
Abstract
Histone deacetylases (HDACs) are a family of deacetylase enzymes that regulate the acetylation state of histones and a variety of other non-histone proteins including key oncogenic and tumor suppressor proteins, which modulates chromatin conformation, leading to regulation of gene expression. HDACs has been grouped into classes I–IV and histone deacetylase 9 (HDAC9) belongs to class IIa which exhibits tissue-specific expression. Recent reports have demonstrated both pro-oncogenic and tumor suppressive role for HDAC9 in different cancers; however, its role in OSCC remains elusive. Here, we investigated the role of HDAC9 in pathogenesis of oral squamous cell carcinoma (OSCC). Our data showed significantly increased mRNA and protein expression of HDAC9 in clinical OSCC samples and UPCI-SCC-116 cells as compared to normal counterpart. Kaplan–Meier analysis showed that the patients with high-level of HDAC9 expression had significantly reduced overall survival than those with low-level of HDAC9 expression (p = 0.034). Knockdown of HDAC9 using siRNA interference suppressed cell proliferation, increased apoptosis, and induced G0/G1 cell cycle arrest in UPCI-SCC-116 cells. Immunofluorescence analysis showed increased nuclear localization of HDAC9 in frozen OSCC sections, and indicative of active HDAC9 that may transcriptionally repress its downstream target genes. Subsequent investigation revealed that overexpression of HDAC9 contributes to OSCC carcinogenesis via targeting a transcription factor, MEF2D, and NR4A1/Nur77, a pro-apoptotic MEF2 target.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oral squamous cell carcinoma (OSCC) is one of the most common malignancies worldwide [1]. In India, OSCC represents a major health problem with highest incidence, poor prognosis, and low survival rate [2]. This necessitates development of newer and more effective therapeutic interventions based on molecular mechanisms of oral carcinogenesis.
Epigenetic modifications such as histone modifications and DNA methylation are important mechanisms which regulate transcriptional activity and gene expression by remodeling the chromatin structure. Among histone modifications, acetylation is one of the important mechanism carried out by enzymes histone acetylases (HATs) and histone deacetylases (HDACs), leading to either gene transcription or gene silencing, respectively [3]. HDACs are a ubiquitous family of enzymes that via deacetylation regulate the transcription of histones, and many non-histone proteins including many transcription factors which lead to chromatin condensation and thereby cause repression of gene expression [4]. Altered expression of HDACs has been linked to carcinogenesis as these molecules regulate the expression and activity of several genes involved in cell cycle, differentiation, apoptosis, angiogenesis, invasion, adhesion, and migration [5, 6]. Indeed, aberrant expression of several HDACs has been reported in different cancers and found to be associated with clinical prognosis [7, 8]. Consequently, HDACs have emerged as promising therapeutic targets for cancer treatment and several HDAC inhibitors are being currently tested for their anti-tumorigenic potential in various solid and hematologic cancers [9, 10]. However, these inhibitors are not HDAC isoform specific as our current knowledge regarding the contributions of different HDACs and understanding regarding key pathways influenced by HDACs in various malignancies remains poor. Thus, research is required to explore the expression patterns and biological role of different HDAC isoforms in different malignancies so that the discrepancy in the literature regarding their contribution in various cancers can be reduced.
HDACs have been grouped into classes I–IV based on their structure and homology [4]. HDAC9 belongs to class IIa and is alternatively spliced to encode multiple protein isoforms due to which it possess distinct biological functions [11]. HDAC9 regulates a wide range of normal physiological processes as like HDAC9 levels were found to be high in cardiac muscle which not impacted normal heart growth. Similarly, mutant mice lacking HDAC9 were sensitized to hypertrophic signals and exhibited stress-dependent cardiomegaly, implying that HDAC9 negatively regulates cardiac hypertrophy [12]. HDAC9 expression is also high in regulatory T cells, and in association with Foxp3, it may control suppressive function of these cells [13]. Other important functions of HDAC9 include a role in limb development by repression of sonic hedgehog signaling pathway [14], modulation of neuron-elicited electrical activity [15], regulation of gene expression and dendritic growth in developing cortical neurons [16], regulation of expression of fatty acid synthase enzyme involved in lipogenesis [17], and control of muscle differentiation [18]. In addition, HDAC9 also regulates numerous pathological processes including tumorigenesis [19, 20]. HDAC9 exhibits tissue-specific expression and recent studies have alluded both pro-oncogenic and tumor suppressive role for HDAC9 in different malignancies; however, its role in OSCC has not been studied till date.
