
Abstract
The implication of oxidative stress as primary mechanism inducing doxorubicin (DOX) cardiotoxicity is still questionable as many in vitro studies implied supra-clinical drug doses or unreliable methodologies for reactive oxygen species (ROS) detection. The aim of this study was to clarify whether oxidative stress is involved in compliance with the conditions of clinical use of DOX, and using reliable tools for ROS detection. We examined the cytotoxic mechanisms of 2 μM DOX 1 day after the beginning of the treatment in differentiated H9c2 rat embryonic cardiac cells. Cells were exposed for 2 or 24 h with DOX to mimic a single chronic dosage or to favor accumulation, respectively. We found that apoptosis was prevalent in cells exposed for a short period with DOX: cells showed typical hallmarks as loss of anchorage ability, mitochondrial hyperpolarization followed by the collapse of mitochondrial activity, and nuclear condensation. Increasing the exposure period favored a shift to necrosis as the cells preferentially exhibited early DNA impairment and nuclear swelling. In either case, measuring the fluorescence lifetime of 1-pyrenebutyric acid or the intensities of dihydroethidium or amplex red showed a consistent pattern in ROS production which was a slight increased level far from representative of an oxidative stress. Moreover, pre-treatment with dexrazoxane provided a cytoprotective effect although it failed to detoxify ROS. Our data support that oxidative stress is unlikely to be the primary mechanism of DOX cardiac toxicity in vitro.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The anthracycline compound doxorubicin (DOX), also called adriamycin, is an anti-cancerous reagent widely used for clinical treatment of many tumors. The mechanisms linked to its anti-tumoral effect were extensively characterized (see [1] for review). Briefly, DOX was reported to impair DNA by interfering with the topoisomerase II (TOP2) enzyme [2] or by forming DOX-DNA adducts [3]; to disrupt mitochondrial activity [4]; or to induce an overproduction of reactive oxygen species (ROS), i.e., oxidative stress, that damages the cells [5, 6]. While its chemotherapeutic efficiency is acknowledged, DOX also induces cardiotoxicity at cumulative doses exceeding 450–600 mg/m2 [1, 6, 7]. Although a matter of debate [1], DOX cardiotoxicity is thought to be due to induction of apoptosis [5, 8–10] resulting from oxidative stress [5, 6, 9, 11, 12]. The clinical use of compounds that detoxify ROS has been advocated as adjunctive therapy [6]. Accordingly, the bisdioxopiperazine agent dexrazoxane (DXR) showed a successful cardioprotection in the clinical setting [5, 6, 13] that was proposed to result from its metabolic bioactivation into rings-opened metal ion-chelating forms that display an antioxidant activity [14, 15].
Nevertheless, the implication of an oxidative stress as a primary mechanism of DOX cardiotoxicity remains questionable. A major issue comes from the inherent limitations of the in vitro cultured cell models for mimicking in vivo conditions. As primary cardiomyocytes require being isolated from sacrificed animals and are difficult to handle in long-term culture, toxicological assays are alternatively carried out in in vitro cardiac cell models. To date, the H9c2 immortalized cell line is the most used cell model for in vitro studies of DOX cardiotoxicity [8–10, 12, 16–19]. These cells are proliferating myoblasts derived from the embryonic rat ventricular myocardium, which have the ability to be differentiated into cardiac muscles [20]. However, concerns have been raised about the suitability of the H9c2 cells as model of myocardium since they are commonly used under proliferation state in most of reported cardiotoxicity studies [8–10, 12, 16–19], while differentiated cardiomyocytes without proliferative activity mainly comprise the adult heart in vivo. Consequently, the relevance of the conclusions made by the in vitro studies on DOX cardiotoxicity is disputed as the cell differentiation state was found to alter dose–responses [21].
Furthermore, discrepancies in experimental procedures make it even more difficult to predict the events that may occur in clinical settings. First, the intravenous administration doses of DOX per treatment cycle in patients are commonly ranging from about 30 to 75 mg/m2 (i.e., ~1.5–3 μM) [7]. Accordingly, studies involving single treatment with supra-clinical concentrations reflect potential DOX effects [17, 22–24], yet they diverge from the conditions of clinical use, and the conclusions of such studies should be thus considered with caution [1]. Second, DOX effects clearly depend on the cell type, the initial dosage, and the frequency of administration. Even so, assessing DOX effects in in vitro cultures imperatively requires taking into consideration the exposure duration as it influences the concentration eventually accumulated within the cells. Finally, the main source of discrepancy lies in the methodologies for ROS detection. Most of the methods that imply oxidative stress in DOX cardiotoxicity use fluorescent probes, even though their use have several pitfalls [25, 26]. The prominent examples are the fluorescein derivatives that were broadly used [8–10, 12, 22, 27] in spite of their lack of specificity and sensitivity for ROS detection [25, 26]. The limitations of the widespread ROS sensors were recently reviewed [28]. Thus, a proper choice of the probes and a thorough understanding of their limitations will reduce artifacts and prevent misinterpretations.
For all these reasons, examining the mechanisms implicated in DOX cardiotoxicity in vitro obliged us to follow as much as possible the conditions of clinical use of the reagent. Here, we investigate the effect of 2 μM DOX, which corresponds to the upper drug level clinically measured in the blood of patients [1, 18]. We used two different exposure durations: a 2-h exposure followed by a 22-h drug-free period that mimics a single chronic dosage in patients, and a 24-h exposure to promote the drug accumulation reflecting either a cumulative lower dose or a supra-clinical acute one. We selected the H9c2 cell line as in vitro cardiac cell model. However, to stay in compliance with the differentiated state of the cardiomyocytes in vivo, we performed experiments on differentiated H9c2 cells as previously reported [29]. We assessed several functional and morphological features to highlight the cell death mechanisms linked to DOX cardiotoxicity. Moreover, clarifying whether oxidative stress is the primary cause of DOX cardiotoxicity requires reliable methodologies and an utmost caution in the experimental procedures. We therefore used three separate determinations for ROS detection: two fluorescence intensity-based methods using dihydroethidium and amplex red sensors [28], and an approach based on the measurement of the fluorescence lifetime of the 1-pyrenebutyric acid probe [30–33]. We demonstrate that short period of drug incubation mediates apoptosis, while longer period favors a shift to necrosis. We reveal that ROS levels increase only mildly reflecting the mitochondrial perturbation rather than an oxidative stress. The cells thus commit to either apoptosis or necrosis independently of ROS levels. Additionally, while the cardioprotective effect of DXR is usually attributed to iron chelation and ROS scavenging [6, 14, 15], here DXR failed to detoxify ROS although still providing cytoprotection. Our data support the view that oxidative stress is not the primary cause of DOX-induced cardiotoxicity in vitro.
Materials and methods
Cell culture and treatment
The H9c2 rat embryonic cardiac cell line (ATCC) was cultivated in DMEM (Flow) supplemented with 10 % heat-inactivated fetal bovine serum (FBS; Gibco), 1 % penicillin and streptomycin (Cambrex Bio Science), and 2 mM l-glutamine (ICN Biomedicals) in 75 cm2 culture flasks at 37 °C, 5 % CO2. Cells were seeded every 3–4 days when they reached 70 % confluency. As the adult heart tissue contains mostly cardiac cells that lost their proliferative activity, i.e., differentiated cardiomyocytes, we induced the cell differentiation to be in line with the in vivo conditions. Therefore, cells were deprived of serum by diminishing the FBS to 1 % [20] to arrest their proliferation prior to the experiments to be performed in accordance with [29]. 24 h later, cells were treated with doxorubicin (DOX; Sigma-Aldrich) up to 24 h prior analyses. DOX effects were monitored from a dose range of 20 nM to 2 μM. Moreover, effects of constant exposure to 2 μM DOX (i.e., 24 h exposure) were compared with a shorter exposure, i.e., 2-h exposure followed by 22 h in drug-free culture medium. Effects were also compared with cells treated with dexrazoxane (DXR; ICRF-187; Novartis). A DXR dose of 20 μM was administrated, which corresponds to the DXR/DOX ratio 10:1 reported to be optimal by clinical investigations [34]. Untreated differentiated cells were pre-incubated with DXR for 30 min prior to DOX treatment according to previous studies [34]. Analyses were then performed 24 h later. Accordingly, this time range was appropriate for the metal ion-chelating forms of DXR to be formed as maximum levels of DXR metabolites were detected within a few hours following the administration of the parent molecule in clinical settings [35].
