
Abstract
Kolaviron is a mixture of biflavonoids found in the nut of the West African edible seed Garcinia kola, and it has been reported to exhibit a wide range of pharmacological activities. In this study, we investigated the effects of kolaviron in neuroinflammation. The effects of kolaviron on the expression of nitric oxide/inducible nitric oxide synthase (iNOS), prostaglandin E2 (PGE2)/cyclooxygenase-2, cellular reactive oxygen species (ROS) and the pro-inflammatory cytokines were examined in lipopolysaccharide (LPS)-stimulated BV2 microglial cells. Molecular mechanisms of the effects of kolaviron on NF-κB and Nrf2/ARE signalling pathways were analysed by immunoblotting, binding assays and reporter assays. RNA interference was used to investigate the role of Nrf2 in the anti-inflammatory effect of kolaviron. Neuroprotective effect of kolaviron was assessed in a BV2 microglia/HT22 hippocampal neuron co-culture. Kolaviron inhibited the protein levels of NO/iNOS, PGE2/COX-2, cellular ROS and the pro-inflammatory cytokines (TNFα and IL-6) in LPS-stimulated microglia. Further mechanistic studies showed that kolaviron inhibited neuroinflammation by inhibiting IκB/NF-κB signalling pathway in LPS-activated BV2 microglia. Kolaviron produced antioxidant effect in BV2 microglia by increasing HO-1 via the Nrf2/antioxidant response element pathway. RNAi experiments revealed that Nrf2 is needed for the anti-inflammatory effects of kolaviron. Kolaviron protected HT22 neurons from neuroinflammation-induced toxicity. Kolaviron inhibits neuroinflammation through Nrf2-dependent mechanisms. This compound may therefore be beneficial in neuroinflammation-related neurodegenerative disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accumulating evidence continues to link chronic neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) with chronic neuroinflammation.
Chronic neuroinflammation is a self-perpetuating response that persists long after an initial injury or insult. It is characterised by prolonged activation of CNS-resident macrophages, the microglia and subsequent sustained release of pro-inflammatory mediators [1]. Microglial activation perpetuates the inflammatory cycle by activating other microglia, resulting in a further release of inflammatory factors [1]. Inflammatory signalling pathways following microglial activation therefore represent a prime target for inhibiting neuroinflammation-induced neurodegeneration.
Oxidative stress has been strongly linked to the aetiology of many neurodegenerative diseases. According to Li et al. [2], accumulation of reactive oxygen and nitrogen species generated by inflammatory cells that is one of the mechanisms through which chronic inflammation contributes to diseases. The Nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2) is a redox-sensitive transcription factor and a critical regulator of endogenous-inducible defence systems in the body [3, 4]. Under physiological conditions, Nrf2 is sequestered in the cytoplasm by the regulatory protein Kelch-like ECH-associated protein 1 (Keap1) [3, 5]. However, in response to oxidative stress, Nrf2 translocates to the nucleus and binds to antioxidant response elements (ARE) in the DNA, to initiate transcription of cytoprotective genes [4].
New information suggest that activation of Nrf2 is an important strategy in blocking neuroinflammation in the microglia. Recent findings indicate that restoration of redox balance through activation of Nrf2 may be a determining factor in restoring the microglia back to its resting inactivated state, and presents as a strategy for modulating neuroinflammation in neurodegenerative disorders [6]. A study by Koh et al. [3] showed that Nrf2 inhibits microglial hyperactivation by suppressing p38 MAPK and NF-κB signalling pathway. Further evidence indicates that the hippocampi of Nrf2 knockout mice were shown to be hypersensitive to the neuroinflammation induced by LPS, as demonstrated by an increase in the inflammation markers iNOS, IL-6 and TNFα, compared with the hippocampi of wild-type littermates [7]. Taken together, it has become clear that measures to activate Nrf2 in the microglia would be a significant strategy in reducing neuroinflammation.
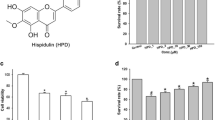
Kolaviron (Fig. 1) is a mixture of biflavonoids found in the nut of the West African edible seed, Garcinia kola. Kolaviron has been widely reported to produce a wide range of pharmacological activities. However, this flavonoid is mostly known for its antioxidant and anti-inflammatory activities. Kolaviron has been reported to prevent hepatotoxicity through inhibition of lipid peroxidation [8–10]. It has also been shown to be neuroprotective, an action that has been attributed to its antioxidant property [11–13]. In vivo and in vitro research have shown that kolaviron exhibited anti-inflammatory effects. In a study reported by Olaleye et al. [14], kolaviron inhibited carrageenan-induced rat paw oedema and suppressed TNFα, NO and PGE2 production in RAW 264.7 macrophages. These results were confirmed by a study reported by Abarikwu [15], who showed that the suppressive effects of kolaviron on the production of pro-inflammatory mediators in macrophages is possibly due to inhibition of NF-κB, Akt and MAPK signalling pathways. Farombi et al. [16] also showed that kolaviron prevented liver injury through inhibition of COX-2 and iNOS protein expressions, possibly by targeting NF-κB and AP-1 transcription factors.

Structural representation of kolaviron
In this study, we have investigated the effect of kolaviron on neuroinflammation in LPS-activated BV2 microglia. We have also shown that kolaviron exhibits antioxidant effect in BV2 microglia through activation of HO-1/Nrf2/ARE cytoprotective mechanisms.
Materials and methods
Extraction of kolaviron
Kolaviron was extracted from the seeds of Garcinia kola as earlier described [17]. The seeds were sliced, air-dried and powdered. The powdered seeds were defatted with n hexane for 24 h. The defatted dried marc was then extracted with methanol. Kolaviron was fractionated from the concentrated methanol extract using chloroform, to give a golden-yellow solid which consists of biflavanones-GB1, GB2 and kolaflavanone [18]. This was purified in a silica gel column with the elution of methanol and then, ethyl acetate, and the final compound has a glittering golden-brown appearance. Kolaviron was dissolved in DMSO for experiments.
Cell culture
BV2 mouse microglia cell line ICLC ATL03001 (Interlab Cell Line Collection, Banca Bilogica e Cell Factory, Italy) were maintained in RPMI 1640 medium with 10 % foetal bovine serum (FBS) (Sigma), 2 mM l-glutamine (Sigma), 100 U/ml penicillin and 100 mg/ml streptomycin (Sigma) in a 5 % CO2 incubator at 37 °C.
