
Abstract
Human adipose-derived stem cells (hASCs) become an appealing source for regenerative medicine. However, with the multi-passage or cryopreservation for large-scale growth procedures in terms of preclinical and clinical purposes, hASCs often reveal defective cell viability, which is a major obstacle for cell therapy. In our study, the effects of induced pluripotent stem cells-derived conditioned medium (iPS-CM) on the proliferation and anti-apoptosis in hASCs were investigated. hASCs at passage 1 were identified by the analysis of typical surface antigens with flow cytometry assay and adipogenic and osteogenic differentiation. The effect of iPS-CM on the proliferation in hASCs was analyzed by cell cycle assay and Ki67/P27 quantitative polymerase chain reaction analysis. The effect of iPS-CM on the anti-apoptosis of hASCs irradiated by 468 J/m2 of ultraviolet C was investigated by annexin v/propidium iodide analysis, mitochondrial membrane potential assay, intracellular reactive oxygen species assay, Western blotting and caspase activity assays. The effect of iPS-CM on the surface antigen expressions of hASCs was analyzed using flow cytometry assay. The levels of Activin A and bFGF in culture supernatant of hASCs with different treatments were also detected by enzyme-linked immunosorbent assay. iPS-CM promoted proliferation and inhibited apoptosis of hASCs. This discovery demonstrates that iPS-CM might be used as one of the available means to overcome the propagation obstacle for hASCs and make for large-scale growth procedures in terms of preclinical and clinical purposes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human adipose-derived stem cells (hASCs) were isolated from adipose tissues, which were obtained in high yields by minimally invasive liposuction techniques [1]. They had the potential to differentiate into many types of cells [2]. There were a large number of literatures which had described the trophic effects of hASCs on the protection, survival, differentiation of endogenous cells/tissues, and immuno-modulatory property owing to their secretome and soluble factors [3]. Given that adipose tissues are easily accessible, hASCs become an appealing source for autologous cell transplantation [4]. Besides, hASCs could avert the clinical obstacles of potential immunological rejection and tumorigenesis, respectively, compared to embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) [5]. Recently, much attention has been focused on clinical application of hASCs in the fields such as regenerative medicine, soft tissue reconstruction, and cosmetic surgery [6]. However, with the multi-passage or cryopreservation for large-scale growth procedures in terms of preclinical and clinical purposes, hASCs often reveal defective cell viability, which is a major trouble for stem cell therapy [7].
iPSCs are a type of pluripotent stem cell that can be generated directly from adult cells [8–10]. iPSCs share the features of ESCs that were capable of self-renewal and differentiation into three germ layers [11]. iPSCs could address immune rejection and ethical issues of the autologous cell transplantation. iPSCs also offered an attractive approach to disease model, pharmaceutical screening, toxicology research, and so on [12, 13]. Conditioned medium could affect cell functions via biologically active components. It was reported that secretome such as basic fibroblast growth factor (bFGF), nerve growth factor (NGF), stem cell factor (SCF), hepatocyte growth factors (HGF), vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF-1), and brain-derived neurotrophic factor (BDNF) could be secreted in supernatant of cultured stem cells, which could encourage the growth of other cells [14–16]. Zhang et al. demonstrated that iPSCs-derived conditioned medium (iPS-CM) could promote proliferation by attenuating G1 phase arrest of cell cycle and suppress apoptosis by inhibiting p53/p21 and p16/pRb pathways in H9C2 cells [17]. Neel and Singla [18] found that iPS-CM would decrease the number of cardiac apoptotic nuclei in the streptozotocin of diabetic cardiomyopathy (SIDC) rat model. Li et al. [9] reported that iPS-CM potentially restored the bronchial microstructure in acute lung injury (ALI). Zhang et al. [19] mentioned that iPS-CM contributed to recovery from the effect of endotoxin-induced ALI in mice. The above studies showed that iPS-CM stimulated cell proliferation and protected them from apoptosis. The stimulating proliferation and anti-apoptosis from iPS-CM were the result of cytokines secretion, which were generally safe and would not produce tumors [17–19].
In this research, we investigated the influence of iPS-CM on the proliferation and the ultraviolet C (UVC)-induced apoptosis of hASCs, and evaluated the effect of iPS-CM on differentiation potential and marker gene expression in hASCs. Moreover, the levels of bFGF and Activin A in supernatant of hASCs with or without iPS-CM treatment were also detected. The main purpose of this study was to explore new way to improve the activity and growth state of apoptotic hASCs. We hoped that the treatment with iPS-CM would promote the application of hASCs in regenerative medicine and tissue engineering.
Materials and methods
Materials
hASCs were cultured in a traditional medium including Low-Glucose Dulbecco’s Modified Eagle’s Medium (DMEM), 100 U/ml penicillin/streptomycin, and 10 % fetal bovine serum (FBS, Gibco, USA), which was abbreviated as DMEM-LG. Unless otherwise stated, all the other reagents were from Sigma (St. Louis, MO, USA). Matrigel, Y-27632, and fluorescein isothiocyanate (FITC/PE)-conjugated monoclonal antibodies including CD29, CD34, CD44, CD45, CD59, CD105, and HLA-DR were purchased from BD Bioscience (Franklin Lakes, NJ, USA). Adipogenic and osteogenic differentiation culture mediums were purchased from Biowit Technologies (Shenzhen, China). Alizarin red S staining solution was purchased from Merk (New Jersey, USA). mTeSR1 medium and trypsin–EDTA were purchased from StemCell (Vancouver, BC, Canada). Cell counting kit-8 (CCK-8) was bought from BestBio (Shanghai, China). TRIzol and reverse-transcriptase reagents kit were bought from Life Technologies (NY, USA). Primers were synthesized by Life Technologies (Shanghai, China). ABI Prism 5,59,6,69-tetrachloro-1,19,3,39-tetraethylbenzimi-dazolylcarbocyanine iodide (JC-1) assay kit, cell cycle kit, and annexin v-FITC/propidium iodide (PI) apoptosis detection kit were bought from KeyGEN BioTECH. Co.,LTD (Nanjing, China). The fluorescence dye 2′7′-dichlorofluorescin diacetate (DCFH-DA) assay kit was got from Qcbio Science & Technologies (Shanghai, China). Caspase-3 and caspase-9 activity assay kit were got from Beyotime Institute of Biotechnology (Haimen, China). Enzyme-linked immunosorbent assay kit was got from Chemicon International, Inc (Temecula, CA, USA) (Table 1).