Cell counting and trypan blue exclusion assay
Cells were seeded in 25 cm2 culture flasks and treated as mentioned above. Floating (i.e., non-adherent) cells were counted using a Beckman Coulter Z2 (Beckman Coulter) to assess the loss in the cell anchorage. Then, adherent cells were harvested with trypsin/EDTA (Invitrogen) and coulter counted separately. The growth rate was calculated as previously reported [30]. The plasma membrane integrity was monitored by trypan blue exclusion assay (Sigma-Aldrich). Adherent and detached cells were separated and stained with trypan blue (0.5 %) in HBSS (Cambrex Bio Science). The proportion of blue-stained cells was evaluated using a Thoma cell counting chamber.
Imaging and multiparametric analysis
Cells that committed to irreversible step of apoptosis were quantified by assessing the mitochondrial disruption as previously reported [33]. Briefly, cells were simultaneously stained 2 h with 10 μM Hoechst 33342 (HO342) and 5 min with 10 μM nile red (<1 % ethanol) to obtain the nuclear and cell boundaries, respectively, and 1 h with 10 μM rhodamine 123 (Rh-123; all Sigma-Aldrich) for labeling the mitochondria. Fluorescent images were acquired using multi-wavelength videomicrofluorometry (widefield), and a semi-automated image processing method was applied for acquisition of both total fluorescence intensity of Rh-123 (tFI-Rh-123) and cell volume (V cell). Apoptotic cells were discriminated according to the drop of their mitochondrial potential that is traduced by the collapse of tFI-Rh-123/V cell ratio values (i.e., <0.1 a.u.) [33, 36].
Necrotic cells were identified by their nuclear swelling and loss in DNA content [37, 38]. Following the image processing method, the cells in the late phase of apoptosis (i.e., tFI-Rh-123/V cell < 0.1 a.u.) were excluded and the data from the remaining cell pool were subjected to a multiparametric analysis (XLSTAT-Pro software; Addinsoft) for classifying the cells into groups reflecting the cell cycle phases: G0/G1, S, G2+M, and polyploid cells (>4 N) [33]. The groups were distributed according to the cell volume, and the fluorescence intensity of HO342 (FI-HO342), which reports the nuclear volume and the DNA content. Necrotic cells were classified into a supplemental sub-group that encompassed cells with larger nuclei (nuclear swelling) and weaker DNA content (DNA degradation) than untreated cells in G0/G1 group. Conversely, the cells representing the earlier step of apoptosis were classified into a distinct sub-group showing weak DNA content, nuclear condensation, and a mitochondrial hyperpolarization [39–41].
Spectrofluorimetry
Levels of radical forms of ROS were monitored according to the oxidation of dihydroethidium (DHE; Invitrogen). Intracellularly, DHE is oxidized by superoxide anion to 2-hydroxyethidium and/or by other radical species to ethidium, which in turn intercalate into nuclear DNA and exhibit similar fluorescence characteristics [28]. Although DHE is mostly used for monitoring superoxide anion levels, here we used the fluorescent assay as an indicator for changes in overall ROS levels. Therefore, the specific detection of 2-hydroxyethidium (i.e., extraction and HPLC analysis [28, 42]) was not required, and we measured the fluorescence signal output without distinguishing between the two fluorescent products (referred hereafter as oxidized DHE). Stock solution of DHE was prepared prior to use in dimethyl sulfoxide (DMSO; Sigma-Aldrich) into degassed atmosphere to avoid contaminating oxidation. Detached and adherent cells were collected separately 24 h after treatment. Cells were suspended in 1 ml PBS (Gibco) at the density of 5 × 105 cells/ml, loaded with 20 μM DHE (<0.2 % DMSO) in the dark at 37 °C for 30 min, and then washed twice prior to analysis [33]. As the maximum fluorescence emissions of ethidium and 2-hydroxyethidium are, respectively, 587 and 568 nm [42], fluorescence spectra of oxidized DHE were recorded within the emission range of 530–700 nm after excitation of the cells at 518 nm using a SAFAS flx spectrofluorimeter (SAFAS). To exclude the background, i.e., intrinsic fluorescence of the cells, and the spectral crosstalk of DOX that fluoresces above 530 nm, the fluorescence spectra of non-stained cells were acquired from both untreated- and DOX-treated populations, respectively. The contributions of each fluorescent component were separated from the complex fluorescent spectrum recorded in stained DOX-treated cells, and the fluorescence emitted by oxidized DHE only was quantified.
Hydrogen peroxide (H2O2) levels were assessed using the amplex red (AmR) hydrogen peroxide/peroxidase assay kit (Invitrogen). AmR is a non-fluorescent probe that does not cross the cell plasma membrane and is commonly used for assessing extracellular H2O2 released from the cells. The reaction between AmR and H2O2 is catalyzed by horseradish peroxidase and generates the stable fluorescent product, resofurin [28]. To ensure about detectable concentrations of H2O2 from differentiated H9c2 cells under our experimental conditions, we lysed the cells for H2O2 to be fully released from intracellular compartments. Stock solution and staining with AmR were performed according to the manufacturer’s instructions. Adherent and detached cells were separately suspended in Krebs–Ringer phosphate buffer at the density of 1.7 × 105 cells/ml and lysed for 5 min with 0.25 % Triton-X 100 (Roche Molecular Biochemicals). Then, 100 μl AmR reaction mixture was added to 100 μl lysates and incubated for 10 min, in the dark to avoid photo-oxidation [43]. Cell lysates were excited at 550 nm, and fluorescence emission was recorded between 560 and 600 nm using SAFAS flx spectrofluorimeter. A H2O2 standard curve was used to convert fluorescence intensities into H2O2 equivalents (μmoles released). As positive controls, we used 10 μM DOX or 1 mM H2O2 solution (Sigma-Aldrich).
Time-resolved microfluorimetry
Fluorescence lifetime of the cell permeable probe 1-pyrenebutyric acid (PBA; Acros Organics) was assessed by time-resolved microfluorimetry to quantify intracellular ROS levels as previously reported [30]. Adherent and detached cells were suspended separately in HBSS and loaded with 0.2 μM PBA (<0.5 % ethanol) for 5 min at 37 °C. After three washing steps, cells were suspended in 300 μl HBSS and placed in Sykes-Moore tissue-culture chambers for analysis. A pulsed laser source excited each individual cell at 337 nm, and a 404-nm filter was applied to the emission path. The fluorescent emission decay curves were recorded and resolved to obtain time constants (i.e., lifetimes) of PBA correlated to ROS concentrations as previously described [30]. ROS levels were expressed as fold change relative to untreated cells (control) for which a value of 1 arbitrary unit (a.u.) was assigned [30]. As negative controls, either NADPH oxidase (NOX) was inhibited with 10 μM diphenyliodonium (DPI; Sigma-Aldrich) (<0.2 % DMSO) [19] or the mitochondrial activity was blocked by means of fixation with Baker solution (10 % paraformaldehyde in 1 % aqueous calcium chloride) [30]. 10 μM DOX and 1 mM H2O2 bolus were used as positive controls.
Statistical analysis
Statistical analyses were performed using two-tailed unpaired Student’s t test. *p ≤ 0.05, significantly different from the control (untreated cells). A two-sample Kolmogorov–Smirnov test was used to determine whether the distribution of ROS production values was different from the control. Data are presented as mean ± SD (standard deviations) and averaged from at least three independent experiments.