HDAC9 has an extended C terminal tail with a long regulatory N-terminal domain through which it interacts with various transcription factors and co-repressors [21]. HDAC9 binds to MADS domain of a transcription factor, myocyte enhancer factor 2D (MEF2D), and represses transcription of MEF2D target genes, such as NR4A1/Nur77 [18, 22]. The MEF2 family of transcription factors which comprise of MEF2A, MEF2B, MEF2C, and MEF2D were initially identified as major transcriptional activators for muscle differentiation [22, 23] but later on they were known to play a key role in development processes including muscle and neural differentiation, cardiac morphogenesis, blood vessel formation, and growth factor responsiveness [24–27]. Recently, MEF2 family members have also been recognized as survival factors in various cell types and may have a central role in carcinogenesis process [28, 29]. MEF2 performs a pro-apoptotic function by stimulating the expression of the orphan nuclear hormone receptor gene NR4A1/Nur77 [28]. NR4A1/Nur77 directly interacts with Bcl-2 or indirectly stimulates other pro-apoptotic proteins such as Bax which in turn may activate cytochrome c-mediated apoptosis [30]. The NR4A subfamily belongs to steroid nuclear hormone receptor superfamily and consists of three members NR4A1, NR4A2, and NR4A3. Unlike other steroid hormone receptors, NR4A1 are constitutively active and possess ligand-independent activation [31]. These receptors have diverse functions which are context and tissue specific [32], and are of integral importance in tumorigenesis as they act molecular switches in numerous processes like proliferation, apoptosis, inflammation, and angiogenesis [30].
In the present study, we investigated the role of HDAC9 in OSCC tumorigenesis. Our results reveal that overexpression of HDAC9 promotes OSCC carcinogenesis as its in vitro silencing significantly reduced cell proliferation, increased apoptosis, and induced G0/G1 cell cycle arrest. We show that pro-tumorigenic effect of HDAC9 is mediated via deregulation of HDAC9-MEF2D axis which has a repressive effect on its pro-apoptotic target gene NR4A1/Nur77. To our knowledge, this is the first report showing oncogenic role of HDAC9 in pathophysiology of OSCC.
Materials and methods
Tissue samples
OSCC tissues were collected from clinically and histologically confirmed primary OSCC patients undergoing surgical resection or routine biopsy procedure in the Department of Otolaryngology and Head and Neck Surgery, PGIMER, Chandigarh, India. Clinical staging and TNM status of OSCC were determined according to the classification of the International Union against Cancer (UICC) convention. Normal oral tissues were obtained from age- and gender-matched subjects, undergoing oral maxillofacial surgery for non-neoplastic diseases at Department of Oral Health Sciences Centre, PGIMER, Chandigarh, India. Tissue samples were collected after obtaining written informed consent from the patients. The study was approved by the Institute Ethics Committee (Approval number: PGI/IEC/2014/769) and conducted according to the guidelines of Declaration of Helsinki (as revised in Seoul, 2008).
Cell culture
OSCC cell line, UPCI-SCC-116 was kindly provided by Dr. Susanne M. Gollin (University of Pittsburgh Graduate School of Public Health, Pittsburgh, USA). Cell line was maintained in Minimal Essential Medium (MEM; Gibco) supplemented with l-glutamine (Gibco) and 10 % fetal bovine serum (FBS; Gibco) at 37 °C in humidified incubator with 5 % CO2.
RNA extraction and real-time PCR
Total RNA was extracted using mirVanaTM kit (Ambion) as per manufacturer’s protocol and RNA purity was evaluated by NanoDrop (ND-1000 spectrophotometer; Thermo) using 260-to-280 nm ratios. Total RNA (500 ng) was used to generate cDNA using RevertAid first strand cDNA synthesis kit (Fermentas Life Sciences), and real-time analysis was performed on StepOne Plus RT-PCR system (Applied Biosystems) using Power SYBR® Green PCR master mix (Applied Biosystems). The oligonucleotide sequence of primers is shown in Table S1 (Supplementary data). The data was normalized to GAPDH and expression levels were determined by \(2^{{ - \Delta C_{\text{t}} }}\) method.
Western blotting
Immunoblotting was performed as described previously [33]. Briefly, protein was extracted by adding RIPA lysis buffer (50 mM Tris–HCl pH 7.6, 150 mM NaCl, 1 mM EGTA, 1 % Triton NP-40, 1 % sodium deoxycholate, 0.1 % SDS, 50 mM NaF, and 1× Protease inhibitor), and concentration was measured using quick startTM Bradford dye reagent, 1X (Bio-Rad). Equal amount of protein was resolved on 10 % SDS-PAGE, transferred onto PVDF membrane at 4 °C, blocked in 5 % BSA, and incubated with primary antibody against HDAC9 (sc-398003; Santa Cruz Biotechnology) followed by HRP-conjugated secondary antibody (Santa Cruz Biotechnology). Detection was performed using EZ-ECL system (Biological Industries) and exposing the membrane to X-ray film. The data were normalized to GAPDH.