HT22 mouse hippocampal neuronal cells were a kind gift from Dr Jeff Davies, and cultured in DMEM supplemented with 10 % FBS, 100 U/ml penicillin and 100 µg/ml streptomycin in a 5 % CO2 incubator at 37 °C.
Cell viability
The colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was performed to determine the viability of BV2 microglia incubated with or without LPS (100 ng/ml) in the presence of kolaviron (5–20 μM) for 24 h. Following LPS stimulation, culture media was replaced with media containing 5 mg/ml MTT solution and incubated for 4 h. Thereafter, 200 μl of medium was removed from each well without disturbing the cell clusters, and 150 μl of DMSO solution was added to wells to dissolve the formazan crystals. Thorough mixing of the preparation was facilitated by shaking the plate for a few seconds before absorbance was read at 540 nm with a plate reader (Infinite F50, Tecan) [19].
Determination of TNFα and IL-6 production in LPS-activated BV2 microglia
BV2 cells were stimulated with LPS (100 ng/ml) in the presence or absence of kolaviron (5–20 µM) for 24 h. Thereafter, culture supernatants were collected and centrifuged. Concentrations of TNFα and IL-6 in cell supernatants were analysed with a commercially available ELISA kit (BioLegend, UK), followed by measurements in a plate reader at a wavelength of 450 nm.
Determination of PGE2 production in LPS-activated BV2 microglia
Following pre-treatment with kolaviron (5–20 μM) and stimulation with LPS (100 ng/ml) for 24 h, levels of PGE2 in BV2 cell culture medium were measured using commercially available enzyme immunoassay kit (Arbor Assays, Ann Arbor, Michigan, USA) according to manufacturer’s instructions. Absorbance was read at 450 nm in a microplate reader (Infinite F50, Tecan).
Determination of NO production in LPS-activated BV2 microglia
Following pre-treatment with kolaviron (5–20 μM) and stimulation with LPS (100 ng/ml) for 24 h, levels of nitrite in culture media were measured using commercially available Griess assay kit (Promega) according to the manufacturer’s instructions. Absorbance was measured at 540 nm in a microplate reader (Infinite F50, Tecan).
Determination of cellular reactive oxygen species (ROS)
The effect of LPS on intracellular ROS levels in BV2 cells was performed using the fluorescent 2′,7′-dichlorofluorescin diacetate (DCFDA)-cellular reactive oxygen species detection assay kit (Abcam). BV2 microglia were incubated with 10 µM DCFDA for 30 min at 37 °C. After removal of excess DCFDA, cells were washed and then pre-treated with kolaviron (5–20 µM) for 30 min followed by stimulation with 100 ng/ml LPS for 4 h at 37 °C. Intracellular production of ROS was measured by the fluorescence detection of dichlorofluorescein (DCF) as the oxidised product of DCFH on a microplate reader with an excitation wavelength of 485 nm and emission wavelength of 535 nm.
Immunoblotting
Following pre-treatment with kolaviron (5–20 μM) and stimulation with LPS (100 ng/ml), cell lysates were prepared by washing cells with PBS, followed by addition of lysis buffer and phenylmethylsulfonyl fluoride (PMSF), and centrifugation for 10 min. Nuclear extracts were prepared using EpiSeeker Nuclear Extraction Kit (Abcam), according to the manufacturer’s instructions. Briefly, cells were washed with cold PBS, followed by the addition of 20 μl of pre-extraction buffer and incubation on ice for 10 min. Thereafter, cells were centrifuged at 12,000 rpm for 1 min. Supernatants were discarded, and 10 μl of extraction buffer was added to the pellet and incubated on ice for 15 min, followed by centrifugation at 13,500 rpm for 15 min at 4 °C. The resulting nuclear extracts in the supernatants were collected.
Twenty-five micrograms of protein was subjected to sodium dodecyl sulphate-polyacrylamide (SDS) gel electrophoresis. Proteins were then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA) for 2 h. Membranes were then blocked at room temperature for 1 h and then incubated with primary antibodies overnight at 4 °C. Primary antibodies used in the experiments were rabbit anti-iNOS (Santa Cruz, 1:500), rabbit anti-COX2 (Santa Cruz, 1:500), rabbit anti-phospho-IκBα (Santa Cruz, 1:250), rabbit anti-total IκBα, rabbit anti-HO-1 (Santa Cruz, 1:500) and rabbit anti-Nrf2 (Santa Cruz, 1:500). Blots were detected with Alexa Fluor® 680 goat anti-rabbit IgG (Life technologies, UK) using the Licor Odyssey infrared imager. Equal protein loading was assessed using rabbit anti-actin antibody (Sigma, 1:1000).
NF-κBp65 ELISA
Cultured BV2 cells were stimulated with LPS (100 ng/ml) for 1 h following treatment with kolaviron (5–20 µM). Thereafter, nuclear extracts were collected and analysed for levels of phospho-NF-κBp65 using ELISA (eBioscience), according to the manufacturer’s instructions. Absorbance was read at 450 nm.
Reporter gene assays
BV2 microglia were cultured as described above. At confluence, the cells were sub-cultured (at a ratio of 1:3) for 24 h before transfection. Thereafter, cells were harvested and re-suspended at 4 × 105 cells/ml in Opti-MEM containing 5 % FBS and 1 % NEAA. Cells were seeded out in a solid white 96-well plates and incubated with pGL4.32 [luc2P/NF-κB-RE/Hygro] vector (Promega, UK), using Fugene 6 (Promega) transfection reagent (diluted 1:3 in serum-free Opti-MEM) and incubated for a further 16 h at 37 °C in 5 % CO2 incubator. Following transfection, media was changed to OPTI-MEM and incubated for another 8 h. Thereafter, transfected cells were treated in kolaviron (5–20 µM) and incubated for 30 min at 37 °C followed by LPS (100 ng/ml) for 6 h. At the end of the stimulation, NF-κB-mediated gene expression was measured using One-Glo luciferase Assay kit (Promega), according to the manufacturer’s instructions.