Results
Short exposure of H9c2 cells with 2 μM DOX induces apoptosis, while constant exposure favors necrosis
We investigated the mechanisms implicated in DOX-induced cardiotoxicity in the rat embryonic cardiac cell line H9c2. To stay in conformity with the in vivo conditions (i.e., differentiated cardiomyocytes), we carried out all our experiments in differentiated H9c2 cells. First, proliferating cells were seeded for 24 h in complete culture medium (10 % FBS). Then, we induced the differentiation by depriving the cells of serum (1 % FBS) to block their growth [20]. 24 h after the beginning of the serum starvation, we treated the cells with 2 μM DOX. Moreover, the arrest of the cell proliferation by serum starvation aims to exclude the interference of the cytostatic effect of DOX in the interpretation of its own cytotoxic mechanisms resulting from the different treatment conditions. Cells were exposed with DOX for a short period (i.e., 2 h with DOX followed by 22 h in drug-free 1 % FBS medium) or longer period (i.e., 24 h with the drug) and then monitored for the cytotoxic effect. We first assessed the DOX effect on the cell growth by coulter counting (Fig. 1a). As expected, reducing the FBS amount to 1 % inhibited the growth rate of untreated cells (0.4 a.u. vs. 1.8 a.u. for cells in 10 % FBS). The slight increase we observed may result from cells that entered into the cell cycle prior to the serum starvation and subsequently underwent the division. Negative values were obtained for growth rates in DOX-treated cells. A significant decrease was even observed for 2-h exposure as DOX cytotoxicity led to the formation of cellular debris that could not be detected by coulter counter.

Cytotoxic effects of DOX on differentiated H9c2 cells 24 h after drug exposure for 2 or 24 h. a Growth rate of cells cultivated for 48 h under 10 or 1 % FBS. 24 h after the beginning of serum starvation (i.e., differentiation), cells were exposed for 24 h with 2 μM DOX (DOX 24 h) or exposed for 2 h followed by 22 h incubation under 1 % FBS only (DOX 2 h) (n = 105 cells/ml per condition). *p ≤ 0.05 versus cells at t = 0 h (Student’s t test). b Loss of cell anchorage after treatment with 0.4 µM or c 2 µM DOX (n = 6 × 105 cells per condition). *p ≤ 0.05 versus control (Student’s t test). d Plasma membrane integrity for untreated cells (control), and 2 or 24 h exposures with 2 μM DOX. Proportions of trypan blue-stained cells were calculated in both adherent and detached cell groups and e for the overall populations (n = 200–300 cells per condition) *p ≤ 0.05 versus control (Student’s t test). All counting were performed in duplicate for three independent experiments. All data are mean ± SD
We further examined by microscopy whether DOX led to a loss of cell anchorage that is typically observable for cells undergoing apoptosis [44]. One could suggest that increasing the exposure duration would enhance the drug efficiency in inducing cell detachment. Such an assumption is confirmed when using the sub-clinical DOX dose of 0.4 μM for which the cell detachment increased linearly with the time of exposure (Fig. 1b). However, we found a reversed pattern when using the clinical drug dose of 2 μM: 2-h exposure led to 80 % of detached cells, whereas 24-h exposure induced only 40 % of detached cells (Fig. 1c). We then monitored the loss of plasma membrane integrity using trypan blue exclusion assay. The proportions of blue-stained adherent cells were comparable between untreated and DOX-treated cells (Fig. 1d). Both DOX treatments enhanced similarly the proportion of blue-stained cells in the detached population (Fig. 1d). In the overall population, 2-h exposure led to twice as much blue-stained cells than 24-h exposure (Fig. 1e). However, one should be cautious about the interpretation of the cell mortality yield one day after treatment as the trypan blue method does not discriminate healthy cells from cells with membrane integrity but losing cell functions. Moreover, primary necrosis (from dramatic damages to the cells) cannot be distinguished from secondary necrosis (latest event in apoptosis) [45].
Next, we investigated the mitochondrial membrane potential disruption, a known marker of apoptotic events [36]. We examined the tFI-Rh-123/V cell ratio that reflects the mitochondrial function [33]. DOX treatment increased the mitochondrial energetic state for most of the adherent cells. A mitochondrial hyperpolarization was reported to be a step preceding mitochondrial disruption during apoptosis [41], even though an increased mitochondrial activity was also observed during necrosis [46, 47]. 24-h exposure with DOX led to a significant increase (1.3-fold) in mitochondrial activity compared to untreated cells. The increase was even higher (1.8 fold) for 2 h exposure (Fig. 2a). DOX treatments also led to a collapse in mitochondrial function (tFI-Rh-123/V cell < 0.1 a.u.) for a few amount of adherent cells (Fig. 2a). However, it is unlikely that such a collapse in adherent cells is mediated by DOX as a similar negligible proportion (~5 %) was found in untreated cells (Fig. 2b). In contrast, 2-h exposure with DOX resulted in collapsed mitochondrial function for 70 % of detached cells versus 17 % for the longer exposure (Fig. 2b). Our data indicate that apoptosis hallmarks (mitochondrial disruption, loss of cell anchorage) are enhanced when cells are exposed to DOX for a short period.

Mitochondrial disruption induced by 2 µM DOX treatments. a Mean values of tFI-Rh-123/V cell ratio for 2- and 24-h exposures with DOX obtained in adherent cells and compared with untreated cells (control) and with a small portion of adherent treated cells for which the mitochondrial activity collapsed (ratio <0.1) (n > 103 cells per condition). b Proportions of adherent and detached treated cells with collapsed mitochondrial activity compared to the overall population of untreated cells (n > 103 cells per condition). For all data, bars are SD and *p ≤ 0.05 versus control (Student’s t test) for three independent experiments
As DOX was also widely reported to target the nucleus and impair DNA [48], we examined whether DNA was altered using imaging and multiparametric analysis [33]. Apoptotic cells (i.e., tFI-Rh-123/V cell < 0.1 a.u.) were first separated from the cell population. Then, the cells from the remaining pool were classified according to their DNA content (i.e., FI-HO342), nuclear volume, and size in sub-groups reflecting the cell cycle phases [33]. Of note, although serum starvation induces cell differentiation, the process requires many genetics and morphological changes and takes several days to be fully completed [20]. Upon 48 h of serum starvation, the cells still share typical morphological features of cells cultivated in 10 % FBS as shown in Fig. S1 (Online Resource 1). So that sub-groups resulting from their classification mimic the typical cell cycle phases. The pattern of the cell cycle distribution was comparable between untreated cells and cells exposed for 2 or 24 h to DOX: ~50 % of cells in G0/G1-like group, ~30 % in S-like phase, and less than 20 % in G2+M-like group (Fig. 3a). Therefore, cell detachment does not depend on the cell cycle phase. A similar distribution was found in cells prior to the serum starvation, i.e., 0 h, as shown in Fig. S2 (Online Resource 2), confirming that serum starvation blocked the cell proliferation.

DNA alteration mediated by DOX treatments. a Cell distributions in different phases of the cell cycle. Cells with collapsed mitochondrial function were excluded prior to the classification was performed. (n > 103 cells per condition). b Percentages of DOX-treated cells with DNA content smaller than cells in G0/G1-like group. The proportion of adherent (in white) and detached cells (in gray) within the populations are included. c Mean values of nuclear volumes (n > 103 cells per condition) and d tFI-Rh-123/V cell ratio (n > 103 cells per condition) for the DOX-treated cells with weak DNA content (white histograms) compared with untreated cells in G0/G1-like group (in dark). e Mean values of nuclear volumes calculated from the total populations of untreated (in dark) and treated cells (white histograms) distributed in the cell cycle (n > 103 cells per condition). f Mean values and g microscopic images of the FI-HO342 for untreated and DOX-treated cells. Values were calculated for each cell cycle group (n > 103 cells per condition). Cells were chemically detached prior to image acquisition. For all data, bars are SD and *p ≤ 0.05 versus control (Student’s t test) for three independent experiments
Interestingly, all the cells did not satisfy the criteria for classification and formed distinct sub-groups of cells with DNA content smaller than cells in G0/G1-like group (Fig. 3b). 2-h exposure with DOX did not induce severe DNA damage as only few cells (4 %) were located within the sub-group (Fig. 3b). In contrast, 24-h exposure led to significant DNA damage as more than 30 % of cells exhibited smaller DNA content (Fig. 3b). These data show that increasing the exposure duration to DOX favors DNA damage. We then monitored nuclear volume and mitochondrial activity of the cells in these two sub-groups. Nuclear volume of the cells exposed to DOX for 2 h was smaller than of the cells in G0/G1-like group (Fig. 3c) indicating a nuclear condensation that is a typical apoptotic feature [40]. These cells could not be distinguished from the other apoptotic cells beforehand as they exhibited high values of tFI-Rh-123/V cell ratio (Fig. 3d). These findings indicate that these cells are committed to earlier apoptotic events, with the drop in mitochondrial function happening later [41]. No change in tFI-Rh-123/V cell values was observed in the cells that have been exposed to DOX for 24 h (Fig. 3d). Remarkably, while showing a smaller DNA content, these cells exhibited nuclear volumes higher than cells in G0/G1-like group (Fig. 3c), indicating a nuclear swelling, which is a common feature of cells undergoing necrosis [37]. We further compared the nuclear volume of cells distributed across the cell cycle (Fig. 3e). 2-h exposure with DOX did not alter nuclear volume, whereas it increased significantly in cells permanently exposed to the drug. This finding confirms that longer exposure to DOX favors a nuclear swelling.
