resulting balance between kinase and phosphatase activity in controlling cardiac ion channel activity and rhythmic Ca2+ cycling.

Hypothesis for S1P inhibition of ISO stress induced abnormal Ca2+ events in rat ventricular myocytes—Modified from Wang et al. [32]. Protein kinase PKA and phosphatase PP2A are associated with key Ca2+ handling and regulatory proteins, which in turn are linked to upstream signalling cascades. A balance of protein kinase and phosphatase activities is required to maintain normal cardiac functions. Pak1 also regulates hypertrophic signalling and gene expression of SERCA2a through other signalling pathways by activating its downstream effectors (e.g. JNK). (NCX: Na+–Ca2+ exchanger, PMCA: Plasma membrane Ca2+ ATPase, JNK: c-Jun N-terminal Kinase)
References
Means CK, Brown JH (2009) Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc Res 82:193–200. doi:10.1093/cvr/cvp086
Payne SG, Milstien S, Spiegel S (2002) Sphingosine-1-phosphate: dual messenger functions. FEBS Lett 531:54–57
Pyne S, Pyne NJ (2000) Sphingosine 1-phosphate signalling in mammalian cells. Biochem J 349:385–402
Spiegel S, Milstien S (2003) Exogenous and intracellularly generated sphingosine 1-phosphate can regulate cellular processes by divergent pathways. Biochem Soc Trans 31:1216–1219. doi:10.1042/bst0311216
Ishii I, Friedman B, Ye X, Kawamura S, McGiffert C, Contos JJ, Kingsbury MA, Zhang G, Brown JH, Chun J (2001) Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. J Biol Chem 276:33697–33704. doi:10.1074/jbc.M104441200
Zhang G, Contos JJ, Weiner JA, Fukushima N, Chun J (1999) Comparative analysis of three murine G-protein coupled receptors activated by sphingosine-1-phosphate. Gene 227:89–99
Graler MH, Bernhardt G, Lipp M (1998) EDG6, a novel G-protein-coupled receptor related to receptors for bioactive lysophospholipids, is specifically expressed in lymphoid tissue. Genomics 53:164–169. doi:10.1006/geno.1998.5491
Im DS, Heise CE, Ancellin N, O’Dowd BF, Shei GJ, Heavens RP, Rigby MR, Hla T, Mandala S, McAllister G, George SR, Lynch KR (2000) Characterization of a novel sphingosine 1-phosphate receptor, Edg-8. J Biol Chem 275:14281–14286
Landeen LK, Dederko DA, Kondo CS, Hu BS, Aroonsakool N, Haga JH, Giles WR (2008) Mechanisms of the negative inotropic effects of sphingosine-1-phosphate on adult mouse ventricular myocytes. Am J Physiol Heart Circ Physiol 294:H736–H749. doi:10.1152/ajpheart.00316.2007
Egom EE, Ke Y, Musa H, Mohamed TM, Wang T, Cartwright E, Solaro RJ, Lei M (2010) FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J Mol Cell Cardiol 48:406–414. doi:10.1016/j.yjmcc.2009.10.009
Egom EE, Mohamed TM, Mamas MA, Shi Y, Liu W, Chirico D, Stringer SE, Ke Y, Shaheen M, Wang T, Chacko S, Wang X, Solaro RJ, Fath-Ordoubadi F, Cartwright EJ, Lei M (2011) Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am J Physiol Heart Circ Physiol 301:H1487–H1495. doi:10.1152/ajpheart.01003.2010
Clair T, Aoki J, Koh E, Bandle RW, Nam SW, Ptaszynska MM, Mills GB, Schiffmann E, Liotta LA, Stracke ML (2003) Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res 63:5446–5453
Ke Y, Wang L, Pyle WG, de Tombe PP, Solaro RJ (2004) Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ Res 94:194–200. doi:10.1161/01.RES.0000111522.02730.56
Ke Y, Lei M, Collins TP, Rakovic S, Mattick PA, Yamasaki M, Brodie MS, Terrar DA, Solaro RJ (2007) Regulation of L-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ Res 100:1317–1327. doi:10.1161/01.RES.0000266742.51389.a4
Sheehan KA, Ke Y, Wolska BM, Solaro RJ (2009) Expression of active p21-activated kinase-1 induces Ca2+ flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am J Physiol Cell Physiol 296:C47–C58. doi:10.1152/ajpcell.00012.2008