Immunoflourescence
Tissues were immediately frozen in OCT (Tissue-tek, Sakura finetek), cut into 4-µm-thick sections on pre-coated polylysine glass slides using cryostat (Leica Biosystems). Sections were washed with PBS and incubated with primary HDAC9 antibody (sc-398003; Santa Cruz Biotechnology) followed by Texas Red-conjugated secondary antibody (Santa Cruz Biotechnology). Hoechst 33342 (Sigma) was used to stain nuclei and sections were mounted with 5 % of phosphate-buffered glycerol. Slides were analyzed under confocal microscope (Olympus FV1000-IX81, Olympus), and images were captured using OLYMPUS Fluoview.Ver.3.0a software.
Silencing of HDAC9 by small interfering RNA
The HDAC9-specific siRNA duplex (Silencer® Select) was synthesized by Ambion; sequence sense: 5′-GCUCAAUGCUUCGAAUUCAtt-3′ and antisense: 5′-UGAAUUCGAAGCAUUGAGCtg-3′. Cy3™ dye-labeled scramble siRNA was purchased from Ambion which was used as a negative control. The siRNA concentrations used for the study were 25, 50, and 75 nM. Cells were transiently transfected with siRNA using transfection reagent Opti-MEM I (Invitrogen) and Lipofectamine2000 (Invitrogen) according to the manufacturer’s instructions. Cy3™ dye-labeled scramble siRNA uptake by UPCI-SCC-116 cells were imaged using an inverted fluorescent microscope (Olympus IX51), and transfection efficiency was determined by flow cytometry (BD FACS CantoTM II, Becton–Dickinson Biosciences).
MTT cell proliferation assay
Cells were harvested after incubating for each indicated time line (0, 24, 48, and 72 h), and 15 µl sterile MTT solution (5 mg/ml in PBS, Himedia) was added for 3 h at 37 °C after which the dark blue formazan product was solubilized by adding 100 µl DMSO. The absorbance was read at 570 nm with reference wavelength 630 nm using ELISA micro-plate reader (Sunrise, TECAN) and analyzed using Magellan software V6.6.
Annexin V/PI apoptosis assay
Apoptosis was measured using Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit (Invitrogen) according to the manufacturer’s instruction. In brief, cells were centrifuged, washed with cold PBS, resuspended in Annexin Binding Buffer and incubated with propidium iodide (PI; 1 mg/ml) and Alexa Fluor 488 labeled Annexin for 15 min at RT. Stained cells were finally passed through BD FACS CantoTM II (Becton–Dickinson Biosciences), and analysis was performed using BD FACSDiva software version 6.1.2 (Becton–Dickinson).
Cell cycle analysis by PI staining and flow cytometry
Cell cycle analysis was performed as described previously [34]. Briefly, cells were centrifuged, washed with cold PBS, resuspended in ice-cold 70 % ethanol, and stored at 4 °C overnight. Cells were then incubated with 50 µg/ml PI and 50 µg/ml DNase-free RNase (Sigma-Aldrich), and stained cells were analyzed by flow cytometry (BD FACS CantoTM II; Becton–Dickinson Biosciences) using BD FACSDiva software version 6.1.2 (Becton–Dickinson).
Statistical analysis
Statistical analysis was performed using Graphpad Prism 5 Version 5.03. Intergroup differences were compared using student’s t test, Mann–whitney U test, ANOVA, and Pearson’s χ 2 test, as appropriate. Analysis of patient survival was performed using Kaplan–Meier analysis and log rank test. Relationship between HDAC9 expression levels and clinicopathological parameters were assessed with Pearson’s χ 2-test and Fisher’s exact test. The median was used to separate samples into low–high expression groups. p values ≤0.05 were considered statistically significant.
Results
Demographic and clinical characteristics of OSCC patients and controls are given in Table 1. No significant difference in age, gender, and mean lifetime exposure to smoking, alcohol consumption, and tobacco chewing was observed between two groups.