Finally, to confirm the implication of DOX in DNA alteration, we quantified the mean FI-HO342 in every phase of the cell cycle. As DOX competes with HO342 for DNA binding sites, the decrease in mFI-HO342 is a good indicator of the extent of DOX binding to DNA and further alteration as previously demonstrated [33]. The mFI-HO342 values correlate with the cell distribution as values for cells in G2+M-like group are 2-fold higher than for cells in G0/G1-like group (Fig. 3f). Overall, 2-h exposure with DOX led to a moderate decrease in mFI-HO342 values (<20 %) whereas a strong reduction (>50 %) was found for 24-h exposure (Fig. 3f). Decreased mFI-HO342 and nuclear swelling for 24-h exposure were confirmed by microscopy (Fig. 3g). These data show that longer than 2-h exposure to DOX is required for DNA to be quenched and deteriorated.
Taken together, the findings demonstrate that 2 μM DOX drives cells into two distinct cell death pathways depending on the exposure duration. 2-h exposure promotes loss of cell anchorage, mitochondrial hyperpolarization followed by collapse of the mitochondrial activity, and nuclear condensation that are typical features of apoptosis [36, 40, 41, 44]. 24-h exposure favors DOX binding to DNA, nuclear swelling, and DNA damage that make necrosis the dominant cell death mechanism in this case [37, 38].
Both 2- and 24-h exposures to 2 μM DOX lead to only a mild increase in ROS level
Many studies support the implication of ROS in the clinical cardiotoxic effect of DOX [9, 11, 12]. We investigated whether ROS production is involved in cell death events mediated by 2 μM DOX using three different methods of ROS detection. We first assessed the changes in the magnitude of the fluorescence intensity spectrum of oxidized DHE in cells treated for 24 h with DOX (Fig. 4). Here we did not aim to examine exclusively the influence of superoxide anion but rather the DOX effect on the overall ROS levels. Therefore, we did not discriminate between the two oxidized forms (i.e., 2-hydroxyethidium and ethidium). To make the ROS quantification reliable, we resolved the complex fluorescence spectra to exclude the strong interference of DOX fluorescence intensity and to measure the fluorescence relying on DHE oxidation only (Fig. 4a–c). Without spectral correction, the DOX fluorescence contribution would lead to overestimation of the amplitude of ROS production. Figure 4d reveals that DOX treatment did not significantly change the ROS levels in adherent cells showed by the mean fluorescence value of oxidized DHE. However, the high standard deviation indicates a fluctuation in ROS level values within the treated population. In contrast, ROS levels decreased in detached cells.

ROS levels assessed by fluorescence intensity-based methods. Cells were loaded with DHE, and then 24 h after treatment with DOX (2 µM), fluorescence spectra were recorded in a detached treated cells and compared to b untreated cells and c adherent treated cells. Experimental fluorescence spectra (thick dark curve); fluorescence spectra of DOX (regular dark curve) and oxidized DHE alone (thin dark curve); differences between experimental and calculated curves (residual curve ×5). d Mean values of both DOX and oxidized DHE intensities were calculated. (n = 5 × 105 cells/ml per condition). e Changes of intracellular H2O2 levels recorded 10 min after AmR loading in cells treated with 2 µM DOX for 24 h. Mean values were compared between lysed detached treated cells, lysed (+TX) and non-lysed (−TX) adherent treated cells, and lysed untreated cells (control). f Further comparison was performed with cells treated for 1 h with 10 μM DOX or 1 mM H2O2. g The duration of AmR incubation with 2 µM DOX-treated cells was extended up to 1 h, and measurements were performed every 10 min. (n = 1.7 × 105 cells/ml per condition). For all data, bars are SD and *p ≤ 0.05 versus control (Student’s t test) for three independent experiments
Additionally, we monitored the changes in H2O2 level using AmR. This non-fluorescent membrane-impermeant probe was initially designed for assessing extracellular H2O2 that is released by viable cells. Although H2O2 is not directly measured within the intact cells, it is admitted that its extracellular level reflects its intracellular accumulation. This fluorescent assay is certainly convenient for assessing H2O2 when detectable levels are commonly secreted by various cell types (e.g., activated leukocytes) or released following plasma membrane injury that is mediated by treatments. When this is not the case, one alternative consists in lysing the cells to artificially release the intracellular H2O2 in its entirety [49]. Cells were lysed prior to AmR loading, then fluorescence intensities were measured each 10 min up to 20 min and values were converted to H2O2 equivalents. We assessed H2O2 level after lysis of untreated cells (Fig. 4e), and we found a weak H2O2 level (~0.05 μM). As control, we monitored the signal resulting from non-lysed cells (-TX) and did not detect any significant H2O2 signal. 24-h treatment with 2 μM DOX led to an increase in H2O2 level up to 0.20 μM in adherent cells (+TX). However, such increased level remains modest when compared to cells treated with the reagents at high cytotoxic doses. Indeed, increases of H2O2 levels up to 0.26 and 0.50 μM resulted from 1-h treatment with 10 μM DOX or 1 mM H2O2 solution, respectively (Fig. 4f). Of note, a discrepancy was observed between the nominal dose of exogenous H2O2 solution added as a bolus (1 mM) and the level detected after cell lysis. Such a difference can be explained by the important elimination of H2O2 from the medium of in vitro cultured cells [50]; the gradient existing between external and cytosolic H2O2 that limits its entrance [51]; and the activation of intracellular sites of H2O2 consumption (e.g., antioxidant defense systems). In any case, the increased H2O2 levels remained below the intracellular steady-state concentrations (~1.0 μM) considered as necessary to induce an oxidative stress [52]. Importantly, the magnitude of the fluorescent signal output needs to be interpreted with caution as a time-dependent amplification of the signal was observed. After 1 h of incubation of AmR, a H2O2 level of 0.5 μM was measured in cells treated for 24 h with 2 μM DOX (Fig. 4g) that may resulted from the photo-reduction of resofurin due to the irradiation performed along the kinetics experiments [43]. This finding suggests that the DOX-mediated ROS production in adherent cells is overestimated and is in reality more modest. In contrast, H2O2 did not accumulate in detached cells: the signal was comparable with lysed untreated cells and non-lysed treated cells, and remained unchanged even despite the time-dependent auto-amplification of the signal (Fig. 4e, g).
Although both DHE and AmR methods revealed an analogous pattern for the ROS levels, we decided to use an additional method to rule out the artifacts inherent to these intensity-based approaches. Accordingly, we examined the fluorescence lifetime decay of PBA as previously reported [30–33]. We first monitored the sensitivity of the method for detecting ROS in H9c2 cells using ROS inhibitors and activators (Fig. 5a). Impeding the cellular activity by cell fixation with Baker solution [30] or inhibiting NOX with DPI [19] resulted in a 2-fold decrease in ROS levels. Conversely, treatments with supra-clinical DOX dose or H2O2 solution led to 1.5- and 2-fold increase, respectively. These findings are consistent with data obtained with AmR (Fig. 4f) and confirm the robustness of the method for ROS detection.