To carry out the ARE-dependent reporter gene assay, BV2 microglia were seeded out and incubated in solid white 96-well at 37 °C for 24 h. A transfection cocktail was made by diluting ARE vector (pGL4.37 [luc2P/ARE/Hygro]; Promega) at a concentration of 1 ng DNA/µl in Fugene 6 transfection reagent diluted in OPTI-MEM. The cocktail was incubated at room temperature for 20 min, followed by the addition of 8 µl to each well, followed by incubation for 18 h at 37 °C. Thereafter, culture medium was changed to OPTI-MEM and incubated for 6 h at 37 °C. Cells were then treated with kolaviron (5–20 µM) and incubated at 37 °C for 18 h. Following incubation, plates were allowed to cool at room temperature for 15 min. Thereafter, 80 µl of luciferase buffer containing luminescence substrate was added to each well and luminescence was read with FLUOstar OPTIMA reader (BMG LABTECH).
DNA binding assays
An ELISA-based DNA binding assay was used to investigate the effects of kolaviron on DNA binding of NF-κB. BV2 microglia were treated with 5, 10 and 20 μM kolaviron 30 min before stimulation with LPS (100 ng/ml). 1 h later, nuclear extracts were prepared using EpiSeeker Nuclear Extraction Kit (Abcam), according to the manufacturer’s instructions. DNA binding assay was carried on nuclear extracts using the TransAM NF-κB transcription factor ELISA kit (Activ Motif, Belgium) according the manufacturer’s instructions. The ELISA kit employs a 96-well plate to which an oligonucleotide containing the NF-κB consensus site (5′-GGGACTTTCC-3′) has been immobilised. Briefly, 30 μl of complete binding buffer was added to each well, followed by 20 μg nuclear extract samples. The plate was covered and rocked (100 rpm) for 1 h at room temperature. This was followed by the addition of NF-κB antibody (1:1000; 1 h) and HRP-conjugated antibody (1:1000; 1 h). Absorbance was read on a Tecan F50 microplate reader at 450 nm.
To investigate DNA binding of Nrf2, BV2 microglia were treated with kolaviron (5–20 μM). Nuclear extracts were added to 96-well plates to which an oligonucleotide containing the ARE consensus binding site (5′-GTCACAGTGACTCAGCAGAATCTG-3′) has been immobilised. Assay procedure was as described for NF-κB, using an Nrf2 antibody (1:1000; 1 h).
Nrf2 gene silencing experiments
Small interfering RNA (siRNA) targeted at Nrf2 (Santa Cruz Biotechnology) was used to knockout Nrf2. BV2 cells were cultured and incubated at 37 °C in a 5 % CO2 incubator until 70–80 % confluent. Thereafter, 2 µl Nrf2 siRNA duplex was diluted into 100 µl of siRNA transfection medium (Santa Cruz Biotechnology). In a separate tube, 2 µl of transfection reagent (Santa Cruz biotechnology) was diluted into 100 µl of siRNA transfection medium. The dilutions were mixed gently and incubated for 30 min at room temperature. Next, cells were incubated in Nrf2 siRNA transfection cocktail for 6 h at 37 °C. Following transfection, media was changed in all cells to complete media and incubated for a further 18 h. Effects of kolaviron (20 μM) on NO, PGE2, TNFα and IL-6 production in LPS-stimulated normal and Nrf2-silenced BV2 cells were then investigated. Nuclear NF-κBp65 ELISA and NF-κB DNA binding assays were conducted in LPS-stimulated normal and Nrf2-silenced BV2 microglia treated with kolaviron (20 μM).
BV2 microglia/HT22 hippocampal neuron co-culture
Neuroprotective effects of kolaviron was investigated using a transwell co-culture system. BV2 cells were cultured at the density of 5 × 104 cells/ml on the transwell insert (pore size 0.4 µm; Corning) in 96-well plate placed above the HT22 neuronal layer. 24 h after establishing co-culture, BV2 cells were pre-treated with kolaviron (5–20 µM) and then stimulated with LPS (1 µg/ml) for 24 h. At the end of the experiment, 200 µl MTT solution (5 mg/ml) was added to each well containing HT22 neurons and incubated at 37 °C for 4 h. Then, 200 μl of medium was removed and 150 μl of DMSO solution was added to wells to dissolve the formazan crystals. Thorough mixing of the preparation was facilitated by shaking the plate for a few seconds before absorbance was read at 540 nm with a plate reader.
Supernatants were also collected from the BV2 microglia layer and analysed for levels of TNFα, IL-6, NO and PGE2.
Cellular DNA fragmentation assay in HT22 hippocampal neurons co-cultured with BV2 microglia
The effect of microglial activation on cellular DNA fragmentation in HT22 cells was conducted using an assay kit (Roche Diagnostics, Mannheim, Germany), according to the manufacturer’s instructions. HT22 neurons were co-cultured with BV2 cells grown in transwells. Then, HT22 layers were labelled with 5-bromo-2′-deoxyuridine (BrdU) for 12 h. Following labelling; BV2 layer was treated with kolaviron (5–20 µM) 30 min prior to LPS (1 µg/ml) stimulation for 24 h. Thereafter, HT22 cells were collected, centrifuged at 250 g for 10 min and supernatants removed. The cells were then lysed with 200 µl of buffer and incubated for 3 min at room temperature. Cells were centrifuged again 250 g for 10 min and supernatants removed. The labelled DNA in the supernatants as a result of DNA fragmentation was measured using ELISA with an anti-BrdU antibody.
Statistical analysis
All experiments were performed at least three times and in triplicates unless otherwise stated. Data in figures and text were expressed as mean ± SEM. Statistical analysis was performed using one-way ANOVA with post hoc Student Newman-Keuls test (multiple comparisons).
Results
Kolaviron did not affect the viability of BV2 microglia
Cell viability experiments were carried out to determine whether concentrations of kolaviron used in this study affected viability of BV2 microglia. Figure 2 shows that kolaviron at concentrations ranging from 5 to 20 µM did not produce cytotoxicity in BV2 microglia after 24 h.