High expression of HDAC9 in clinical OSCC samples and OSCC cells
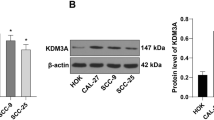
We measured the mRNA levels of 11 HDAC genes in 60 primary OSCC tissues and 30 matched normal tissues. The qRT-PCR analysis showed significantly elevated mRNA levels of HDAC9 (3.6-fold, Fig. 1a, b) and significantly reduced levels of HDACs 5, 6, 8, 10, and 11 in OSCC tissues in comparison to corresponding normal tissues (Fig. 1a). No statistically significant change was observed in expression of HDACs 1, 2, 3, 4, and 7 (Fig. 1a). Since HDAC9 mRNA levels were observed to be significantly increased among all HDACs, thus we focused on HDAC9, explored the clinical relevance of this overexpression, and attempted to understand HDAC9 function at molecular level in OSCC, which remains undefined. We performed Western blot analysis to test the HDAC9 protein levels in clinical samples, and our data revealed HDAC9 protein levels to be markedly upregulated in OSCC tissues as compared to non-cancerous tissues (11-fold, Fig. 1c). We also measured the expression levels of HDAC9 in OSCC cell line, UPCI-SCC116 cells. The data showed that mRNA (4-fold, Fig. 1d) and protein levels (3.8-fold, Fig. 1e) of HDAC9 were significantly elevated in UPCI-SCC-116 cells in comparison to normal counterpart. Collectively, our results show HDAC9 overexpression in OSCC tissues and cell line.

HDAC9 is overexpressed in OSCC clinical specimens and OSCC cells. a qRT-PCR analysis of 11 HDAC genes in OSCC (n = 60) and normal oral tissues (n = 30). b Scatter plot representation of HDAC9 expression in OSCC (n = 60) and normal oral tissues (n = 30). c Protein expression of HDAC9 in clinical specimens (n = 3/group) by Western blotting. d Kaplan–Meier survival curve of HDAC9 levels and median overall survival of OSCC patients with high (n = 10) or low (n = 10) HDAC9 expression. mRNA and protein expression of HDAC9 in UPCI-SCC cells determined by e qRT-PCR and f Western blotting, respectively. The mRNA levels are expressed as ratio of target gene to GAPDH; protein levels are expressed as ratio of target protein to GAPDH, normalized to normal group. Error bars show mean ± SEM; *p < 0.05 or **p < 0.01 or ***p < 0.001 versus normal group
Increased expression of HDAC9 correlates with reduced overall survival of OSCC patients
To examine the effect of HDAC9 expression on overall patient survival, we performed Kaplan–Meier survival analysis in a subgroup of twenty OSCC patients as we were unable to follow-up other patients due to change in contact details. The survival analysis showed that the patients with high expression of HDAC9 had lower median overall survival than those with low expression of HDAC9 (p = 0.034; Fig. 1f), thus indicating positive association of HDAC9 expression with overall patient survival. We found no significant correlation between HDAC9 expression levels and various clinicopathological parameters of OSCC patients (Supplementary Table S2).
HDAC9 knockdown inhibited cell proliferation of UPCI-SCC-116 cells
To investigate whether HDAC9 plays a pro-oncogenic role in OSCC development, we knocked down HDAC9 in UPCI-SCC-116 cells using specific siRNA against HDAC9. Fluorescence microscopic images displayed efficient delivery of Cy3™ dye-labeled scramble siRNA into UPCI-SCC-116 cells (Fig. 2a), and flowcytometric analysis revealed 94–98 % uptake of Cy3™ dye-labeled scramble siRNA by UPCI-SCC-116 cells (Fig. 2b). To confirm HDAC9 knockdown in transfected cells, mRNA and protein expression of HDAC9 was measured by qRT-PCR analysis and Western blotting, respectively. We observed a dose and time-dependent decrease in HDAC9 mRNA expression in UPCI-SCC-116 cells treated with HDAC9 siRNA in concentrations of 25, 50, and 75 nM for 48 and 72 h as compared to untreated or scramble siRNA-treated cells (Fig. 2c). We observed maximum inhibition of HDAC9 mRNA expression in 75 nM HDAC9 siRNA group after 72 h of transfection (96 % inhibition) when compared with untreated cells. Similarly, a significant decrease in HDAC9 protein expression (95 % inhibition) was observed in 75 nM HDAC9 siRNA group after 72 h of treatment as compared to scramble siRNA-treated cells (Fig. 2d).