Changes in ROS levels examined by time-resolved microfluorimetry. a ROS levels (fold control) in cells treated with DPI, Baker solution, DOX, or H2O2 bolus (n = 70–140 cells per condition). *p ≤ 0.05 versus control (Student’s t test) for three independent experiments. Data are mean ± SD. b Scatter plots of ROS levels in cells exposed for 2 h or continually with 2 μM DOX (n = 50–100 cells per condition) *p ≤ 0.05: distribution of values significantly different from untreated cells (Kolmogorov–Smirnov test) for three independent experiments
We then assessed the effect of various DOX doses on ROS production. The drug was left in contact with the cells and analyses were done from few hours to several days of treatment (Table 1). Small doses that slow down the cell growth (20–70 nM) did not affect ROS levels. Moreover, no significant change in the mean values of ROS levels was found from doses inhibiting cell growth (0.1–0.9 μM) to doses inducing cell death after several days (1–1.4 μM) or hours (1.7–2 μM) except for the supra-clinical dose of 10 μM DOX. However, the standard deviations increased which reflect a large variation in ROS levels inside the populations. We further monitored the effect of 2 μM DOX on ROS levels in both adherent and detached cells 24 h after the beginning of the treatment (Fig. 5b). Interestingly, while 2-h and continuous exposures to DOX led to distinct cell death processes, a comparable slight increase in ROS levels was found (1.23 vs. 1.12 fold, respectively). While significant, such increased ROS levels appear low when compared to levels reached already after 1 h of treatment with 10 μM DOX or 1 mM H2O2 solution (Fig. 5a). Conversely, ROS levels strongly decreased in detached cells for both DOX treatment conditions (Fig. 5b). Therefore, the three methods used for ROS detection show a consistent pattern of ROS production mediated by 2 μM DOX: a strong reduction of ROS production in detached cells and a mild increase in adherent cells. Even in adherent cells, ROS production is modest compared to cells treated with lethal doses of DOX or H2O2 bolus that are known to cause oxidative stress in cardiac cells [23, 53].
To confirm that ROS does not play a key role in DOX cardiotoxicity, we monitored the effects of DXR pre-treatment. We assumed that DXR would act as antioxidant since it has been reported that its metal ion-chelating forms resulting from its metabolic hydrolysis protect cardiac cells from DOX by favoring ROS scavenging [14, 15]. Prior to DOX treatment, cells were pre-incubated for 30 min with DXR at the DXR/DOX ratio 10:1 [34], and analysis was performed 24 h later. We found that DXR alone did not induce any cytotoxic effect and that the growth rate was comparable with untreated cells. In the presence of DXR, the growth rate was less reduced for 2 h exposure with DOX and even slightly increased for 24-h exposure when compared with cells treated with DOX alone (Fig. 6a). DXR pre-treatment did not alleviate apoptosis mediated by 2-h exposure with DOX as the proportion of cells with collapsed mitochondrial activity (Fig. 6b) or with smaller DNA content and nuclear condensation (Fig. 6c) remained unchanged. In contrast, DXR reduced the necrotic yield. The proportion of cells with smaller DNA content and nuclear swelling due to 24 h exposure with DOX was decreased (Fig. 6c). Such cytoprotection is independent of DOX binding to DNA, as changes in mFI-HO342 (Fig. 6d) were comparable with cells treated with DOX alone (Fig. 3f). Finally, we assessed the effect of DXR on ROS levels (Fig. 6e). DXR alone led to a slight increase in ROS production that corroborates previous findings reporting its weak prooxidant property [54]. Importantly, although DXR attenuated cell death, it failed to modify the ROS production pattern resulting from DOX treatments in both detached and adherent cells (Fig. 6e).

Influences of DXR pre-treatment on DOX effects. Cells were pre-treated with 20 μM DXR and further exposed for 2 or 24 h with 2 μM DOX. a Growth rates (n = 105 cells/ml per condition), b proportions of cells with collapsed mitochondrial activity (n = 500–1000 cells per condition), or c weak DNA content (n = 500–1000 cells per condition), and d mean values of the FI-HO342 for each cell cycle group (n = 500–1000 cells per condition) were calculated 24 h after the beginning of the DOX treatment and compared with cells treated with DOX only, DXR only, and untreated cells (control). All data are mean ± SD. *p ≤ 0.05 (Student’s t test) for three independent experiments. e Box plots representing the changes in ROS levels monitored by time-resolved microfluorimetry. Cell treatments are indicated below the x-axis (n = 50–100 cells per condition). *p ≤ 0.05: distribution of values significantly different from untreated cells (Kolmogorov–Smirnov test) for three independent experiments. ns. no significant difference between two compared conditions
Taken together, our findings show that both short and continuous exposures to 2 μM DOX induce similar changes in ROS levels, while they lead to distinct cytotoxic mechanisms. Our findings also support that ROS do not play a key role in inducing DOX cardiotoxicity in vitro. Instead, the strict correlation of the ROS pattern with the mitochondrial activity implies that changes in ROS levels may just occur due to the mitochondrial perturbations mediated by DOX treatment.
Discussion
Oxidative stress is commonly reported to induce DOX cardiotoxicity [5, 6, 9, 11, 12]. However, its implication as primary cause is still questionable because of uncertainties inherent to the experimental procedures used in most of the reports [8–10, 12, 17, 22–24, 27]. Hence, in this study, we aimed to clarify in vitro the mechanisms of DOX-induced cardiotoxicity in compliance with the clinical settings. Firstly, the clinical relevant dose of 2 μM was selected, as it corresponds to a recommended dose (~45 mg/m2) intravenously administered per treatment cycle [7, 18]. Secondly, we took into consideration the potential influence of the exposure duration of the cells with the drug. Thus, DOX effects were examined 1 day after the beginning of the treatment according to two time periods of cell exposure. We postulated that a short period of exposure (2 h) would recapitulate the mechanisms resulting from a single chronic dosage. In contrast, a longer period (24 h) may favor DOX accumulation and mimic effects of either cumulative lower concentrations or acute high dose that are both likely to transiently enhance the DOX plasma levels of patients.
Importantly, while we used the H9c2 cell line as in vitro cardiac cell model, we assessed DOX effects in differentiated H9c2 cells to remain as much as possible in conformity with the in vivo context since the adult heart tissue mostly includes differentiated cardiomyocytes, i.e., cardiac cells without proliferative activity. Indeed, concerns have been raised about the appropriateness of H9c2 cells for mimicking cardiac cells and investigating cardiac toxicity as they are commonly used under their proliferation state [8–10, 12, 16–19]. Accordingly, the reliability of the results obtained in the proliferating cells when compared to primary cardiomyocytes was challenged since dose–responses were found being altered by the cell differentiation state [21]. While inducing the H9c2 differentiation is a pertinent approach to elude the above-mentioned limitation, one should mention that it may also add constraints in the experimental procedure. On the one hand, it has been suggested that serum deprivation may induce apoptosis in the H9c2 cell culture independently of the DOX treatment and therefore may lead to erroneous interpretation [16]. However, we did not find any change in the baseline viability after serum depletion under our experimental conditions. On the other hand, long-term differentiation of H9c2 myoblasts does not lead only to the formation of cardiac muscles but also to cells exhibiting skeletal muscle phenotype [20]. Moreover, while the administration of all-trans-Retinoic acid during the differentiation forces the cells to acquire a cardiac muscle phenotype [20], the cell population still remains heterogeneous that makes fastidious any quantitative analyses as each phenotype requires to be discriminated. To overcome this issue, we opted for a short-term differentiation [29]. Cells were differentiated up to two days prior to analyses to be performed. Thus, while the differentiation process was not fully completed [20], we believe that our approach makes suitable the use of H9c2 cells as in vitro cardiac cell model for DOX cardiotoxicity study as it provides the following advantages: (i) arrest of the proliferative activity in accordance with the in vivo context; (ii) removal of the risk of alteration of the dose–response that is dependent on the differentiation state; and (iii) exclusion of the potential interference of the drug cytostatic effect in the interpretation of the cytotoxic effects.
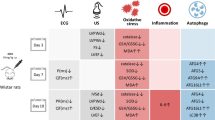
Morphological and functional features were examined to elucidate, which cell death pathway is implicated in DOX cardiotoxicity. We revealed that the clinical DOX dose of 2 μM drives cells into two distinct cell death pathways depending on the exposure duration with DOX. The mechanisms are outlined in Fig. 7. We observed that apoptosis is the predominant mechanism in cells exposed to DOX for a short period of time. Although DNA fragmentation is a marker of apoptosis, it occurs in the later phase [55]. Therefore, the absence of considerable nuclear damage in spite of DOX binding to DNA is not surprising. Nonetheless, we found that almost all cells lost their adherence capacity [44]. Moreover, mitochondrial activity was disrupted in two steps: a hyperpolarization occurred as early event [41] prior to the loss of adherence ability, and preceded a collapse of the mitochondrial function [36] found in detached cells. Finally, cells underwent a nuclear condensation [40].