Ethics statement
Subcutaneous adipose tissues were harvested from six healthy female donors with a mean age of 35-year old carrying out liposuction. Written informed consent was obtained from all study participants, and the documents of participant consent are preserved in each patient’s medical casebook. The study protocols were approved by the institutional ethical review board of First Affiliated Hospital of Jinan University and carried out according to the principles of the Declaration of Helsinki.
Isolation and culture of hASCs
The fresh adipose tissues were instantly washed 5 times with sterile phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.4) to remove the debris of blood vessels and connective tissues, and then were transferred into DMEM containing 0.1 % type I collagenase and incubated for 40 min at 37 °C shaking table. Then cells were centrifuged (300×g, 8 min), and the supernatant was removed. After that, cells were resuspended in 0.3 % NaCl for 10 min and subsequently centrifuged at 300×g for 5 min again. The suspension was filtered through 100-µm nylon membrane. The cells at the concentration of 1 × 104 cells/ml were directly seeded into a 25 cm2 plastic culture flask and incubated at 37 °C, 5 % CO2 incubator. The culture medium was changed twice a week. Cells were passaged every week by 0.25 % trypsinization.
Flow cytometry assay for cell surface antigens
Flow cytometry assay was performed according to the reported method with minor modification [20]. Briefly, hASCs with iPS-CM or without iPS-CM treatment were harvested using 0.25 % trypsin–EDTA and centrifuged at 300×g for 10 min. Then cells were resuspended with PBS. The total of 3 × 105 hASCs were darkly incubated for 30 min with FITC/PE-conjugated monoclonal antibodies: CD29, CD34, CD44, CD45, CD59, CD105, and HLA-DR. Then cells were washed with PBS for 3 times, centrifuged (400×g, 5 min), and suspended in 200 μl PBS. CD surface antigens were analyzed on a FACS Calibur flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA).
Adipogenic differentiation in vitro
Adipogenic induction of hASCs was followed the reported procedure with slight modification [21]. Briefly, cells at P2 were obtained using 0.25 % trypsin–EDTA and then seeded at the density of 5 × 104 cells/ml in a 6-well plate. At 90–100 % confluence, the medium was replaced with adipogenic differentiation culture medium. The medium was changed every 3 days. The staining of Oil red O solution was conducted at day 14. Cells were washed twice with PBS and fixed in 4 % formaldehyde for 10 min at room temperature, and then washed with PBS twice again. Staining for accumulated cytoplasmic lipid droplets was conducted with filtered Oil red O solution for 30 min at 37 °C, 5 % CO2 incubator. Finally, cells were washed twice again with PBS and then photographed under an inverted microscope (Olympus, Tokyo, Japan).
Osteogenic differentiation in vitro
Osteogenic induction of hASCs was performed according to a reported method with minor modifications [21]. Briefly, cells at P2 were obtained using 0.25 % trypsin–EDTA and then seeded at the density of 5 × 104 cells/ml in a 6-well plate. At 70–80 % confluence, the medium was replaced with osteogenic differentiation culture medium. The medium was changed every 3 days. Alizarin red S staining was implemented at day 21. Cells were washed twice with PBS and fixed with 4 % formaldehyde for 15 min, and then washed with PBS twice again. Staining for calcium deposit was conducted with filtered 0.2 % Alizarin red S solution for 30 min at 37 °C, 5 % CO2 incubator. Lastly, cells were washed 3 times with PBS and then photographed using an inverted microscope.
The culture of iPSCs and preparation of iPS supernatant
Human iPS cell line was obtained from Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences. These iPSCs were generated from the umbilical cord matrix and amniotic membrane mesenchymal cells by transducting retroviral factors, embracing Oct4, Sox2, c-Myc, and Klf4 as previously described [22]. iPSCs were cultured according to the previous protocols [23, 24]. Briefly, the dishes were coated with 1 % Matrigel half an hour in advance, and then iPSCs were cultured in mTeSR1 medium in a 37 °C incubator of 5 % CO2. They were subcultured every 6 days with 0.05 % trypsin–EDTA at 37 °C for 5 min, and then they were seeded onto 1 % Matrigel-coated culture plate. The ROCK inhibitor Y-27632 (10 mM) was added to each well for the first day after each passage. They demonstrated positive gene expressions including Sox2 and Oct4, and positive protein expressions such as Nanog, SSEA-4, and TRA-1–60 [22–24]. iPS supernatant was derived by collecting the medium from iPSCs cultured in mTeSR1 medium every 24 h. The collected medium was filtered (0.22 μm) to remove dead cells and stored at −80 °C for at least 2 weeks. iPS-CM was iPS supernatant mixed with DMEM-LG at a ratio of 1:2.
CCK-8 assay
Cell counting kit-8 (CCK-8) was used to detect the proliferation of hASCs with different treatments. Briefly, 100 μl of 1 × 105 cells/ml were plated into each well of 96-well plates. Then cells were treated with different mediums. Then cells were incubated for 48 h at 37 °C, 5 % CO2 incubator. Subsequently, the each well was supplemented with 10 μl CCK-8 solution and incubated for 4 h at 37 °C. At last, the absorbance at 450 nm was measured with a microplate reader (Thermo, MA, USA).
Semiquantitative reverse transcription PCR (RT-PCR) assay and quantitative real-time PCR (qPCR)
Total cellular RNA was extracted from hASCs in DMEM-LG (control medium) and iPS-CM (conditioned medium) using TRIzol, and the concentration of RNA was quantified by measuring OD at 260 nm. cDNA was synthesized using an reverse-transcriptase reagent kit according to the manufacturer’s instructions. The cDNA (1 μg) was synthesized for RT-PCR of CD90, CD44, CD29, CD13, and GAPDH genes. The mixture was denatured at 94 °C for 2 min, then amplified for 40 cycles (94 °C, 30 s; 59 °C, 30 s; 72 °C, 30 s) using an authorized thermal cycler (Eppendorf, Hamburg, GER). Finally, 6 μl of PCR product and 2 μl of 6× loading buffer were mixed and electrophoresed in a 2 % agarose gel. Gels were photographed and scanned.
Total cDNA was used to perform qPCR to analyze the expression levels of GAPDH, CD90, CD44, CD29, CD13, Ki67, and P27 genes using the ABI Prism 7500™ instrument (Applied Biosystems, Shanghai, China). The reaction mixture consisted of 10 μl SYB Green Mix, 1 μl forward, 1 μl reverse primers, 1 μg diluted cDNA and 3–6 μl ddH2O. The reaction process was 95 °C for 2 min, followed by 40 cycles of 95 °C for 10 s, 59 °C for 30 s. GAPDH gene was used as the internal control. The quality of PCR amplification of each sample was calculated according to the \(2^{{ - {\Delta} {\Delta} C_{\text{t}} }}\) method.