Efficient siRNA delivery and high transfection efficiency in UPCI-SCC-116 cells. a Representative fluorescent microscopy images of UPCI-SCC-116 cells transfected with Cy3™ dye-labeled scramble siRNA for 48 h (red). b Transfection efficiency of Cy3™ dye-labeled scramble siRNA in UPCI-SCC-116 cells measured by flow cytometry; P3 indicates the percent of cells harboring siRNA. mRNA and protein levels of HDAC9 after HDAC9 knockdown in UPCI-SCC-116 cells assessed by c qRT-PCR and d Western blotting, respectively. The mRNA levels are expressed as ratio of target gene to GAPDH; protein levels are expressed as ratio of target protein to GAPDH, normalized to scramble siRNA group. Error bars show mean ± SEM; *p < 0.05 or ***p < 0.001 versus untreated or scramble siRNA group. (Color figure online)
To examine the role of HDAC9 knockdown on cell growth and proliferation, we measured cell proliferation by MTT assay. Compared with untreated and scramble siRNA-treated cells, HDAC9 knockdown cells exhibited significantly lower growth rates for all three concentrations 25, 50, and 75 nM of HDAC9 siRNA after 48 and 72 h of treatment (Fig. 3a). The growth inhibition by HDAC9 siRNA was not observed after 24 h of treatment and was more after 72 than 48 h of treatment in all three groups.

HDAC9 silencing inhibited cell proliferation and arrested cell cycle at G0/G1 phase in UPCI-SCC cells. a The cell proliferation measured by MTT absorbance at 570 nm at the indicated times in untreated or scramble or HDAC9-treated groups. b Cell cycle analysis of PI-stained untreated or scramble or HDAC9-treated cells for 72 h by flow cytometry. Histogram peaks represents G1/G0, S, and G2/M phases of the cell cycle. c Bar graph representation of distribution and percentages of cells in different phases. Error bars show mean ± SEM. *p < 0.05 or **p < 0.01 or ***p < 0.001 versus untreated group
HDAC9 knockdown arrested UPCI-SCC-116 cell cycle
To investigate the mechanism by which HDAC9 silencing suppressed OSCC cell growth, we assessed whether growth inhibition was associated with cell cycle changes. Therefore, we carried out flowcytometric analysis of PI-stained DNA to monitor cell cycle progression in UPCI-SCC-116 cells. The analysis showed a dose-dependent increase in the percentage of G0/G1-phase cells in 25, 50, and 75 nM HDAC9 siRNA groups from 62.3 to 74.2, 78.7, and 79.5 % and decrease in the S-phase cells from 25.3 to 17.9, 12.8, and 13.9 %, respectively, after 72 h of treatment (Fig. 3b, c). These results indicate that HDAC9 siRNA-mediated inhibition of cell proliferation was mainly at G0/G1 phase of the cell cycle.
HDAC9 knockdown induced apoptosis in UPCI-SCC-116 cells
We further examined the effect of HDAC9 knockdown on cell apoptosis in HDAC9 or scramble siRNA-treated UPCI-SCC-116 cells. We found a dose-dependent increase in percent apoptotic cells from 2.24 to 18.78, 27.17, and 29.80 % in 25, 50, and 75 nM HDAC9 siRNA groups, respectively, after 72 h of treatment compared to untreated group. HDAC9 knockdown induced both early and late apoptosis in UPCI-SCC-116 cells in comparison to untreated or scramble siRNA group (Fig. 4a, b).

HDAC9 knockdown induced both early and late apoptosis in UPCI-SCC-116 cells. a Apoptosis in annexin-alexa flour/PI-stained untreated or scramble or HDAC9-treated cells for 72 h by flow cytometry. The upper left quadrant (Q1) indicates Annexin V−/PI+ necrotic cells, upper right quadrant (Q2) depicts Annexin V+/PI+ late apoptotic cells, lower left quadrant (Q3) represents Annexin V−/PI− viable cells, and lower right quadrant (Q4) shows Annexin V+/PI− early apoptotic. b Graphical representation of flow cytometry results. Error bars show mean ± SEM; **p < 0.01 or ***p < 0.001 versus untreated group
Increased nuclear localization and activity of HDAC9 in OSCC
Shuttling of class IIa HDACs between nucleus and cytoplasm is an important mechanism through which their activity is regulated, and nuclear export of class IIa HDACs promotes transcriptional repression [35]. Thus, we performed immunofluorescence analysis to visualize cellular localization of HDAC9 in frozen sections of OSCC and normal oral tissues. The confocal images displayed stronger florescent signals as well as intense nuclear staining for HDAC9 in OSCC as compared to normal sections (Fig. 5a, b).