Schematic representation of effects of DOX on cells depending on the exposure time of the drug and leading to cardiotoxicity
Despite DOX being commonly linked to induction of apoptosis [5, 8–10], increasing its exposure duration favored the cell commitment to necrosis. One could argue that fewer cells lost their membrane integrity, while it is a typical late feature for necrotic cells [56]. However, one should also point out that the trypan blue assay cannot discriminate between primary and secondary necrosis, or healthy cells and cells that kept their membrane integrity, but that are functionally impaired [45]. Therefore, it is likely that the cells from the population undergoing primary necrosis did not broadly loose their membrane integrity one day after the beginning of the treatment. Nevertheless, several other characteristic features sustain that necrosis was predominant. Cells presented substantial DNA impairment that is a typical early event in necrosis [38]. Such DNA damage resulted from a larger DOX binding to DNA as the drug was left in contact with the cells for a longer period and highly accumulated. In addition, a major part of the cell population presented nuclear swelling [37]. Furthermore, we also observed a mitochondrial perturbation that is likely to happen during necrosis although a strong disruption is frequently assimilated with apoptosis. It is widely recognized that when the mitochondrial activity is rapidly perturbed, the cellular energy level becomes promptly deficient and drives the cells into necrosis. Conversely, when the disruption is slower, the apoptogenic proteases can be activated and the cellular energetic state has time to culminate to high level required for driving the apoptotic processes [46, 47]. These observations are in line with our findings that a highly increased mitochondrial activity preceded a collapse of the mitochondrial function in apoptotic cells [41]. In contrast, the increase in mitochondrial activity was modest in necrotic cells as they did not require massive energy source for the death events to be processed [47]. Finally, the majority of the cells did not lose their adherence capacity, as only few were found detached. Necrotic and apoptotic processes can occur simultaneously and overlap within cells [47, 57], and their inverse relationship was known for many years [58]. Accordingly, it is likely that commitment to apoptosis still occurred albeit less frequently and that necrosis prevailed. The shift in the balance between apoptosis and necrosis is influenced by the onset and magnitude of mitochondrial disruption and DNA damage, which depend on DOX accumulation within the cells. The stronger and faster the mitochondrial function and nuclear content are altered, the most probable the cells commit to necrosis. As the time period of cell incubation with DOX influences the drug concentration accumulated within the cells, our findings corroborate previous reports that low DOX doses induce apoptosis whereas higher doses favor necrosis [18].
The quinone moiety of DOX gives it the ability to generate ROS through a series of reactions known as redox cycling [1, 5], which lead to an oxidative stress that is believed to be the cause for the cardiotoxic effect of the drug [5, 6, 9, 11, 12]. As DOX cardiotoxicity is dose dependent [1, 18], one could expect that prolonging the incubation time with DOX, and thus, the drug accumulation within cells would result in enhanced ROS production. Nevertheless, our findings challenge the current insights about the prominence of oxidative stress in DOX cardiotoxicity. Our first discrepancy is based on the fact that short and long exposure durations with DOX-induced comparable changes in ROS levels, but distinct cell death mechanisms. This finding supports the view that the ROS generation mediated by clinical dose of DOX does not play a primary role in the cell decision to commit either to apoptosis or necrosis. Furthermore, our second discrepancy with the current insights relies on the magnitude of ROS production resulting from DOX treatment. The increase in ROS levels is too small to be assigned to an oxidative stress, but rather it is just reflecting the perturbation in the mitochondrial electron transport chain. Our observation can be explained by the methodologies used for ROS detection. Commonly applied methods are sources of many artifacts connected to the lack in sensitivity of the fluorescent probes for detection of ROS [25, 26, 28]. The epitome of inherent limitations of such methodologies may lie in the use of fluorescein derivatives. These probes have been widely used in studies linking oxidative stress to DOX cardiotoxicity [8–10, 12, 22, 27] although they are currently considered as unsuitable for ROS detection [28]. Accordingly, the involvement of ROS in cardiotoxicity is still ambiguous, and their detection requires caution to avoid misinterpretations. Here, we used AmR that was proposed as an alternative to fluorescein derivatives [28] and found an increase in ROS levels too weak to be considered as a characteristic feature of an oxidative stress. Nevertheless, interpretations from this method alone were excluded as we also monitored a time-dependent auto-amplification of the fluorescent signal reported to be connected with the photo-reduction of the probe [43]. The results acquired with another ROS sensor, DHE [28], however, confirmed the absence of ROS overproduction and challenge the reports that used this probe to detect an oxidative stress [9, 27]. Here again, a cautious use of the probe was required as an inappropriate correction of the signal, i.e., no correction for the DOX fluorescence contribution would have led to misinterpretation. In parallel, we assessed the fluorescence lifetime of PBA as this method does not have common artifacts inherent to the intensity-based approaches [30–33]. We confirmed a similar ROS pattern, i.e., a slight ROS increase far from representative of an oxidative stress.
Additionally, our data regarding the DXR pre-treatment confirmed that DOX cardiac toxicity is not primarily triggered by an oxidative stress. DXR is currently used for cardioprotection in patients clinically treated with anthracyclines (see for reviews [1, 6, 13]). Its cardioprotective effect is usually attributed to its rings-opened metabolites resulting from enzymatic hydrolysis, which exhibit iron chelation properties [6]. We used here DXR for its presumed antioxidant activity as it is believed that DXR metabolites attenuate the iron-dependent oxidative stress mediated by anthracyclines. Indeed, DXR has been reported to scavenge ROS in in vitro solution systems [14, 15] and to improve in vivo the plasma antioxidant activity of patients treated with epirubicin [14]. However, other findings challenge the cardioprotective role of DXR metabolites as metal ion chelator in vitro. It has been reported that DXR reduced apoptosis induced by 1.5 µM DOX, while its metal ion-chelating forms failed to protect cardiomyocytes although exhibiting a more efficient chelation activity [59]. In addition, unlike other iron chelators, DXR failed to protect H9c2 cells from oxidative stress and injury mediated by treatment with catecholamines [60]. These observations argue for the involvement of other mechanisms than ion chelation in the cardioprotective effect of DXR. Our data support that DXR does not protect in vitro cultured cells from DOX treatment by acting as antioxidant. Indeed, in spite of its optimal administration 30 min prior to DOX treatment at the DXR/DOX ratio 10:1 [34], DXR failed to detoxify the mild increase in ROS levels mediated by both short and long period of incubation of differentiated H9c2 cells with 2 µM DOX, although it still exerted a cytoprotective effect. Further, our findings contrast with studies reporting that DXR prevents DOX-induced mitochondrial damages [22] and attenuates apoptosis mediated by DOX in cardiomyocytes [59]. Indeed, we did not detect any attenuation of DOX-mediated mitochondrial membrane potential disruption, and the apoptotic cell yield remained unchanged. Remarkably, we found that DXR rather protected the cells from necrotic cell death by relieving DOX-mediated DNA damage, indicating that DXR is involved in a mechanism occurring within the nuclear compartment.
Although an iron-catalyzed oxidative stress is the mechanism the most frequently linked to DOX-mediated cardiotoxicity (see for reviews [5, 6]), many other mechanisms have also been suggested including dysregulation of intracellular calcium homeostasis [5]; mitochondrial DNA degradation [61]; alteration of cardiac muscle gene expression [62]; perturbation of cytoskeletal and extracellular matrix proteins [63]; interference with cell survival signaling, e.g., neuregulin [64]. In connection with the nuclear compartment, DOX is widely recognized to act as topoisomerase 2 (TOP2) poison [2]. Interestingly, DXR has also been described to interact with TOP2 [65], and raising evidences support that DXR cardioprotection may rather be linked to its interference with DOX on their shared nuclear molecular target TOP2. The works carried out by Lyu et al. suggested that proteasomal degradation of TOP2β induced by DXR abolishes DOX-mediated DNA damage in H9c2 cardiomyoblasts [66]. Next, Martin et al. compared the cardioprotective effect of DXR with ICRF-161, a DXR derivative that is not capable of inhibiting TOP2. While both compounds exhibited comparable efficiency in iron-chelating activity in primary cardiomyocytes, ICRF-161 failed to protect the myocardium of a rat model of chronic anthracycline-induced cardiomyopathy unlike DXR [67]. Recently, Deng et al. confirmed in differentiated H9c2 cells the inefficiency of ICRF-161 in reducing DNA impairment from DOX treatment. Their results support that DXR antagonizes DOX-mediated double strand breaks by depleting TOP2β [68]. Although DXR mechanisms have not been thoroughly investigated here, our findings support that it is more likely that DXR cardioprotective effect on differentiated H9c2 cells is linked to its interference with DOX on TOP2-mediated DNA cleavage rather than through a metal ion-chelating activity.