Kolaviron (5–20 µM) did not affect the viability of BV2 microglia with or without LPS (100 ng/ml) for 24 h. Results are expressed as mean ± SEM of three independent experiments
Kolaviron prevented the production of TNFα and IL-6 in LPS-activated BV2 microglia
To investigate the effects of kolaviron on the secretion of pro-inflammatory cytokines, BV2 cells were treated with LPS in the presence or absence of kolaviron for 24 h. Thereafter, levels of TNFα and IL-6 were measured. Expectedly, stimulation with LPS induced a marked increase in the levels of TNFα and IL-6. We observed that kolaviron dose-dependently reduced the secretion of TNFα and IL-6 (Fig. 3a, b).

Kolaviron suppressed a TNFα and b IL-6 production in LPS-activated BV2 cell line. Cells were treated with or without kolaviron (5–20 µM) followed by LPS (100 ng/ml) treatment for 24 h. Production of TNFα and IL-6 was assessed in culture supernatants using an ELISA kit. Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of three independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001)
Kolaviron produced iNOS-mediated reduction of NO secretion in LPS-activated BV2 microglia
Microglial activation during neuroinflammation results in increased production of nitric oxide, which reacts with oxygen to produce the highly reactive peroxinitrite (ONOO•) anion. As expected, there was an increase in the levels of nitrite (a marker of NO production) following stimulation of BV2 microglia with LPS (100 ng/ml). LPS stimulation of BV2 microglia caused an increase in NO production. Pre-treatment with kolaviron (5–20 µM) resulted in a significant and concentration-dependent reduction in nitrite production (Fig. 4a). Encouraged by this observation, we used Western blot to evaluate the effects of kolaviron on protein levels of iNOS in LPS-activated microglia and showed that the compound significantly reduced iNOS protein at 5 µM (~1.3-fold, p < 0.001), 10 µM (~2-fold, p < 0.001) and 20 µM (~3.3-fold, p < 0.001) (Fig. 4b).

Kolaviron inhibited LPS-induced iNOS-mediated NO production by in BV2 microglia. Cells were pre-treated with indicated concentrations of kolaviron for 30 min prior adding LPS (100 ng/ml) for 24 h. NO accumulation in the supernatants was measured using Griess assay kit (a). Levels of iNOS protein was determined using western blotting (b). Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of 3 independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001)
Kolaviron produced COX-2-mediated reduction of PGE2 secretion in LPS-activated BV2 microglia
Studies have shown that PGE2 is a common mediator in brain inflammation [20]. It has also been shown that PGE2 is released following stimulation of microglia with LPS [21]. Hence we investigated whether kolaviron could suppress PGE2 production in LPS-activated BV2 microglia. Expectedly, LPS stimulation of BV2 microglia produced elevated levels of PGE2 in cell supernatants (Fig. 5a), an outcome that was reduced by 5 µM (~1.4-fold, p < 0.01), 10 µM (~2-fold, p < 0.001) and 10 µM (~2.2-fold, p < 0.001) kolaviron. As shown in Fig. 5b, kolaviron also produced 1.3–2.0-fold reduction in elevated levels of COX-2 protein in LPS-activated BV2 microglia.

Kolaviron suppressed LPS-induced PGE2 production (a) by inhibiting COX-2 protein (b) in BV2 microglia. Cells were pre-treated with indicated concentrations of kolaviron for 30 min prior to adding LPS (100 ng/ml) for 24 h. Supernatants were subjected to EIA to evaluate PGE2 levels. Cell lysates were subjected to Western blot to determine COX-2 expression. Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of 3 independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001)
Cellular ROS production was suppressed by kolaviron
Microglia-mediated inflammation is known to induce oxidative damage [22, 23]. To test the effects of kolaviron on cellular ROS generation, the intracellular ROS was determined by the DCFDA fluorescence detection. Stimulating BV2 cells with LPS (100 ng/ml) induced a characteristic increase in cellular ROS generation (Fig. 6). This increase was significantly (p < 0.001) inhibited by kolaviron (5–20 µM).

Kolaviron reduced LPS-induced cellular ROS generation in BV2 microglia. Cellular ROS levels were measured using DCFDA-cellular reactive oxygen species detection assay kit. Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of 3 independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001)
Kolaviron inhibited neuroinflammation by targeting IκB/NF-κB signalling pathway
The genes encoding inflammatory proteins produced in response to microglial activation, such as the pro-inflammatory cytokines, PGE2/COX-2, NO/iNOS and the ROS is under the transcriptional control of NF-κB. Consequently, we explored the NF-κB signalling pathway as a possible mechanistic target for the inhibitory action of kolaviron in neuroinflammation.
In unstimulated microglia NF-κB exists complexed to the inhibitory IκB. Upon microglial activation by LPS, IκB is phosphorylated and undergoes proteasomal degradation. This results in nuclear translocation of NF-κBp65 subunit and subsequent transcriptional activity.
Our first line of investigation in this respect therefore focused on IκB phosphorylation and degradation. We showed that LPS induced a characteristic increase in phospho-IκB following stimulation with LPS (Fig. 7a). Following pre-treatment with kolaviron, levels of phospho-IκB protein was significantly (p < 0.001) reduced in a concentration-dependent fashion (5 µM, ~1.4-fold; 10 µM, ~1.7-fold; 20 µM, ~2-fold). Similarly, LPS treatment of BV2 microglia-induced degradation of IκB (Fig. 7b), a phenomenon that was significantly and dose-dependently attenuated by pre-treatment with kolaviron. Figure 7c shows the effect of kolaviron on nuclear translocation of NF-κBp65 subunit following stimulation with LPS. Evaluation of nuclear phospho-NF-κBp65 using ELISA revealed that prior incubation with kolaviron significantly (p < 0.001) prevented translocation of NF-κBp65 to the nucleus (5 µM, ~1.4-fold; 10 µM, ~2.1-fold; 20 µM, ~2.2-fold). These results suggest that kolaviron modulates NF-κB signalling by interfering with the IκB/NF-κB complex.