Cell cycle assay
hASCs at P2 were planted in a 6-well plate at a density of 2 × 105 cells/ml and incubated 24 h. Then cells were washed with PBS and cultured with DMEM-LG (control medium) and iPS-CM (conditioned medium) for 48 h at 37 °C, 5 % CO2 incubator. Then cells were collected and fixed in 75 % cold ethanol at 4 °C overnight. The next day, the fixed cells were washed with PBS and stained by PI in the dark for 30 min at room temperature. The stained cells were analyzed using flow cytometry.
Annexin v and propidium iodide (PI) assay
hASCs were seeded into 6-well plates at the density of 2.5 × 106 cells/ml in DMEM-LG for 24 h. Then cells were washed with PBS and pretreated with DMEM-LG (control medium) or iPS-CM (conditioned medium) for 1 h. Then hASCs were irradiated with 468 J/m2 UVC. After that, cells in control medium were changed with DMEM-LG, while cells in conditioned medium were changed with iPS-CM for 24 h at 37 °C, 5 % CO2 incubator. To evaluate early and lately apoptotic activities, an annexin v-FITC/PI apoptosis detection kit was used according to the manufacturer’s instructions. hASCs were harvested and washed with cold PBS. Then cells were resuspended in 200 μl annexin v-binding buffer. After cells were stained with 5 μl of FITC-labeled annexin v and 5 μl of PI, they were instantly measured using flow cytometry.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was evaluated with 5,59,6,69-tetrachloro-1,19,3,39-tetraethylbenzimi-dazolylcarbocyanine iodide (JC-1) assay kit. Approximately, 1.5 × 105 cells/ml hASCs were plated into every well of 6-well plate and incubated in DMEM-LG at 37 °C, 5 % CO2 incubator for 24 h. Then cells were washed with PBS and added DMEM-LG (control medium) or iPS-CM (conditioned medium) for 1 h. Then hASCs were exposed to UVC at the dose of 468 J/m2. Then cells in control medium were changed with DMEM-LG, while cells in conditioned medium were changed with iPS-CM for 24 h at 37 °C, 5 % CO2 incubator. Subsequently, medium was removed and washed with cold PBS twice. hASCs were gathered applying trypsin digestion method. Cells were incubated for 30 min at 37 °C with 500 μl JC-1 (5 mM). Then cells were collected by centrifugation at 300×g for 5 min and washed twice again with 1× incubation buffer. Cells were then resuspended with 500 μl 1× incubation buffer. Red and green fluorescence emissions were analyzed by flow cytometry using an excitation wavelength of 488 nm and emission wavelengths of 530 nm.
Measurement of cellular UVC-induced ROS
Reactive oxygen species (ROS) production was measured with the fluorescence dye 2′7′-dichlorofluorescin diacetate (DCFH-DA) assay kit. Briefly, 1.5 × 105 cells/ml hASCs at P2 were planted into 6-well plate and cultured at 37 °C, 5 % CO2 incubator. After 24 h, the adherent cells were washed with PBS and added DMEM-LG (control medium) or iPS-CM (conditioned medium) for 1 h. And then cells in control medium and conditioned medium were both exposed to 468 J/m2 UVC. After that, cells in control medium were changed with DMEM-LG, while cells in conditioned medium were changed with iPS-CM for 2 h at 37 °C, 5 % CO2 incubator. Then cells were gathered and suspended with 200 μl DCFH-DA for 20 min at 37 °C in the dark. Then cells were washed twice with PBS and fluorescence intensity was measured by flow cytometry.
Western blotting analysis
hASCs were washed with cold PBS and then lysed in a RIPA buffer (Beyotime Institute of Biotechnology, Haimen, China) supplemented with a Protease Inhibitor Cocktail (Amyjet Scientific Co., Ltd, Wuhan, China). Lysates were centrifuged at 12,000×g for 15 min at 4 °C. The supernatant protein concentration was measured using a BCA assay kit (Takara Bio Inc., Shiġa, Japan) according to the manufacturer’s instructions. Subsequently, 40 μg of proteins were subjected to 10 % SDS-PAGE and transferred into a polyvinylidene fluoride (PVDF) membrane. After being blocked with 5 % nonfat milk in tris-buffered saline containing 0.05 % Tween 20 (TBST) overnight at 4 °C, the membrane was incubated with antibody against Bcl-2, Bax (1:1000, ProteinTech Group, Chicago, IL, USA) and GAPDH (1:3000, Cell Signaling Technology, Danvers, MA, USA) overnight at 4 °C. The membrane was then washed 5 times with TBST and incubated with horseradish peroxidase-conjugated secondary antibody (Beyotime Institute of Biotechnology, Haimen, China) for 2 h at room temperature. Signals were visualized with enhanced chemiluminescence (ECL) (Pierce Chemical Co., Rockford, IL, USA) and quantified using Image J. The ratio of the expression of target proteins was determined after normalizing the individual GAPDH level.
Caspase activity assay
Caspase-3 and caspase-9 activity assay kits were used to detect caspase-3 and caspase-9 activities. Approximately 1.5 × 105 cells/ml hASCs were plated into every well of 6-well plates and incubated in DMEM-LG at 37 °C, 5 % CO2 incubator for 24 h. Then cells were washed with PBS and added DMEM-LG (control medium) or iPS-CM (conditioned medium) for 1 h, and hASCs were exposed to UVC at the dose of 468 J/m2. After that, cells in control medium were changed with DMEM-LG, while cells in conditioned medium were changed with iPS-CM for 12 h at 37 °C, 5 % CO2 incubator. Later medium were removed, and adherent cells were washed in cold PBS twice. Samples were collected, and the activities of caspase-3 and caspase-9 were subsequently analyzed using a microplate reader at a wavelength of 405 nm.
Enzyme-linked immunosorbent assay (ELISA)
The levels of Activin A and bFGF in different supernatants of hASCs were detected with commercially available enzyme-linked immunosorbent assay kits according to the manufacturer’s protocols. Briefly, 200 μl samples and 50 μl assay diluent were added to 96-well plates. The plates were incubated for 2 h at room temperature and washed 5 times with washing buffer. 100 μl peroxidase-conjugated IgG anti-Activin A or anti-bFGF solution was added into each well for 2 h at room temperature. Then plates were washed 5 times again with washing buffer. Later 100 μl substrate buffers were added into each well and darkly incubated for 30 min at room temperature. Lastly, the enzyme reaction was stopped with 50 μl stop solution. Assay results were measured using a microplate reader at the wavelength of 450 nm as a test wavelength and 570 nm as the reference one.