In summary, our study aimed to elucidate the implication of oxidative stress in DOX-induced cardiac toxicity in vitro. Our investigations were conducted by following as much as possible the clinical settings. We assessed clinical relevant doses of DOX and took into consideration the exposure duration of the drug that influences in vitro the cell uptake and the final drug concentration accumulated within the cells. Moreover, we used H9c2 cells under differentiation state to stay in compliance with the absence of proliferative activity of the cardiomyocytes in vivo. Accordingly, assuming that short exposure of in vitro cultured cells with DOX is comparable with a single dose in the clinical setting, our findings support that apoptosis is predominantly induced by this type of treatment. Moreover, if one admits that longer drug administration promotes intracellular drug accumulation, necrosis may also occur and lead to cumulative dose-dependent cardiomyopathy under clinical conditions. Next, our data support that oxidative stress does not act as primary mechanism in DOX-induced cardiotoxicity and corroborate previous findings [69]. It has been reported that DOX induces a persistent stimulation in ROS production a long time after the injection of the drug [70]. In this connection, it could be that the slightly increased ROS production we showed may be enhanced with cumulative DOX administrations and may eventually favor oxidative damages and further sustain the irreversible cardiomyopathy observed in clinical settings. In addition, our data underline the importance of using reliable methodologies for in vitro studies of ROS generation to avoid any artifacts during the data acquisition. Finally, here we support that the thorough cautions we applied on the experimental procedure and conditions made the differentiated H9c2 cells as a fair alternative to primary cardiomyocytes cultivated in vitro and therefore suitable for their use as in vitro cardiac cell model. However, further in vivo studies on mammalian animals are required to unequivocally confirm the above-mentioned mechanisms described in in vitro cultured cells.
References
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L (2010) Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56:185–229
Pommier Y, Leo E, Zhang H, Marchand C (2010) DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 17:421–433
Cutts SM, Nudelman A, Rephaeli A, Phillips DR (2005) The power and potential of doxorubicin-DNA adducts. IUBMB Life 57:73–81
Jung K, Reszka R (2001) Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev 49:87–105
Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns H, Moens AL (2012) Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 52:1213–1225
Štěrba M, Popelová O, Vávrová A, Jirkovský E, Kovaříková P, Geršl V, Šimůnek T (2013) Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 18:899–929
Alberts DS, Hess LM, Von Hoff DD, Dorr RT (2009) Pharmacology and therapeutics in gynecologic cancer. In: Barakat RR, Markman M, Randall ME (eds) Principles and practice of gynecologic oncology, 5th edn. Lippincott Williams and Wilkins, Philadelphia, pp 409–462
Kluza J, Marchetti P, Gallego MA, Lancel S, Fournier C, Loyens A, Beauvillain JC, Bailly C (2004) Mitochondrial proliferation during apoptosis induced by anticancer agents: effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene 23:7018–7030
Spallarossa P, Garibaldi S, Altieri P, Fabbi P, Manca V, Nasti S, Rossettin P, Ghigliotti G, Ballestrero A, Patrone F, Barsotti A, Brunelli C (2004) Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol 37:837–846
Tan X, Wang DB, Lu X, Wei H, Zhu R, Zhu SS, Jiang H, Yang ZJ (2010) Doxorubicin induces apoptosis in H9c2 cardiomyocytes: role of overexpressed eukaryotic translation initiation factor 5A. Biol Pharm Bull 33:1666–1672
Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U (2006) New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. J Mol Cell Cardiol 41:389–405
Ma J, Wang Y, Zheng D, Wei M, Xu H, Peng T (2013) Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc Res 97:77–87
Lipshultz SE, Sambatakos P, Maguire M, Karnik R, Ross SW, Franco VI, Miller TL (2014) Cardiotoxicity and cardioprotection in childhood cancer. Acta Haemathol 132:391–399
Galetta F, Franzoni F, Cervetti G, Regoli F, Fallahi P, Tocchini L, Carpi A, Antonelli A, Petrini M, Santoro G (2010) In vitro and in vivo study on the antioxidant activity of dexrazoxane. Biomed Pharmacother 64:259–263
Junjing Z, Yan Z, Baolu Z (2010) Scavenging effects of dexrazoxane on free radicals. J Clin Biochem Nutr 47:238–245
Green PS, Leeuwenburgh C (2002) Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. BBA Mol Basis Dis 1588:94–101
Choi EH, Chang HJ, Cho JY, Chun HS (2007) Cytoprotective effect of anthocyanins against doxorubicin-induced toxicity in H9c2 cardiomyocytes in relation to their antioxidant activities. Food Chem Toxicol 45:1873–1881
Bernuzzi F, Recalcati S, Alberghini A, Cairo G (2009) Reactive oxygen species-independent apoptosis in doxorubicin-treated H9c2 cardiomyocytes: role for heme oxygenase-1 down-modulation. Chem-Biol Interact 177:12–20
Gilleron M, Marechal X, Montaigne D, Franczak J, Neviere R, Lancel S (2009) NADPH oxidases participate to doxorubicin-induced cardiac myocyte apoptosis. Biochem Biophys Res Commun 388:727–731
Pereira SL, Ramalho-Santos J, Branco AF, Sardão VM, Oliveira PJ, Carvalho RA (2011) Metabolic remodeling during H9c2 myoblast differentiation: relevance for in vitro toxicity studies. Cardiovasc Toxicol 11:180–190
Branco AF, Sampaio SF, Moreira AC, Holy J, Wallace KB, Baldeiras I, Oliveira PJ, Sardão VA (2012) Differentiation-dependent doxorubicin toxicity on H9c2 cardiomyoblasts. Cardiovasc Toxicol 12:326–340
Hasinoff BB, Schnabl KL, Marusak RA, Patel D, Huebner E (2003) Dexrazoxane (ICRF-187) protects cardiac myocytes against doxorubicin by preventing damage to mitochondria. Cardiovasc Toxicol 3:89–99
Corna G, Santambrogio P, Minotti G, Cairo G (2004) Doxorubicin paradoxically protects cardiomyocytes against iron-mediated toxicity: role of reactive oxygen species and ferritin. J Biol Chem 279:13738–13745
Ludke A, Sharma AK, Bagchi AK, Singal PK (2012) Subcellular basis of vitamin C protection against doxorubicin-induced changes in rat cardiomyocytes. Mol Cell Biochem 360:215–224
Gomes A, Fernandes E, Lima JLFC (2005) Fluorescence probes used for detection of reactive oxygen species. J Biochem Biophys Methods 65:45–80
Bartosz G (2006) Use of spectroscopic probes for detection of reactive oxygen species. Clin Chim Acta 368:53–76
Gao J, Yang G, Pi R, Li R, Wang P, Zhang H, Le K, Chen S, Liu P (2008) Tanshinone IIA protects neonatal rat cardiomyocytes from adriamycin-induced apoptosis. Transl Res 151:79–87
Kalyanaraman B, Darley-Usmar V, Davies KJA, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, Ischiropoulos H (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 52:1–6
Shan PR, Xu WW, Huang ZQ, Pu J, Huang WJ (2014) Protective role of retinoid X receptor in H9c2 cardiomyocytes from hypoxia/reoxygenation injury in rats. World J Emerg Med 5:122–127
Rharass T, Vigo J, Salmon JM, Ribou AC (2006) Variation of 1-pyrenebutyric acid fluorescence lifetime in single living cells treated with molecules increasing or decreasing reactive oxygen species levels. Anal Biochem 357:1–8
Moné Y, Ribou AC, Cosseau C, Duval D, Théron A, Mitta G, Gourbal B (2011) An example of molecular co-evolution: reactive oxygen species (ROS) and ROS scavenger levels in Schistosoma mansoni/Biomphalaria glabrata interactions. Int J Parasitol 41:721–730