Kolaviron inhibits IκB/NF-κB signalling pathway in LPS-activated BV2 microglia. Cells were pre-treated with kolaviron (5–20 µM) for 30 min and incubated prior to stimulation with LPS (100 ng/ml) for a further 30 min. Protein levels of phospho-(a) and total IκB (b) were determined with Western blot. Nuclear extracts were prepared from BV2 microglia pre-treated with indicated concentrations of kolaviron 30 min before stimulation with LPS for 60 min, and analysed for p-NF-κBp65 using an ELISA kit (c). DNA binding activity was evaluated with nuclear extracts in an ELISA-based DNA binding assay (d). e Effect of kolaviron on NF-κB-luc reporter gene activity. Cells transfected with the reporter plasmid (luc2P/NF-κB-RE/Hygro) were treated with kolaviron (5, 10, and 20 μM) with or without LPS (100 ng/ml) for 6 h, and the reporter gene assay was performed. Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of 3 independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001)
Encouraged by these results, we decided to assess the effect of kolaviron on binding of NF-κBp65 to an immobilised 5′-GGGACTTTCC-3′ oligonucleotide, which confers the NF-κB binding site. This ELISA method provides specific and quantitative data on DNA binding of nuclear NF-κB. Using the method, we could demonstrate that LPS induced a marked DNA binding of NF-κB in BV2 microglia (Fig. 7d). We however observed that nuclear extracts from kolaviron-treated cells showed significantly reduced (p < 0.001) DNA binding by NF-κB. There was a ~4 and ~5-fold inhibition of DNA binding by 10 and 20 µM concentrations of kolaviron, respectively.
Finally, we measured NF-κB activity directly using reporter genes controlled by an NF-κB response element. Due to the rapid activation and inactivation of NF-κB following activation, the luciferase reporter assay was used in our experiments. BV2 microglia were transfected with the pGL4.32 [luc2P/NF-κB-RE/Hygro] vector and then stimulated with LPS (100 ng/ml). Our results showed that kolaviron produced a concentration-dependent and significant (p < 0.001) inhibition of luciferase activity, demonstrating inhibition of NF-κB activity directly (Fig. 7e).
Kolaviron activated HO-1/Nrf2/ARE antioxidant protective mechanism in BV2 microglia
The expression of the antioxidant protein HO-1 has been shown to mediate the resolution of neuroinflammation [24]. We were therefore interested in evaluating whether activation of HO-1 might be contributing to the anti-neuroinflammatory effects of kolaviron. Using Western blot, we showed that treatment of BV2 microglia with kolaviron resulted in significant increase in the protein expression of HO-1 (Fig. 8a).

Kolaviron increased HO-1 via activation of Nrf2/ARE pathway in BV2 microglia. a Western blot analysis shows the effects of kolaviron on HO-1 protein expression. Cell lysates were obtained from BV2 cells treated with kolaviron (5, 10 and 20 μM) for 24 h. Quantification data are shown in the graph (n = 3). b Western blot for Nrf2. Nuclear extracts were prepared from BV2 cells treated with kolaviron (5, 10 and 20 μM) for 1 h. Quantification data are shown in the graph (n = 3). c Effect of kolaviron on ARE-luc reporter gene activity. Cells transfected with the reporter plasmid (ARE-luc) were treated with kolaviron (5, 10 and 20 μM) for 6 h, and reporter gene assay was performed. d EMSA for Nrf2. Nuclear extracts were prepared from BV2 cells treated with kolaviron for 1 h and incubated with the Nrf2 oligonucleotide. Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of 3 independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001)
HO-1 expression is controlled by Nrf2, which binds to a specific DNA sequence (ARE) present in the promoter region of phase II enzymes such as HO-1, and enhances their transcription. Based on our observation that kolaviron increases the protein expression of HO-1, we further determined whether this effect was mediated by Nrf2. Western blot analysis showed that kolaviron increased levels of Nrf2 in the nucleus, when compared with untreated (control) cells (Fig. 8b). Treatment with 5, 10 and 20 µM kolaviron were shown to induce ~2.5-, ~3- and ~4-fold increase in nuclear Nrf2.
We decided to explore the effect of kolaviron further using a luciferase reporter which is under the control of a promoter containing the ARE consensus. Using this reporter assay, we observed that kolaviron produced a dose-dependent increase in ARE luciferase activity (Fig. 8c). This seems to suggest that kolaviron increases the transcriptional activity of Nrf2, which might explain its effect on HO-1. We confirmed these observations with an Nrf2 binding assay, which employs immobilised oligonucleotide containing the ARE binding site 5′-GTCACAGTGACTCAGCAGAATCTG-3′. This experiment revealed that kolaviron (5–20 µM) produced significant and dose-dependent activation, and hence DNA binding of Nrf2 (Fig. 8d).
Inhibition of neuroinflammation by kolaviron is dependent on Nrf2
Based on our results showing that kolaviron inhibits neuroinflammation and activates the HO-1/Nrf2 antioxidant mechanisms, we hypothesised that activation of Nrf2 contributed to the inhibitory effect of the flavonoid on neuroinflammation. Consequently, Nrf2 siRNA duplex was transfected into BV2 microglia, followed by stimulation with LPS in the presence or absence of kolaviron (20 µM). After 24 h, supernatants were collected and analysed for levels of secreted TNFα, IL-6, NO and PGE2.
Expectedly, production of NO, PGE2, TNFα and IL-6 in Nrf2-silenced BV2 microglia was significantly (p < 0.05) increased, in comparison with control cells (Fig. 9a–d). In control cells, pre-treatment with kolaviron (20 µM) resulted in significant (p < 0.05) reduction in pro-inflammatory mediators. However, silencing Nrf2 resulted in almost complete reversal of the anti-inflammatory effect of kolaviron, with significant increases in the production of NO, PGE2, TNFα and IL-6, when compared to control cells (Fig. 9a–d).

Anti-inflammatory activity of Kolaviron is dependent on Nrf2. BV2 microglia were transfected with Nrf2 siRNA and treated with kolaviron (20 µM), followed by LPS (100 ng/ml) for 24 h. Supernatants were analysed for levels of NO (a), PGE2 (b), TNFα (c) and IL-6 (d). Nuclear extracts (LPS stimulation, 1 h) were also analysed for nuclear p-NF-κBp65 subunit using ELISA (e), and DNA binding (f). Western blot experiments on nuclear extracts showing efficiency of Nrf2 siRNA transfection (g). Statistical analysis was performed using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). (*p < 0.05, **p < 0.01, ***p < 0.001), (θθθ p < 0.001), (&&& p < 0.001)
In order to confirm the role of Nrf2 in the anti-inflammatory effect of kolaviron, we compared its actions on protein levels of nuclear phospho-NF-κBp65 following LPS stimulation in control and Nrf2-silenced BV2 cells. We observed that protein expression of phospho-NF-κBp65 in nuclear extracts of Nrf2-silenced BV2 microglia was significantly increased, in comparison with control cells (Fig. 9e). Interestingly, reduction in LPS-induced increase in nuclear phospho-NF-κBp65 by kolaviron (20 µM) was completely reversed following transfection of BV2 microglia with Nrf2 siRNA (Fig. 9E). Similar results were obtained in experiments to investigate the effects of kolaviron (20 µM) on DNA binding of NF-κB. Following Nrf2 knockout, we observed that inhibition of LPS-induced DNA binding by kolaviron (20 µM) was completely reversed (Fig. 9f). Our results suggest a loss in the anti-inflammatory activity of kolaviron in BV2 microglia in the absence of the Nrf2 gene.