Statistical analysis
Data were analyzed using the SPSS 16.0. The statistical significance comparing multiple sample sets with the control was analyzed with a one-way ANOVA followed by the Dunnett’s test. Comparisons between the two groups were analyzed using Student’s t tests. Values were presented as mean ± SD, the P < 0.05 was considered to be statistically significant. The P > 0.05 was not considered to be statistically significant.
Results
Identification of hASCs
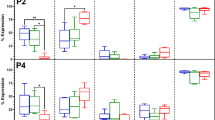
In order to characterize the cells derived from adult adipose tissue, we performed flow cytometry. The result revealed that hASCs positively expressed mesenchymal stem cell markers such as CD29, CD105, CD44, and CD59, while negatively expressed hematopoietic cell markers including CD34, CD45, and HLA-DR (Fig. 1a, b). Meanwhile, these cells could be successfully differentiated into osteogenic and adipogenic lineages (Fig. 1c). These results confirmed that cells derived from adipose tissue were hASCs.

Identification of hASCs. a Flow cytometry analysis of surface phenotypes of hASCs. b The geometric mean of cell surface antigens. c The adipogenic and osteogenic inductions of hASCs. a Negative control and b positive group for adipogenic differentiation. c Negative control and d positive group for osteogenic differentiation
The effects of iPS-CM on hASCs characteristics
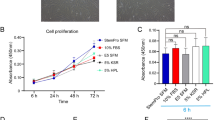
The proliferation of hASCs was detected by CCK-8 to explore the optimal proportion of iPS supernatant/DMEM-LG. hASCs were treated with different proportions of iPS supernatant/DMEM-LG (1:0, 1:1, 1:2, 2:1, 0:1) for 48 h. The result of CCK-8 assay showed that the proliferation of hASCs was the most obvious in iPS supernatant/DMEM-LG of 1:2 compared with other proportions (Fig. 2a). Based on above result, we considered the proportion as our iPS-CM to treat hASCs in later studies.

The effects of iPS-CM on hASCs. a Exploration the optimal proportion of iPS supernatant/DMEM-LG on the proliferation of hASCs by CCK-8 assay. b The semiquantitative gene expressions of CD90, CD44, CD29, CD13, and GAPDH of hASCs in control medium and conditioned medium by RT-PCR. c The quantitative gene expressions of CD90, CD44, CD29, CD13, and GAPDH of hASCs in control medium and conditioned medium by qPCR. d The effect of iPS-CM on the adipogenic and osteogenic differentiation of hASCs. a Negative control and b positive group for adipogenic differentiation. c Negative control and d positive group for osteogenic differentiation. Bars represented the mean ± SD of three independent experiments. *P < 0.05 (1:1 and 2:1 vs 1:0 and 0:1); **P < 0.01 (1:2 vs 1:0 or 0:1); # P > 0.05 (control medium vs conditioned medium)
To detect the effect of iPS-CM on the marker gene expressions of hASCs, we next performed semiquantitative RT-PCR and qPCR assays to detect the gene expression levels of GAPDH, CD90, CD44, CD29, and CD13 of hASCs in control medium and conditioned medium. The semiquantitative RT-PCR assay showed that the ratio of CD90/GAPDH, CD44/GAPDH, CD29/GAPDH, and CD13/GAPDH of hASCs in control medium had no significant difference with conditioned medium (Fig. 2b). The qPCR assay displayed that hASCs in control medium had no obvious difference with conditioned medium in the gene expression levels of CD13, CD29, CD44, and CD90 (Fig. 2c). These results demonstrated that iPS-CM would have no significant effect on marker gene expressions of CD90, CD44, CD29, and CD13 in hASCs.
In order to explore the effect of iPS-CM on the differentiation potential of hASCs, adipogenic and osteogenic inductions were conducted for hASCs treated with iPS-CM. The results demonstrated that the differentiation potential of hASCs would not be affected by iPS-CM (Fig. 2d).
Effect of iPS-CM on the proliferation of hASCs
hASCs cultured in DMEM-LG or iPS-CM for 1 day and 3 days were imaged under an inverted contrast microscope, respectively. The number of cells in conditioned medium increased dramatically compared with control medium on day 3 (Fig. 3a). The qPCR assay was conducted to quantitatively compare the gene expression levels of P27 and Ki67 of hASCs in conditioned medium and control medium. The gene of P27 of hASCs in conditioned medium was down-regulated about 7.1 fold than control medium, but the gene of Ki67 was up-regulated nearly 1.9 fold (Fig. 3b). P27 is an inhibitory protein of the cell cycle, while Ki67 is a marker protein of the cell cycle. When cells begin to proliferate, the gene of P27 will be down-regulated and the gene of Ki67 will be up-regulated [25]. This result suggested that the cell viability in conditioned medium was better than control medium.

iPS-CM promoted hASCs propagation. a The bright field of hASCs in control medium and conditioned medium on day 1 and day 3. b The qPCR analysis of P27 and Ki67 mRNA expression levels of hASCs in control medium and conditioned medium. c The cell cycle of hASCs in control medium and conditioned medium. d Cell cycle distribution is represented as mean ± SD of three independent experiments. *P < 0.05 (control medium vs conditioned medium)
The cell cycle assay was used to detect the effect of iPS-CM on the proliferation of hASCs in control medium and conditioned medium. Compared with control medium, the cell cycle of hASCs in conditioned medium was promoted obviously (Fig. 3c). The percentage of cells entering the S and G2 phases in conditioned medium was (28.1 + 22.43) % while in control medium was (11.5 + 10.2) % (Fig. 3d). The assay showed that the cell proliferation in conditioned medium had (16.6 + 12.23) % increase than control medium. The result further demonstrated that iPS-CM could promote the proliferation of hASCs.
Effects of iPS-CM on anti-apoptosis of hASCs
hASCs were exposed to various dose of UVC (0, 117, 234, 468, 702, and 936 J/m2), and then were incubated with DMEM-LG for 24 h. The detached cells were washed away with PBS. The attached cells were imaged using a standard inverted phase contract light microscope (Fig. 4a). We examined the morphological features of UVC-induced cell apoptosis. The hallmark features of apoptosis such as cell detachment and cytoplasmic condensation, would worsen due to increasing ultraviolet exposure dosage. The performance was consistent with the result of CCK-8 assay (Fig. 4b). The results indicated that UVC at the dose of 468 J/m2 was optimal for the apoptosis model.