Egom EE, Ke Y, Musa H, Mohamed TM, Wang T, Cartwright E, Solaro RJ, Lei M (2010) FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J Mol Cell Cardiol 48:406–414. doi:10.1016/j.yjmcc.2009.10.009
Mazurais D, Robert P, Gout B, Berrebi-Bertrand I, Laville MP, Calmels T (2002) Cell type-specific localization of human cardiac S1P receptors. J Histochem Cytochem 50:661–670
Himmel HM, Meyer Zu Heringdorf D, Graf E, Dobrev D, Kortner A, Schuler S, Jakobs KH, Ravens U (2000) Evidence for Edg-3 receptor-mediated activation of I(K.ACh) by sphingosine-1-phosphate in human atrial cardiomyocytes. Mol Pharmacol 58:449–454
Nakajima N, Cavalli AL, Biral D, Glembotski CC, McDonough PM, Ho PD, Betto R, Sandona D, Palade PT, Dettbarn CA, Klepper RE, Sabbadini RA (2000) Expression and characterization of Edg-1 receptors in rat cardiomyocytes: calcium deregulation in response to sphingosine 1-phosphate. Eur J Biochem 267:5679–5686
Liu SJ, Kennedy RH (2003) Positive inotropic effect of ceramide in adult ventricular myocytes: mechanisms dissociated from its reduction in Ca2+ influx. Am J Physiol Heart Circ Physiol 285:H735–H744. doi:10.1152/ajpheart.01098.2002
Relling DP, Hintz KK, Ren J (2003) Acute exposure of ceramide enhances cardiac contractile function in isolated ventricular myocytes. Br J Pharmacol 140:1163–1168. doi:10.1038/sj.bjp.0705510
Guo J, MacDonell KL, Giles WR (1999) Effects of sphingosine 1-phosphate on pacemaker activity in rabbit sino-atrial node cells. Pflugers Arch 438:642–648
Oral H, Dorn GW 2nd, Mann DL (1997) Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian cardiac myocyte. J Biol Chem 272:4836–4842
Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 106:951–961. doi:10.1172/JCI10905
Ancellin N, Hla T (1999) Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J Biol Chem 274:18997–19002
Okamoto H, Takuwa N, Yatomi Y, Gonda K, Shigematsu H, Takuwa Y (1999) EDG3 is a functional receptor specific for sphingosine 1-phosphate and sphingosylphosphorylcholine with signaling characteristics distinct from EDG1 and AGR16. Biochem Biophys Res Commun 260:203–208. doi:10.1006/bbrc.1999.0886
Taha TA, Argraves KM, Obeid LM (2004) Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta 1682:48–55. doi:10.1016/j.bbalip.2004.01.006
Bokoch GM (2003) Biology of the p21-activated kinases. Annu Rev Biochem 72:743–781. doi:10.1146/annurev.biochem.72.121801.161742
Hofmann C, Shepelev M, Chernoff J (2004) The genetics of Pak. J Cell Sci 117:4343–4354. doi:10.1242/jcs.01392
Guo D, Tan YC, Wang D, Madhusoodanan KS, Zheng Y, Maack T, Zhang JJ, Huang XY (2007) A Rac-cGMP signaling pathway. Cell 128:341–355. doi:10.1016/j.cell.2006.11.048
Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA (2000) Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev 52:375–414
Wang Y, Tsui H, Bolton EL, Wang X, Huang CL, Solaro RJ, Ke Y, Lei M (2015) Novel insights into mechanisms for Pak1-mediated regulation of cardiac Ca(2+) homeostasis. Front Physiol 6:76. doi:10.3389/fphys.2015.00076
Acknowledgement
The work was supported by the Medical Research Council (G10002647: Dr Lei), the British Heart Foundation (PG11/59/29006, PG/12/21/29473: Dr. Lei).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Egom, E.EA., Bae, J.S., Capel, R. et al. Effect of sphingosine-1-phosphate on L-type calcium current and Ca2+ transient in rat ventricular myocytes. Mol Cell Biochem 419, 83–92 (2016). https://doi.org/10.1007/s11010-016-2752-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2752-8