In order to confirm that Nrf2 gene was efficiently silenced in these experiments, nuclear extracts were prepared from both control and Nrf2-silenced BV2 cells. Western blot for Nrf2 showed basal levels of Nrf2 protein in control cells. In cells transfected with Nrf2 siRNA, levels of Nrf2 protein were significantly reduced, suggesting that the gene encoding this protein was effectively silenced (Fig. 9g).
Kolaviron protected HT22 hippocampal neurons from neuroinflammation-induced neuronal death
During neuroinflammation, microglial activation by LPS results in significant release of pro-inflammatory mediators such as the ROS, NO, PGE2, TNFα and IL-6 which are toxic to adjacent neurons.
To investigate the neuroprotective effect of kolaviron in microglia-mediated neurotoxicity, we established a BV2 microglia/HT22 neuron transwell co-culture. The microglia layer was then stimulated with LPS (1 µg/ml) in the presence or absence of kolaviron (5–20 µM), as usual. After 24 h, viability of HT22 hippocampal neurons was determined using the MTT assay. Figure 10a shows that LPS stimulation of BV2 microglia layer resulted in significant reduction in neuronal viability, when compared with unstimulated control. However, monolayers of HT22 neurons co-cultured with kolaviron pre-treated BV2 microglia showed significant (p < 0.05) increased viability, thus demonstrating neuroprotection.

Kolaviron produced neuroprotective effect in microglia-mediated HT22 hippocampal neuron death. BV2 microglia were seeded in transwell inserts and the inserts were placed on top of the wells containing the HT22 neuronal culture. The microglia layer was treated with kolaviron (5–20 µM) for 30 min prior to stimulation with LPS (1 µg/ml) for 24 h. a Viability of HT22 hippocampal neuron was assessed using MTT assay. Supernatants were collected from the microglia monolayer and assessed for levels of TNFα, IL-6, NO and PGE2 (b–e). Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of 3 independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001)
To confirm the roles of released pro-inflammatory mediators in neurotoxicity/neuroprotection, supernatants were taken from the microglia layer and analysed for NO, PGE2, TNFα and IL-6. Results showed elevated levels of NO, PGE2, TNFα and IL-6 in the supernatants of LPS-stimulated microglia (Fig. 10b–e). Expectedly, pre-treatment with kolaviron (5–20 µM) resulted in dose-dependent and significant (p < 0.05) reduction in NO, PGE2 and TNFα. Reduction in IL-6 was achieved with 10 and 20 µM kolaviron.
Following apoptotic death of neurons, DNA fragments are released from the cytoplasm of apoptotic cells after lysis with a non-ionic detergent. A colorimetric DNA fragmentation assay was conducted on the HT22 neurons. There was ~1.5-fold increase in DNA fragmentation of HT22 neurons co-cultured with LPS-stimulated BV2 microglia, when compared with neurons co-cultured with unstimulated BV2 cells (Fig. 11). However, neuronal DNA fragmentation was significantly reduced when BV2 cells were pre-treated with kolaviron prior to LPS (5 µM, ~1.2-fold; 10 µM, ~1-fold; 20 µM, ~0.7-fold, compared with unstimulated cells).

Kolaviron prevented DNA fragmentation of HT22 hippocampal neurons exposed to LPS (1 µg/ml)-activated BV2 microglia. BV2 microglia were seeded in transwell inserts and the inserts were placed on top of the wells containing the HT22 neuronal culture. This was followed by labelling of neuronal layer with BrdU for 12 h. Thereafter, microglia layer was pre-incubated with kolaviron (5–20 µM) for 30 min followed by LPS (1 µg/ml) treatment for 24 h. Thereafter labelled DNA from lysates were collected and analysed using ELISA. Statistical analyses were carried out using one-way ANOVA with post hoc Student Newman–Keuls test (multiple comparisons). Results are expressed as mean ± SEM of 3 independent experiments, (*p < 0.05, **p < 0.01, ***p < 0.001)
Discussion
The release of neurotoxic pro-inflammatory mediators from microglia, in response to activators such as LPS results in the death of adjacent neurons. For example, excessive NO synthesis under neuroinflammation is now widely accepted to result in the formation of reactive nitrogen species and neuronal cell death [25]. Similarly, PGE2 has been shown to induce neuronal death through activation of EP2 receptors [26]. Pro-inflammatory cytokines such as TNFα and IL-6, as well as ROS have been implicated in neuronal death which characterise neurodegenerative diseases [27]. In this study we showed that kolaviron inhibited the production of pro-inflammatory cytokines (TNFα and IL-6), NO, PGE2 and cellular ROS in BV2 microglia stimulated with LPS. We further showed that the effect of kolaviron on NO and PGE2 production, were mediated through inhibition of iNOS and COX-2 proteins, respectively. To our knowledge, this is the first study showing inhibition of neuroinflammation by kolaviron in BV2 microglia.
Similar studies in peripheral cells have shown anti-inflammatory activity by kolaviron. Using RAW 264.7 murine macrophages, studies have shown that kolaviron inhibited PGE2, NO and TNFα release following LPS stimulation [14]. Interestingly, another study by Abarikwu [15] reported that kolaviron inhibited LPS-induced secretion of IL-6 but not TNFα in RAW 264.7 macrophages. Kolaviron was also shown to suppress the expressions of TNFα, IL-6 and IL-1α genes in LPS-stimulated 93RS2 sertoli cells [28].