The apoptosis model of UVC-induced hASCs. a The bright field of hASCs exposed to various dose of UVC under an inverted microscope. b The viability effects of iPS-CM on hASCs exposed to UVC at various dose by CCK-8 assay. Bars represented the mean ± SD of three independent experiments. *P < 0.05 (468, 702 and 936 vs 0 J/m2)
Annexin v and PI assay was used to detect the apoptosis of hASCs in control medium, control medium + UVC, and conditioned medium + UVC. Phosphatidylserine (PS) externalization in living cells was a hallmark of early phase of apoptosis. Annexin v, harboring a strong and specific affinity for PS, was used to detect early stage of apoptosis. In the late stage of apoptosis, the damaged cell membranes enabled annexin v and PI to enter into cells. In addition, the membranes of dead cells are only permeable for PI. Hence, hASCs in early apoptosis were annexin v positive and PI negative. Cells in late apoptosis were both annexin v and PI positive, and dead cells were PI positive. Our result showed that iPS-CM could dramatically reduce the ratio of apoptotic in hASCs irradiated with UVC. hASCs in control medium almost had no apoptosis cell (0.6 ± 0.01) %. The apoptotic rate of hASCs in conditioned medium + UVC (21.41 ± 1.39) % was much lower than control medium + UVC (7.74 ± 1.24) % (Fig. 5a). This result demonstrated that iPS-CM could reduce the apoptosis of hASCs exposed to UVC.

iPS-CM inhibited UVC-induced hASCs apoptosis, the loss of ΔΨ m and the generation of ROS. a Annexin v and propidium iodide assay analyzed the apoptosis of hASCs in control medium, control medium + UVC, and conditioned medium + UVC. b Mitochondrial membrane potential assay analyzed the loss of ΔΨ m of hASCs in control medium, control medium + UVC, and conditioned medium + UVC. c Intracellular reactive oxygen species assay analyzed the generation of ROS of hASCs in control medium, control medium + UVC and conditioned medium + UVC. Bars represented the mean ± SD of three independent experiments. **P < 0.01 (control medium + UVC or conditioned medium + UVC vs control medium); ## P < 0.01 (control medium + UVC vs conditioned medium + UVC)
Because the loss of mitochondrial membrane potential (ΔΨ m) might be associated with early apoptosis, the ΔΨ m of hASCs in control medium and conditioned medium treated with UVC were assessed by JC-1. The ΔΨ m in living cells was higher than that in apoptosis cells. JC-1 was the probe that could specially enter the mitochondria. When the ΔΨ m was high, JC-1 would form polymer and glow red fluorescent. When the ΔΨ m was low, it would form the monomer and glow green fluorescence. The ratio of JC-1-red/green indirectly reflected the result of the apoptosis of cells. High ratio presented less apoptosis than low ratio. The result showed that the ratio of hASCs in control medium was (87.78 ± 2.67) %, control medium + UVC (44.65 ± 1.15) %, and conditioned medium + UVC (68.35 ± 2.12) % (Fig. 5b). This result showed that iPS-CM could inhibit the loss of ΔΨ m in hASCs exposed to UVC.
DCFH-DA was used to detect the level of ROS production of hASCs in control medium, control medium + UVC, and conditioned medium + UVC. In the generation of ROS, hASCs in control medium + UVC had approximately a nine-fold increase, while hASCs in conditioned medium + UVC just had almost a four-fold increase compared with control group (Fig. 5c). This result demonstrated that iPS-CM significantly decreased the levels of ROS in hASCs exposed to UVC.
Molecular mechanisms of iPS-CM on anti-apoptosis of hASCs
To elucidate the potential molecular mechanisms involved in the protection of iPS-CM on hASCs with UVC treatment, the levels of apoptotic signaling molecules in the mitochondria including Bcl-2 and Bax were detected by Western blotting after treatment with 468 J/m2 UVC. Compared with control medium + UVC, iPS-CM significantly inhibited the expression of Bax of hASCs in conditioned medium + UVC (Fig. 6a). While the expression level of Bcl-2 of hASCs was much higher in conditioned medium + UVC than control medium + UVC (Fig. 6b). The expression level of Bax of hASCs in control medium was the least, while the expression level of Bcl-2 was the highest (Fig. 6a, b). To further analyze the anti-apoptotic effect of iPS-CM on hASCs irradiated with UVC, the activities of caspase-9 and caspase-3 were detected by caspase colorimetric assay. The data showed that the activities of caspase-3 and caspase-9 of hASCs in conditioned medium + UVC were significantly down-regulated than control medium + UVC. hASCs in conditioned medium + UVC displayed 60 and 40 % decreases in the levels of the caspase-3 and caspase-9, respectively, compared with control medium + UVC. The levels of the caspase-3 and caspase-9 of hASCs in control medium were the least (Fig. 6c, d). These results demonstrated that iPS-CM could obviously reduce the apoptosis of hASCs exposed to UVC, which was related to Bax down-regulation and Bcl-2 up-regulation.

iPS-CM suppressed UVC-induced Bcl-2 down-regulation, Bax up-regulation, caspase-3, and caspase-9 activities in hASCs treated with UVC. The protein expressions of a Bax and b Bcl-2 of hASCs in control medium, control medium + UVC, and conditioned medium + UVC using Western blotting analysis. The activities of c caspase-3 and d caspase-9 of hASCs in control medium, control medium + UVC, and conditioned medium + UVC using ELISA. Bars represented the mean ± SD of three independent experiments. *P < 0.05 (control medium + UVC vs conditioned medium + UVC); **P < 0.01 (control medium + UVC vs control medium)
The levels of bFGF and Activin A in supernatants of hASCs
To investigate the effect of iPS-CM on cell membrane antigen changes of hASCs, we analyzed the expression of CD29, CD34, CD44, CD45, CD59, CD105, and HLA-DR of hASCs (P2) through flow cytometry (Fig. 7a). The result showed that hASCs with or without iPS-CM treatments could positively express high levels of CD29 and CD44, low levels of CD59 and CD105, and negatively express CD34, CD45 and HLA-DR. These data demonstrated that there was no significant difference of membrane antigen expression in hASCs treated with or without iPS-CM (Fig. 7b).