Increased nuclear localization of HDAC9 in frozen OSCC sections. Representative immunofluorescence micrographs stained for HDAC9 using Texas Red-conjugated secondary antibody (red) (n = 2/group), original magnification a X20 b X40; (i) Epithelial cells of renal tubules as positive control (ii) Normal oral section (iii) OSCC section. Nuclei are stained with Hoechst 33342 (blue). Scale bar denote 30um. White arrows show nuclear localization of HDAC9 in OSCC sections. (Color figure online)
HDAC9 promotes OSCC growth by targeting molecules, MEF2D, and NR4A1/Nur77
HDAC9 interacts with MEF2D transcription factor and represses MEF2-dependent genes, such as NR4A1/Nur77, a pro-apoptotic gene [18, 22]. To further gain insights into the oncogenic property of HDAC9, we determined the expression of MEF2D and NR4A1/Nur77 in OSCC and corresponding normal tissues by qRT-PCR. We observed significantly increased expression of MEF2D (Fig. 6a) and decreased expression of Nur77/NR4A1 (Fig. 6b) in OSCC than normal group. Further, we examined whether siRNA-mediated HDAC9 knockdown reverses the repressive influence of HDAC9 on pro-apoptotic MEF2 target gene, NR4A1/Nur77 in UPCI-SCC-116 cells. Our results demonstrated 2-fold increase in expression of NR4A1/Nur77 in HDAC9 knockdown cells compared to that of untreated or scramble siRNA group (Fig. 6c).

HDAC9 promotes OSCC tumorigenesis by targeting molecules, MEF2D and NR4A1/Nur77. Relative mRNA expression of a MEF2D and b NR4A1/Nur77, downstream targets of HDAC9 in OSCC and normal oral tissues (n = 20/group). c Expression of NR4A/Nur77 after HDAC9 knockdown in UPCI-SCC-116 cells by qRT-PCR. The mRNA levels are expressed as ratio of target to GAPDH. Error bars show mean ± SEM; *p < 0.01 or ***p < 0.001 versus normal group
Discussion
In the present study, we elucidated a crucial role of HDAC9 in OSCC. Initially, we determined transcriptomic profile of classic HDACs (HDAC1-HDAC11) in OSCC, and our data reveal that various HDACs are differentially expressed in OSCC. Our results are supported by earlier studies showing that expression of different isoforms of HDACs varies in different tumors, and the expression pattern of a specific HDAC isoform is associated to a specific tumor type [8, 36, 37]. HDAC9 is mapped on chromosome 7p21, a region that is related to several human cancers including bowel cancer [38], fibrosarcoma [39], childhood ALL [40], Wilms’ tumor [41], and peripheral nerve sheath tumor [42]. HDAC9 has been also shown to associate with a variety of non-histone targets such as forkhead box protein 3, ataxia telangiectasia group D-complementing protein (ATDC), and glioblastoma 1 protein, molecules directly implicated in various cancer pathways [13, 35, 43]. In addition, HDAC9 has been demonstrated to bind with co-repressors such as mSin3A, mSin3B, and N-CoR, whose activities have been implicated in the mechanism of action of many cancers [44]. Yuan et al. reported that HDAC9 positively regulated the transcriptional activity of p53 via deacetylation of ATDC, a p53 binding protein, and the suppression of HDAC9 function might impair p53 activity which may aid in tumor promotion and progression [43]. Further, a recent study has also unveiled that HDAC9 may promote growth of glioblastoma through TAZ-mediated EGFR pathway activation [20]. Altogether, this suggests that HDAC9 has a potential role in carcinogenesis.
Our initial finding reveals HDAC9 to be highly expressed in clinical OSCC samples and in an OSCC cell line, and observes reduced overall survival of patients with high levels of HDAC9. Earlier reports have also shown increased expression of HDAC9 in various other cancers including glioblastoma [20], medulloblastoma [19], childhood ALL [37], and cervical cancer [45], and found association with poor prognosis. However, Okudela et al. reported lower expression levels of HDAC9 in lung cancer and suggested HDAC9 to be a tumor suppressor which indicates a dual role of HDAC9 in various cancers [46]. Thus, aberrant expression of HDAC9 may impact different types of tumors in a different way, and the altered levels of HDAC9 may directly affect the physiological regulation of transcription of several genes, leading to promotion of malignant transformation. In light of previous reports and our observations, we hypothesized that overexpressed HDAC9 might be playing an important role in OSCC carcinogenesis.