Ribou AC, Reinhardt K (2012) Reduced metabolic rate and oxygen radicals production in stored insect sperm. Proc R Soc B Biol Sci 279:2196–2203
Savatier J, Rharass T, Canal C, Gbankoto A, Vigo J, Salmon JM, Ribou AC (2012) Adriamycin dose and time effects on cell cycle, cell death, and reactive oxygen species generation in leukaemia cells. Leukemia Res 36:791–798
Imondi AR (1998) Preclinical models of cardiac protection and testing for effects of dexrazoxane on doxorubicin antitumor effects. Semin Oncol 25:22–30
Schroeder PE, Jensen PB, Sehested M, Hofland KF, Langer SW, Hasinoff BB (2003) Metabolism of dexrazoxane (ICRF-187) used as a rescue agent in cancer patients treated with high-dose etoposide. Cancer Chemother Pharmacol 52:167–174
Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssière JL, Petit PX, Kroemer G (1995) Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med 181:1661–1672
Zhivotosky B, Orrenius S (2001) Assessment of apoptosis and necrosis by DNA fragmentation and morphological criteria. Curr Protoc Cell Biol 12:18.3:18.3.1–18.3.23
Napirei M, Wulf S, Mannherz HG (2004) Chromatin breakdown during necrosis by serum Dnase1 and the plasminogen system. Arthritis Rheum US 50:1873–1883
Sakahira H, Enari M, Nagata S (1998) Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391:96–99
Toné S, Sugimoto K, Tanda K, Suda T, Uehira K, Kanouchi H, Samejima K, Minatogawa Y, Earnshaw WC (2007) Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp Cell Res 313:3635–3644
Perl A, Gergely PJ, Nagy G, Koncz A, Banki K (2004) Mitochondrial hyperpolarization: a checkpoint of T-cell life, death and autoimmunity. Trends Immunol 25:360–367
Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vásquez-Vivar J, Kalyanaraman B (2005) Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA 102:5727–5732
Zhao B, Summers FA, Mason RP (2012) Photooxidation of amplex red to resorufin: implications of exposing the amplex red assay to light. Free Radic Biol Med 53:1080–1087
Grossmann J, Walther K, Artinger M, Kiessling S, Schölmerich J (2001) Apoptotic signaling during initiation of detachment-induced apoptosis (“anoikis”) of primary human intestinal epithelial cells. Cell Growth Differ 12:147–155
McCarthy NJ, Evan GI (1997) Methods for detecting and quantifying apoptosis. In: de Pablo F, Ferrús A, Stern CD (eds) Cellular and molecular procedures in developmental biology, vol 36., Current topics in developmental biologyAcademic Press, San Diego, pp 259–278
Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G (1997) The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15:1573–1581
Lemasters JJ (1999) Mechanisms of hepatic toxicity. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol 276:G1–G6
Yang F, Teves SS, Kemp CJ, Henikoff S (2014) Doxorubicin, DNA torsion, and chromatin dynamics. BBA Rev Cancer 1845:84–89
Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS (2007) The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177:1029–1036
Gülden M, Jess A, Kammann J, Maser E, Seibert H (2010) Cytotoxic potency of H2O2 in cell cultures: impact of cell concentration and exposure time. Free Radic Biol Med 49:1298–1305
Antunes F, Cadenas E (2000) Estimation of H2O2 gradients across biomembranes. FEBS Lett 475:121–126
Antunes F, Cadenas E (2001) Cellular titration of apoptosis with steady state concentrations of H2O2: submicromolar levels of H2O2 induce apoptosis through fenton chemistry independent of the cellular thiol state. Free Radic Biol Med 30:1008–1018
Horwitz LD, Leff JA (1995) Catalase and hydrogen peroxide cytotoxicity in cultured cardiac myocytes. J Mol Cell Cardiol 27:909–915
Kaiserová H, den Hartog GJM, Šimůnek T, Schröterová L, Kvasničková E, Bast A (2006) Iron is not involved in oxidative stress-mediated cytotoxicity of doxorubicin and bleomycin. Br J Pharmacol 149:920–930
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Majno G, Joris I (1995) Apoptosis, oncosis and necrosis. An overview of cell death. Am J Pathol 146:3–15
Zeiss CJ (2003) The apoptosis-necrosis continuum: insights from genetically altered mice. Vet Pathol 40:481–495
Arends MJ, McGregor AH, Wyllie AH (1994) Apoptosis is inversely related to necrosis and determines net growth in tumors bearing constitutively expressed myc, ras, and HPV oncogenes. Am J Pathol 144:1045–1057
Hasinoff BB, Schroeder PE, Patel D (2003) The metabolites of the cardioprotective drug dexrazoxane do not protect myocytes from doxorubicin-induced cytotoxicity. Mol Pharmacol 64:670–678
Hašková P, Koubková L, Vávrová A, Macková E, Hrušková K, Kovaříková P, Vávrová K, Šimůnek T (2011) Comparison of various iron chelators used in clinical practice as protecting agents against catecholamine-induced oxidative injury and cardiotoxicity. Toxicology 289:122–131
Lebrecht D, Kokkori A, Ketelsen UP, Setzer B, Walker UA (2005) Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J Pathol 207:436–444
Ito H, Miller SC, Billingham ME, Akimoto H, Torti SV, Wade R, Gahlmann R, Lyons G, Kedes L, Torti FM (1990) Doxorubicin selectively inhibits muscle gene expression in cardiac muscle cells in vivo and in vitro. Proc Natl Acad Sci USA 87:4275–4279
Dudnakova TV, Lakomkin VL, Tsyplenkova VG, Shekhonin BV, Shirinsky VP, Kapelko VI (2003) Alterations in myocardial cytoskeletal and regulatory protein expression following a single doxorubicin injection. J Cardiovasc Pharmacol 41:788–794
Horie T, Ono K, Nishi H, Nagao K, Kinoshita M, Watanabe S, Kuwabara Y, Nakashima Y, Takanabe-Mori R, Nishi E, Hasegawa K, Kita T, Kimura T (2010) Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc Res 87:656–664
Classen S, Olland S, Berger JM (2003) Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc Natl Acad Sci U S A 100:10629–10634
Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF (2007) Topoisomerase IIβ-mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 67:8839–8846
Martin E, Thougaard AV, Grauslund M, Jensen PB, Bjorkling F, Hasinoff BB, Tjørnelund J, Sehested M, Jensen LH (2009) Evaluation of the topoisomerase II-inactive bisdioxopiperazine ICRF-161 as a protectant against doxorubicin-induced cardiomyopathy. Toxicology 255:72–79
Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Gödtel-Armbrust U, Wojnowski L (2014) Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer 14:842
Lai R, Long Y, Li Q, Zhang X, Rong T (2011) Oxidative stress markers may not be early markers of doxorubicin-induced cardiotoxicity in rabbits. Exp Ther Med 2:947–950
Zhou S, Palmeira CM, Wallace KB (2001) Doxorubicin-induced persistent oxidative stress to cardiac myocytes. Toxicol Lett 121:151–157
Acknowledgments
This paper is dedicated to the memory of our friend and colleague Jean Vigo who passed away in July 2015. His enthusiasm and tireless activity on improving instruments for fluorescence lifetime detection will remain a model. We would like to thank Christoph Grunau for proof reading the article. This work was supported by funds from the French “Ligue Nationale Contre le Cancer” (Comités des Pyrénées-Orientales et du Gard) to T.R., A.G., C.C., A.B., J.V. and A.C.R., the ERASMUS program to G.K., and by Helmholtz Young Investigator Program to T.R. and D.P.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rharass, T., Gbankoto, A., Canal, C. et al. Oxidative stress does not play a primary role in the toxicity induced with clinical doses of doxorubicin in myocardial H9c2 cells. Mol Cell Biochem 413, 199–215 (2016). https://doi.org/10.1007/s11010-016-2653-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2653-x