The surface antigen expressions of hASCs and the levels of bFGF and Activin A in hASCs with different treatments. a Flow cytometry analysis of surface antigens of hASCs with or without iPS-CM treatment. b The geometric mean of cell surface antigens of hASCs with or without iPS-CM treatment. c The levels of bFGF in control medium, conditioned medium, control medium + UVC, and conditioned medium + UVC. d The levels of Activin A in control medium, conditioned medium, control medium + UVC, and conditioned medium + UVC. Bars represented the mean ± SD of three independent experiments. **P < 0.01 (control medium vs conditioned medium or control medium + UVC vs conditioned medium + UVC); # P > 0.05 (control medium vs conditioned medium)
In order to explore the expression level changes of bFGF and Activin A in supernatants of hASCs within control medium, conditioned medium, control medium + UVC, and conditioned medium + UVC, we detected the supernatants of hASCs in these groups by ELISA on day 2. The level of bFGF in supernatant of hASCs in conditioned medium (35.99 ± 1.58) ng/ml was higher than control medium (22.19 ± 1.13) ng/ml, and that in conditioned medium + UVC (26.36 ± 1.37) ng/ml was higher than control medium + UVC (12.98 ± 1.04) ng/ml (Fig. 7c). Almost the same tendency was with Activin A. The level of Activin A of hASCs in conditioned medium (825.68 ± 8.36) pg/ml was higher than control medium (577.84 ± 4.22) pg/ml, and that in conditioned medium + UVC (708.08 ± 6.37) pg/ml was higher than control medium + UVC (498.52 ± 3.74) pg/ml (Fig. 7d). These data showed that the levels of bFGF and Activin A in supernatants of hASCs with iPS-CM treatment were higher than DMEM-LG. The effects of iPS-CM on the proliferation and anti-apoptosis of hASCs might be partly related to bFGF and Activin A.
Discussion
Stem cell therapy has been becoming a promising approach for many diseases [2, 26]. Among the different stem cell populations, specific attention has been focused on hASCs due to the potential of secreting bioactive molecules and multi-differentiation [3, 4]. However, despite the encouraging potential in the therapeutic regard, the capacity of hASC restricted proliferation owing to shorter telomerase, DNA mutations with age and extrinsic factors (niche alterations), has limited their use [27]. Moreover, the preservation and subculture of cells in numbers that are sufficient for transplantation are necessary for cell-based therapy. But the techniques may cause cells to lose their specific phenotypes and decrease their proliferative potential [28]. The processes of multi-passage or cryopreservation invariably led these cells into adverse growth status, which affected their viability, morphology, and differentiation potential [29, 30]. These characteristics became the main obstacles to clinical applications of hASCs.
Many literatures implied that the beneficial effects of hASC proliferation in vitro are related to soluble factors supplemented in culture medium. For example, the reports depicted that Rehmannia glutinosa oligosaccharide (RGO), lactoferrin (LF), periostin, exogenous nucleosides, tumor-secreted factors, human insulin-like growth factor-I (IGF-I), bone morphogenetic protein-2 (BMP-2), vascular endothelial growth factor (VEGF) and cerebrospinal fluid (CSF) could be beneficial to expandability of hASCs [31–36].
iPSCs were almost same as ESCs in proliferative abilities, morphology, gene expression, surface antigens, telomerase activity, and epigenetic status of pluripotent cell-specific genes [37]. iPSCs could secrete cytokines, chemokines, growth factors and metabolites, and bioactive lipids. A wide range of products from iPSC secretion could encourage the growth of other cells [38]. iPS-CM could enhance alveolar epithelial regeneration in vivo partially due to containing hepatocyte growth factor [39]. iPS-CM could reduce apoptosis, oxidative stress, and fibrosis, as well as improve cardiac function in diabetic model of rats [18]. High-tidal-volume-induced ventilator induced lung injury could also be suppressed by iPS-CM [9]. In this study, we also found that the survival of hASCs with or without UVC irradiation could be promoted by iPS-CM. Besides, our results showed that the expression level of P27 was down-regulated, while the expression level of Ki67 was up-regulated when hASCs treated with iPS-CM. The percentage of cells entering the S and G2 phases in hASCs treated with iPS-CM was also more than counterparts without iPS-CM. Ki67, a cell proliferation marker and a nuclear non-histone protein, was present during the active phases of cell proliferation (G1, S, G2, and mitosis), but was absent from resting cells (G0) [40]. On the contrary, P27, the cyclin-dependent kinase inhibitor inhibited the G1 to S transition in a variety of cell types [41, 42]. These results indicated that iPS-CM could significantly induce the propagation of hASCs.
Apoptosis, as a basic character of all animal cells, is necessary for normal development and tissue homeostasis [43]. UVC-induced apoptosis is a highly complex process, which is involved in distinct molecular pathways and different cellular structures [44]. Identifying the apoptotic mechanism of hASCs induced by UVC irradiation and the anti-apoptotic mechanism affected by iPS-CM will contribute to hASC applications. In the present study, UVC-induced apoptotic changes in hASCs were demonstrated by up-regulation of Bax and down-regulation of Bcl-2, formation of ROS, loss of mitochondrial membrane potential, and activation of caspase-3 and caspase-9 (Fig. 8). First, the signaling pathway involved in UVC-induced apoptosis was regulated by Bcl-2 family members [45]. Bcl-2, an anti-apoptotic protein in the Bcl-2 family, lies in the cytoplasmic of the outer mitochondrial membrane, nuclear envelope, and endoplasmic reticulum [23, 46]. It was demonstrated that Bcl-2 in fibroblasts was closely related to mitochondrial homeostasis and cell viability [47]. Conversely, Bax, a pro-apoptotic and pore-forming cytoplasmic protein in the Bcl-2 family, translocates to the outer mitochondrial membrane and affects permeability from the inter-membrane space into the cytosol, which later causes cell death [48]. Glover et al. [49] have mentioned that p53 transcriptional activation by UVC exposure led to the imbalance of pro-apoptotic versus anti-apoptotic Bcl-2 family proteins and then brought about mitochondrial membrane depolarization and apoptotic cascade initiation. Second, one striking feature of UVC-induced cell apoptosis is the generation of ROS, followed by a decline in mitochondrial membrane potential and activation of caspase-9 [44, 50]. Third, mitochondrial membrane potential and caspase cascade then participated in UVC-induced apoptosis. Chathoth et al. [51] have reported that DNA damage induced by UVC irradiation activated pro-apoptotic Bcl-2 members and subsequently promoted activation of caspase cascade through cytochrome c release from mitochondria. Fritsch et al. [52] have also reported that the members of the Bcl-2 family of proteins regulated mitochondrial integrity. Caspase-9 activation induced by cytochrome c release from mitochondria was regulated by Bcl-2 down-regulation or Bax hyperexpression and then activated downstream caspase-3, which finally caused cell death [53]. The activation of caspase-9 by apoptosome could further activate the downstream caspases-3, which in turn cleave many intracellular substrates and cause nuclei condensation and fragmentation, thereby forming ROS circle amplification [44]. Among the various molecules that take part in the apoptotic process, caspase plays an important role during the initiation and effector phases of apoptotic cell death. In our previous study, iPSCs irradiated with UVC showed a significant increase in caspase-3 and caspase-9 activities compared with control group. Caspase-3 and caspase-9 are involved in iPSC apoptosis induced by UVC [54]. UVC-induced apoptosis of hASCs in our study was consistent with these results. However, hASCs treated with iPS-CM significantly counteracted the apoptosis. One report demonstrated that genistein pretreatment could reduce I/R injury in rabbit myocardium by reducing Bax expression and by increasing the Bcl-2/Bax ratio [55]. Bax knockout was associated with reduced activation of caspase-3 [56]. It was reported that catalpol decreased the chemia-induced cell death in PC12 cells by up-regulation of Bcl-2, which might block cytochrome c release, suppress mitochondrial membrane potential loss, and inactivate caspase-3 activity [57]. Qiu et al. [58] have revealed that miR-210 suppressed neuronal apoptosis by inhibiting caspase activity and regulating the balance of the levels between Bcl-2 and Bax. Inhibition of caspase activity by the use of physiological or pharmacological agents has been known to reduce apoptosis [49]. Our results strongly suggested that iPS-CM could protect hASCs from apoptosis. The anti-apoptotic role of iPS-CM acted by reducing the activation of ROS, Bax, caspase-3, and caspase-9, while enhancing the activation of Bcl-2 and mitochondrial membrane potential (Fig. 8).