Subsequently, we knocked down HDAC9 using siRNA interference and evaluated cell proliferation, apoptosis, and cell cycle in UPCI-SCC116 cells. Our data showed siRNA-mediated HDAC9 knockdown effectively suppressed cell proliferation of UPCI-SCC-116 cells suggesting that HDAC9 may contribute to OSCC cell proliferation. Further, HDAC9 contribution in OSCC cell proliferation was supported by the finding that HDAC9 knockdown induced G0/G1 phase cell cycle arrest in OSCC cells. Our findings also unveiled that HDAC9 knocked down OSCC cells exhibited abnormally increased both early and late apoptosis. Altogether, our data suggest that HDAC9 downregulation suppresses tumor growth in OSCC via deregulating pro-tumorigenic processes such as cell proliferation, cell cycle, and apoptosis, indicating oncogenic role of HDAC9 in OSCC.
Our findings revealed that HDAC9 was nuclear localized in frozen OSCC sections, suggesting increased HDAC9 activity in OSCC and in nucleus by modulating the acetylation state HDAC9 may repress transcription of its target genes including many cancer relevant genes. To further demonstrate the molecules involved in HDAC9-mediated OSCC carcinogenesis, we evaluated the expression of HDAC9 downstream targets, MEF2D, and NR4A1/Nur77 in OSCC tissues. We found significantly elevated mRNA levels of MEF2D and decreased levels of pro-apoptotic NR4A1/Nur77 gene in OSCC and in vitro silencing of HDAC9 augmented expression of NR4A1/Nur77 gene, suggesting repressive effect of upregulated HDAC9 on apoptosis and its involvement in OSCC tumorigenesis. Consistent with our results, MEF2D overexpression has been reported to promote survival and tumorigenesis in hepatocellular carcinoma [47]. Our findings are also in accordance to those reported by Clocchiatti et al. who demonstrated that class IIa HDACs silencing augmented NR4A1/Nur77gene expression in breast cancer cell line, MCF7 ER+ cells, and proposed that class IIa HDACs have a pro-survival function [48]. Thus our data implies that HDAC9, a class IIa HDAC member, may contribute to oral carcinogenesis by targeting MEF2D and its target gene, NR4A1/Nur77.
In conclusion, our study show a central role of overexpressed HDAC9 in promoting OSCC carcinogenesis by targeting MEF2D and NR4A1/Nur77 resulting in alteration of several pro-oncogenic processes such as cell proliferation, cell cycle, and apoptosis. Our data implies HDAC9 to be a novel promising target for therapeutic intervention in OSCC.
References
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74–108
Coelho KR (2012) Challenges of the oral cancer burden in India. J Cancer Epidemiol 2012:701932
Yang X-J, Seto E (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26:5310–5318
Thiagalingam S, Cheng K-H, Lee HJ et al (2003) Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci 983:84–100
Witt O, Deubzer HE, Milde T, Oehme I (2009) HDAC family: what are the cancer relevant targets? Cancer Lett 277:8–21
Ropero S, Esteller M (2007) The role of histone deacetylases (HDACs) in human cancer. Mol Oncol 1:19–25
Weichert W (2009) HDAC expression and clinical prognosis in human malignancies. Cancer Lett 280:168–176
Ozdağ H, Teschendorff AE, Ahmed AA et al (2006) Differential expression of selected histone modifier genes in human solid cancers. BMC Genom 7:90
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5:769–784
Kim DH, Kim M, Kwon HJ (2003) Histone deacetylase in carcinogenesis and its inhibitors as anti-cancer agents. J Biochem Mol Biol 36:110–119
Petrie K, Guidez F, Howell L et al (2003) The histone deacetylase 9 gene encodes multiple protein isoforms. J Biol Chem 278:16059–16072
Zhang CL, McKinsey TA, Chang S et al (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110:479–488
Tao R, de Zoeten EF, Ozkaynak E et al (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 13:1299–1307
Morrison BE, D’Mello SR (2008) Polydactyly in mice lacking HDAC9/HDRP. Exp Biol Med Maywood NJ 233:980–988
Méjat A, Ramond F, Bassel-Duby R et al (2005) Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci 8:313–321
Sugo N, Oshiro H, Takemura M et al (2010) Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur J Neurosci 31:1521–1532
Wong RHF, Chang I, Hudak CSS et al (2009) A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 136:1056–1072
Haberland M, Arnold MA, McAnally J et al (2007) Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol Cell Biol 27:518–525
Milde T, Oehme I, Korshunov A et al (2010) HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res 16:3240–3252
Yang R, Wu Y, Wang M et al (2015) HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget 6:7644–7656