During neuroinflammation, NF-κB activation controls the expression of pro-inflammatory cytokines, iNOS/NO, PGE2/COX-2 and the ROS to trigger a self-perpetuating process resulting in progressive neuronal damage. We therefore aimed to identify the role of this critical transcription factor in the anti-neuroinflammatory activity of kolaviron. Firstly, we determined the effect of kolaviron on phosphorylation and degradation of IκB protein, a process that is critical to NF-κB activation. We showed that kolaviron produced dual inhibition of phosphorylation and degradation of NF-κB, as well as nuclear translocation of the p65 subunit, suggesting that this biflavonoid acts to stabilise NF-κB in the microglial cytoplasm following activation by LPS.
In order to determine whether kolaviron acts by interfering with the transcriptional activity of NF-κB, or by inhibiting NF-κB activation upstream of its nuclear activity, we analysed the DNA binding activity of NF-κB in LPS-activated BV2 microglia and evaluated the modulatory effect of kolaviron in this process. Our experiments revealed that kolaviron inhibited the activation and binding of NF-κBp65 subunit to its target DNA. We also employed a luciferase reporter gene under the control of a promoter containing the NF-κB consensus sequence, to demonstrate the inhibitory effect of kolaviron on NF-κB activation in LPS-stimulated BV2 microglia. These results provide confirmatory evidence to show that kolaviron blocks nuclear activation of NF-κB, as well as the upstream events which occur prior to nuclear activation.
In RAW264.7 macrophages, higher concentrations of kolaviron (50 and 100 µM) than those used in this study produced a similar effect on LPS-induced phosphorylation and degradation of IκB [15]. It therefore appears that BV2 microglia are more sensitive to the actions of this biflavonoid than peripheral macrophages such as the RAW264.7 cells. Studies in rats have reported that kolaviron blocked the DNA binding activity of NF-κB induced by dimethyl nitrosamine [16]. LPS is known to trigger NF-κB activation through TLR4 signalling. However, studies have shown that dimethyl nitrosamine could be activating the PI3 K-Akt/PKB pathway in immune cells, which then contributes to the activation of NF-κB [29].
Several studies have employed various models to demonstrate the antioxidant property of kolaviron and suggested that this property might be responsible for its neuroprotective effects [11–13]. In view of the role played by the activation of the Nrf2/ARE antioxidant protective mechanism in restoring the microglia to its resting inactivated state, we investigated the effect of kolaviron on the activation of Nrf2/ARE in BV2 microglia. Firstly, we were able to show that the protein level of the phase II enzyme HO-1 was significantly increased by kolaviron. There have been several suggestions in the scientific literature, indicating that HO-1 expression helps in mediating the resolution of inflammation, including neuroinflammation [24, 30].
As HO-1 expression is under the control of Nrf2, we further investigated the effects of kolaviron on the activation of this transcription factor. In the presence of kolaviron, there was an increase in nuclear accumulation of Nrf2 protein in BV2 microglia. This effect was accompanied by an increase in the transcriptional activity and DNA binding of Nrf2 in cells treated with kolaviron. These observations indicate that the antioxidant activity of kolaviron, through activation of Nrf2/HO-1 axis may be contributing to its anti-neuroinflammatory activity. To our knowledge, this is the first study demonstrating that kolaviron could activate the Nrf2/HO-1 system in the microglia. Similar effects have been shown for natural antioxidants such as tiliroside [31] and other small molecule activators of the Nrf2-HO-1 [32].
Nrf2 mediates anti-neuroinflammatory activity through a direct inhibition of NF-κB signalling pathway [3, 7]. We therefore thought that it might be interesting to determine whether Nrf2 is a requirement for the anti-inflammatory effect of kolaviron in BV2 microglia. To achieve this we stimulated both control and Nrf2 siRNA-transfected microglia with LPS in the presence and absence of kolaviron (20 µM). Levels of pro-inflammatory proteins were then measured in culture supernatants. Our results clearly indicate that silencing Nrf2 in BV2 microglia resulted in the reversal of suppressive effects on NO, PGE2, TNFα and IL-6 production in BV2 microglia. Similarly, inhibition of nuclear accumulation of p65 subunit as well as DNA binding by kolaviron were reversed by silencing Nrf2 in the microglia. Taken together, it is suggested that Nrf2 activation is a critical requirement for the inhibition of neuroinflammation by kolaviron. This observation appears to explain, at least in part, the molecular mechanism of action of kolaviron in neuroinflammation.
It is well established that microglial cells can be toxic to neurons through cell–cell contact-dependent mechanisms [33]. A transwell co-culture system was therefore used to study neuroprotection by kolaviron against microglia-induced toxicity. To achieve neurotoxicity, a high concentration (1 µg/ml) of LPS was used to activate microglia. Results showed that kolaviron protected neurons from death induced by microglial activation. In order to confirm the role of released mediators in neurotoxicity, supernatants from the microglial layer were analysed for levels of PGE2, NO, TNFα and IL-6. Interestingly, levels of released inflammatory mediators correlated with neuronal survival, suggesting that kolaviron prevented neuronal death by inhibiting neuroinflammation in the microglia.
Conclusions
The present study demonstrates that kolaviron inhibits neuroinflammation and activates the Nrf2/HO-1 protective mechanism. We also suggest that the inhibitory effect of this biflavonoid compound is dependent on Nrf2.