Diagrams of mechanism related to apoptosis and anti-apoptosis. The solid line represents the apoptotic mechanism of hASCs induced by UVC irradiation. The broken line represents the anti-apoptotic mechanism of hASCs affected by iPS-CM. The arrow indicates the up-regulation effect. The minus sign means down-regulation effect
The characterization of hASCs after expansion in different culture is pivotal to determining if the cells maintain the mesenchymal pluripotency required for cell-based therapy and testing purposes. We conducted flow cytometry and discovered that there was no significant difference for cell membrane antigen expressions such as CD29, CD34, CD44, CD45, CD59, CD105, and HLA-DR before or after hASCs treated with iPS-CM. These findings were consistent with our RT-PCR and q-PCR results. At the same time, hASCs were able to maintain specific phenotypes and differentiated potential of adipogenic and osteogenic lineages after iPS-CM treatment.
It was known that stem cells depended on cell–cell interactions as well as para/autocrine signals [59]. hASCs cultured in iPS-CM guaranteed the robust growth. We supposed that some increased soluble paracrine factors released from iPSCs might be related to such phenomenon. Our results revealed that whether or not hASCs were irradiated by UVC, bFGF, and Activin A levels in supernatants of hASCs in iPS-CM were higher than DMEM-LG. Activin A, belonging to a member of the transforming growth factor-β (TGF-β) superfamily of proteins, is homo or hetero dimeric protein embracing of two β subunits [46]. It harbors multitude functions such as regulating a broad range of cellular processes including proliferation, differentiation, and apoptosis [36]. It was reported that Activin A could promote proliferation of different types of cells including IL-9–secreting T cells (TH9), estrogen-unresponsive cells (HEC-50), rat primary granulosa cells, pancreatic cell, B50 nerve cell line, and chick neural retina cells [60–62]. bFGF is essential for activation of proliferation and to inhibit apoptosis of intestinal stem/progenitor cells [63, 64]. The reports demonstrated that bFGF could promote the propagation of various cells including myoblasts, astrocytes, melanocytes, vascular smooth muscle cells (VSMC), myoblasts, satellite cells, smooth muscle cells, and endothelial cells [65–72]. Our previous report showed that the levels of bFGF and Activin A of single iPS cells in iPS-CM were higher than fresh mTeSR1 medium [24]. The fact that hASCs also secreted bFGF and ActivinA had been reported. Taléns-Visconti et al. [73] have showed that adipose mesenchymal stem cells were able to secrete in vitro consistent amounts of bFGF. Villageois et al. [74] have found that Activin A, secreted by hASCs isolated from various fat depots of donors of different ages, promotes human multipotent adipose-derived stem (hMADS) cell proliferation, and adipocyte differentiation. Therefore, in our study, bFGF and Activin A in control medium were secreted by hASCs, while the much higher levels of bFGF and Activin A in conditioned medium were secreted by iPS and hASCs. The effects of iPS-CM on the proliferation and anti-apoptosis of hASCs might be associated with increased Activin A and bFGF.
In conclusion, we reported that iPS-CM dramatically stimulated propagation and protected from UVC-induced apoptosis in hASCs. The mechanism of anti-apoptotic effect from iPS-CM was correlated with reduction of ROS formation and remission of mitochondrial membrane potential loss, inactivation of caspase-3 and caspase-9, down-regulation Bax, and up-regulation Bcl-2. Subsequently, iPS-CM did not affect the special differentiated potential or marked gene expressions in hASCs. This study first demonstrated the beneficial effects of iPS-CM on hASCs. Such improved growth in hASCs was partly related to the elevated levels of bFGF and Activin A in supernatant. iPS-CM can be used as one of the available means that stimulates the proliferation, improves the activity, and enhances the growth state of hASCs, which will be conductive to overcoming the propagation obstacle for hASC therapy and make for large-scale growth procedures in terms of preclinical and clinical purposes. However, there are still many problems to be solved before application. For example, the risk of tumor formation is still a concern in stem cell therapy, and the long-term effect of iPS-CM cell therapy may deserve to be followed up. The promotion mechanism of bFGF and Activin A in hASCs should be explored. A secretome analysis should be applied to provide a large picture of the secretory products from iPS-CM, which can make us a better understanding and applying of iPS-CM.