Parra M, Verdin E (2010) Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol 10:454–460
McKinsey TA, Zhang CL, Olson EN (2002) MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci 27:40–47
Molkentin JD, Olson EN (1996) Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc Natl Acad Sci USA 93:9366–9373
Potthoff MJ, Olson EN (2007) MEF2: a central regulator of diverse developmental programs. Development 134:4131–4140
Lin Q, Schwarz J, Bucana C, Olson EN (1997) Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276:1404–1407
Arnold MA, Kim Y, Czubryt MP et al (2007) MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell 12:377–389
Hayashi M, Kim S-W, Imanaka-Yoshida K et al (2004) Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest 113:1138–1148
Mao Z, Bonni A, Xia F et al (1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286:785–790
Gaudilliere B, Shi Y, Bonni A (2002) RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem 277:46442–46446
Mohan HM, Aherne CM, Rogers AC et al (2012) Molecular pathways: the role of NR4A orphan nuclear receptors in cancer. Clin Cancer Res 18:3223–3228
Deutsch AJA, Angerer H, Fuchs TE, Neumeister P (2012) The nuclear orphan receptors NR4A as therapeutic target in cancer therapy. Anticancer Agents Med Chem 12:1001–1014
Li Q-X, Ke N, Sundaram R, Wong-Staal F (2006) NR4A1, 2, 3 an orphan nuclear hormone receptor family involved in cell apoptosis and carcinogenesis. Histol Histopathol 21:533–540
Raut SK, Kumar A, Singh GB et al (2015) miR-30c mediates upregulation of Cdc42 and Pak1 in diabetic cardiomyopathy. Cardiovasc Ther 33:89–97
Gao D-J, Xu M, Zhang Y-Q et al (2010) Upregulated histone deacetylase 1 expression in pancreatic ductal adenocarcinoma and specific siRNA inhibits the growth of cancer cells. Pancreas 39:994–1001
Clocchiatti A, Florean C, Brancolini C (2011) Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis. J Cell Mol Med 15:1833–1846
Lucio-Eterovic AKB, Cortez MAA, Valera ET et al (2008) Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer 8:243
Moreno DA, Scrideli CA, Cortez MAA et al (2010) Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol 150:665–673
Aragane H, Sakakura C, Nakanishi M et al (2001) Chromosomal aberrations in colorectal cancers and liver metastases analyzed by comparative genomic hybridization. Int J Cancer J Int Cancer 94:623–629
Schmidt H, Taubert H, Würl P et al (2002) Gains of 12q are the most frequent genomic imbalances in adult fibrosarcoma and are correlated with a poor outcome. Genes Chromosomes Cancer 34:69–77
Jarosová M, Holzerová M, Jedlicková K et al (2000) Importance of using comparative genomic hybridization to improve detection of chromosomal changes in childhood acute lymphoblastic leukemia. Cancer Genet Cytogenet 123:114–122
Powlesland RM, Charles AK, Malik KT et al (2000) Loss of heterozygosity at 7p in Wilms’ tumour development. Br J Cancer 82:323–329
Schmidt H, Würl P, Taubert H et al (1999) Genomic imbalances of 7p and 17q in malignant peripheral nerve sheath tumors are clinically relevant. Genes Chromosomes Cancer 25:205–211
Yuan Z, Peng L, Radhakrishnan R, Seto E (2010) Histone deacetylase 9 (HDAC9) regulates the functions of the ATDC (TRIM29) protein. J Biol Chem 285:39329–39338
Guidez F, Zelent A (2001) Role of nuclear receptor corepressors in leukemogenesis. Curr Top Microbiol Immunol 254:165–185
Choi Y-W, Bae SM, Kim Y-W et al (2007) Gene expression profiles in squamous cell cervical carcinoma using array-based comparative genomic hybridization analysis. Int J Gynecol Cancer 17:687–696
Okudela K, Mitsui H, Suzuki T et al (2014) Expression of HDAC9 in lung cancer–potential role in lung carcinogenesis. Int J Clin Exp Pathol 7:213–220
Ma L, Liu J, Liu L et al (2014) Overexpression of the transcription factor MEF2D in hepatocellular carcinoma sustains malignant character by suppressing G2-M transition genes. Cancer Res 74:1452–1462
Clocchiatti A, Di Giorgio E, Ingrao S et al (2013) Class IIa HDACs repressive activities on MEF2-dependent transcription are associated with poor prognosis of ER+ breast tumors. FASEB J 27:942–954
Acknowledgments
Bhawna Rastogi is a PhD student in the Department of Otolaryngology and Head and Neck Surgery, PGIMER, and fellowship was provided by Indian Council of Medical Education and Research, (3/1/3/JRF-2011/HRD 25), New Delhi, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rastogi, B., Raut, S.K., Panda, N.K. et al. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Mol Cell Biochem 415, 183–196 (2016). https://doi.org/10.1007/s11010-016-2690-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2690-5