References
Polazzi E, Monti B (2010) Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol 92:293–315
Li W, Khor TO, Xu C, Shen G, Jeong WK, Yu S, Kong AN (2008) Activation of Nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochem Pharmacol 76:1485–1489
Koh K, Kim J, Jang YJ, Yoon K, Cha Y, Lee HJ, Kim J (2011) Transcription factor Nrf2 suppresses LPS-induced hyperactivation of BV-2 microglial cells. J Neuroimmunol 233:160–167
Sandberg M, Patil J, D’Angelo B, Weber SG, Mallard C (2014) Nrf2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 79:298–306
Kim J, Cha YN, Surh YJ (2010) A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res 690:12–23
Innamorato NG, Lasters-Becker I, Cuadrado A (2009) Role of microglial redox balance in modulation of neuroinflammation. Curr Opin Neurol 22:308–314
Innamorato NG, Rojo AI, García-Yagüe AJ, Yamamoto M, de Ceballos ML, Cuadrado A (2008) The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol 181:680–689
Farombi EO, Tahnteng JG, Agboola AO, Nwankwo JO, Emerole GO (2000) Chemoprevention of 2-acetylaminofluorene-induced hepatotoxicity and lipid peroxidation in rats by kolaviron-a Garcinia kola seed extract. Food Chem Toxicol 38:535–541
Farombi EO (2000) Mechanisms for the hepatoprotective action of kolaviron: studies on hepatic enzymes, microsomal lipids and lipid peroxidation in carbon tetrachloride-treated rats. Pharmacol Res 42:75–80
Nwankwo JO, Tahnteng JG, Emerole GO (2000) Inhibition of aflatoxin B1 genotoxicity in human liver-derived HepG2 cells by kolaviron biflavonoids and molecular mechanisms of action. Eur J Cancer Prev 9:351–361
Abarikwu SO, Farombi EO, Pant AB (2011) Biflavanone-kolaviron protects human dopaminergic SH-SY5Y cells against atrazine induced toxic insult. Toxicol In Vitro 25:848–858
Igado OO, Olopade JO, Adesida A, Aina OO, Farombi EO (2012) Morphological and biochemical investigation into the possible neuroprotective effects of kolaviron (Garcinia kola bioflavonoid) on the brains of rats exposed to vanadium. Drug Chem Toxicol 35:371–380
Olajide OJ, Enaibe BU, Bankole OO, Akinola OB, Laoye BJ, Ogundele OM (2015) Kolaviron was protective against sodium azide (NaN3) induced oxidative stress in the prefrontal cortex. Metab Brain Dis 31:25–35
Olaleye SB, Onasanwo SA, Ige AO, Wu KK, Cho CH (2010) Anti-inflammatory activities of a kolaviron-inhibition of nitric oxide, prostaglandin E2 and tumor necrosis factor-alpha production in activated macrophage-like cell line. Afr J Med Med Sci 39:41–46
Abarikwu SO (2014) Kolaviron, a natural flavonoid from the seeds of Garcinia kola, reduces LPS-induced inflammation in macrophages by combined inhibition of IL-6 secretion, and inflammatory transcription factors, ERK1/2, NF-κB, p38, Akt, p-c-JUN and JNK. Biochim Biophys Acta 1840:2373–2381
Farombi EO, Shrotriya S, Surh YJ (2009) Kolaviron inhibits dimethyl nitrosamine-induced liver injury by suppressing COX-2 and iNOS expression via NF-kappaB and AP-1. Life Sci 84(2009):149–155
Iwu MM (1985) Antihepatoxic constituents of Garcinia kola seeds. Experientia 41:699–700
Iwu MM, Igboko OA, Okunji CO, Tempesta MS (1990) Antidiabetic and aldose reductase activities of biflavanones of Garcinia kola. J Pharm Pharmacol 42:290–292
Okorji UP, Olajide OA (2014) A semi-synthetic derivative of artemisinin, artesunate inhibits prostaglandin E2 production in LPS/IFNγ-activated BV2 microglia. Bioorg Med Chem. 22:4726–4734
Fiebich BL, Akter S, Akundi RS (2014) The two-hit hypothesis for neuroinflammation: role of exogenous ATP in modulating inflammation in the brain. Front Cell Neurosci 8:260
Olajide OA, Kumar A, Velagapudi R, Okorji UP, Fiebich BL (2014) Punicalagin inhibits neuroinflammation in LPS-activated rat primary microglia. Mol Nutr Food Res 58:1843–1851
Dumont M, Beal MF (2011) Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med 51:1014–1026
Gao HM, Zhou H, Hong JS (2012) NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci 33:295–303
Syapin PJ (2008) Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br J Pharmacol 155:623–640
Yuste JE, Tarragon E, Campuzano CM, Ros-Bernal F (2015) Implications of glial nitric oxide in neurodegenerative diseases. Front Cell Neurosci 9:322
Miyagishi H, Kosuge Y, Yoneoka Y, Ozone M, Endo M, Osada N, Ishige K, Kusama-Eguchi K, Ito Y (2013) Prostaglandin E2-induced cell death is mediated by activation of EP2 receptors in motor neuron-like NSC-34 cells. J Pharmacol Sci 121:347–350
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98
Abarikwu SO (2015) Anti-inflammatory effects of kolaviron modulate the expressions of inflammatory marker genes, inhibit transcription factors ERK1/2, p-JNK, NF-κB, and activate Akt expressions in the 93RS2 Sertoli cell lines. Mol Cell Biochem 401:197–208
Ratajczak-Wrona W, Jablonska E, Garley M, Jablonski J, Radziwon P, Iwaniuk A, Grubczak K (2014) PI3 K-Akt/PKB signaling pathway in neutrophils and mononuclear cells exposed to N-nitrosodimethylamine. J Immunotoxicol 11:231–237
Lee S, Suk K (2007) Heme oxygenase-1 mediates cytoprotective effects of immunostimulation in microglia. Biochem Pharmacol 74:723–729
Velagapudi R, Aderogba M, Olajide OA (2014) Tiliroside, a dietary glycosidic flavonoid, inhibits TRAF-6/NF-κB/p38-mediated neuroinflammation in activated BV2 microglia. Biochim Biophys Acta 1840:3311–3319
Foresti R, Bains SK, Pitchumony TS, de Castro Brás LE, Drago F, Dubois-Randé JL, Bucolo C, Motterlini R (2013) Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 76:132–148
Zujovic V, Taupin V (2003) Use of cocultured cell systems to elucidate chemokine-dependent neuronal/microglial interactions: control of microglial activation. Methods 29:345–350
Acknowledgments
The authors would like to thank the Physiological Society for funding this study, and for the award of an International Junior Research Grant (IJRG) to Dr Samuel A. Onasanwo.
Authors’ contributions
OAO designed the study and wrote the manuscript. SAO, RV, AE and OAO performed the experiments. OAO and RV analysed the data. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Samuel A. Onasanwo and Ravikanth Velagapudi have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Onasanwo, S.A., Velagapudi, R., El-Bakoush, A. et al. Inhibition of neuroinflammation in BV2 microglia by the biflavonoid kolaviron is dependent on the Nrf2/ARE antioxidant protective mechanism. Mol Cell Biochem 414, 23–36 (2016). https://doi.org/10.1007/s11010-016-2655-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2655-8