References
Takeda K, Sowa Y, Nishino K, Itoh K, Fushiki S (2015) Adipose-derived stem cells promote proliferation, migration, and tube formation of lymphatic endothelial cells in vitro by secreting lymphangiogenic factors. Ann Plast Surg 74:728–736
Chang KA, Lee JH, Suh YH (2014) Therapeutic potential of human adipose-derived stem cells in neurological disorders. J Pharmacol Sci 126(4):293–301
Salgado AJ, Reis RL, Sousa NJ, Gimble JM (2010) Adipose tissue derived stem cells secretome: soluble factors and their roles in regenerative medicine. Curr Stem Cell Res Ther 5:103–110
Pal R, Hanwate M, Jan M, Totey S (2009) Phenotypic and functional comparison of optimum culture conditions for upscaling of bone marrow-derived mesenchymal stem cells. J Tissue Eng Regen Med 3:163–174
Liu S, Yuan M, Hou K, Zhang L, Zheng X, Zhao B, Sui X, Xu W, Lu S, Guo Q (2012) Immune characterization of mesenchymal stem cells in human umbilical cord Wharton’s jelly and derived cartilage cells. Cell Immunol 278:35–44
Santiago LY, Nowak RW, Peter Rubin J, Marra KG (2006) Peptide-surface modification of poly(caprolactone) with laminin-derived sequences for adipose-derived stem cell applications. Biomaterials 27:2962–2969
Zhou J, Lu P, Ren H, Zheng Z, Ji J, Liu H, Jiang F, Ling S, Heng BC, Hu X, Ouyang H (2014) 17β-Estradiol protects human eyelid-derived adipose stem cells against cytotoxicity and increases transplanted cell survival in spinal cord injury. J Cell Mol Med 18:326–343
Solari C, Losino N, Luzzani C, Waisman A, Bluguermann C, Questa M, Sevlever G, Miriuka S, Barañao L, Guberman A (2011) Induced pluripotent stem cells’ self-renewal and pluripotency is maintained by a bovine granulosa cell line-conditioned medium. Biochem Biophys Res Commun 410:252–257
Li LF, Liu YY, Yang CT, Chien Y, Twu NF, Wang ML, Wang CY, Huang CC, Kao KC, Hsu HS, Wu CW, Chiou SH (2013) Improvement of ventilator-induced lung injury by IPS cell-derived conditioned medium via inhibition of PI3 K/Akt pathway and IP-10-dependent paracrine regulation. Biomaterials 34:78–91
Firas J, Liu X, Nefzger CM, Polo JM (2014) GM-CSF and MEF-conditioned media support feeder-free reprogramming of mouse granulocytes to iPS cells. Differentiation 87:193–199
Kitazawa A, Shimizu N (2011) Differentiation of mouse induced pluripotent stem cells into neurons using conditioned medium of dorsal root ganglia. N Biotechnol 28:326–333
Kasuda S, Tatsumi K, Sakurai Y, Kato J, Taminishi S, Takeda T, Ohashi K, Okano T, Hatake K, Shima M (2011) Expression of coagulation factors from murine induced pluripotent stem cell-derived liver cells. Blood Coagul Fibrinolysis 22:271–279
Lin J, Fernandez I, Roy K (2011) Development of feeder-free culture systems for generation of ckit + sca1 + progenitors from mouse iPS cells. Stem Cell Rev 7:736–747
Pluchino S, Cossetti C (2013) How stem cells speak with host immune cells in inflammatory brain diseases. Glia 61:1379–1401
Ribeiro CA, Fraga JS, Grãos M, Neves NM, Reis RL, Gimble JM, Sousa N, Salgado AJ (2012) The secretome of stem cells isolated from the adipose tissue and Wharton jelly acts differently on central nervous system derived cell populations. Stem Cell Res Ther 3:18
Ranganath SH, Levy O, Inamdar MS, Karp JM (2012) Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 10:244–258
Zhang Y, Wang D, Cao K, Chen M, Yang X, Tao Y (2014) Rat induced pluripotent stem cells protect H9C2 cells from cellular senescence via a paracrine mechanism. Cardiology 128(1):43–50
Neel S, Singla DK (2011) Induced pluripotent stem (iPS) cells inhibit apoptosis and fibrosis in streptozotocin-induced diabetic rats. Mol Pharm 8:2350–2357
Zhang Y, Deng C, Qian J, Zhang M, Li X (2014) Improvement of radiotherapy-induced lacrimal gland injury by induced pluripotent stem cell-derived conditioned medium via MDK and inhibition of the p38/JNK pathway. Int J Mol Sci 15:18407–18421
Zeng G, Lai K, Li J, Zou Y, Huang H, Liang J, Tang X, Wei J, Zhang P (2013) A rapid and efficient method for primary culture of human adipose-derived stem cells. Organogenesis 9:287–295
Vishnubalaji R, Al-Nbaheen M, Kadalmani B, Aldahmash A, Ramesh T (2012) Comparative investigation of the differentiation capability of bone-marrow- and adipose-derived mesenchymal stem cells by qualitative and quantitative analysis. Cell Tissue Res 347:419–427
Cai J, Li W, Su H, Qin D, Yang J, Zhu F, Xu J, He W, Guo X, Labuda K, Peterbauer A, Wolbank S, Zhong M, Li Z, Wu W, So KF, Redl H, Zeng L, Esteban MA, Pei D (2010) Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells. J Biol Chem 285:11227–11234
Zhao Z, Yu R, Yang J, Liu X, Tan M, Li H, Chen J (2012) Maxadilan prevents apoptosis in iPS cells and shows no effects on the pluripotent state or karyotype. PLoS ONE 7:e33953
Guo X, Lian R, Guo Y, Liu Q, Ji Q, Chen J (2015) bFGF and Activin A function to promote survival and proliferation of single iPS cells in conditioned half-exchange mTeSR1 medium. Hum Cell 28:122–132
Unek G, Ozmen A, Mendilcioglu I, Simsek M, Korgun ET (2014) The expression of cell cycle related proteins PCNA, Ki67, p27 and p57 in normal and preeclamptic human placentas. Tissue Cell 46:198–205
Zhang Y, Wang Y, Wang L, Zhang Y, Qin Y, Chen T, Han W, Chen G (2012) Effects of Rehmannia glutinosa oligosaccharide on human adipose-derived mesenchymal stem cells in vitro. Life Sci 91:1323–1327
Li Y, Yang X, Nie FF, Zhao X, Qin ZL, Li JN (2013) Biological characteristics of human adipose-derived stem cells and their response to periostin in vitro. Chin Med J (Engl) 126:1491–1497
Minonzio G, Corazza M, Mariotta L, Gola M, Zanzi M, Gandolfi E, De Fazio D, Soldati G (2014) Frozen adipose-derived mesenchymal stem cells maintain high capability to grow and differentiate. Cryobiology 69:211–216
Freimark D, Pino-Grace P, Pohl S, Weber C, Wallrapp C, Geigle P, Pörtner R, Czermak P (2010) Use of encapsulated stem cells to overcome the bottleneck of cell availability for cell therapy approaches. Transfus Med Hemother 37:66–73
Renzi S, Lombardo T, Dotti S, Dessì SS, De Blasio P, Ferrari M (2012) Mesenchymal stromal cell cryopreservation. Biopreserv Biobank 10:276–281
Zhu Y, Liu T, Ye H, Song K, Ma X, Cui Z (2010) Enhancement of adipose-derived stem cell differentiation in scaffolds with IGF-I gene impregnation under dynamic microenvironment. Stem Cells Dev 19:1547–1556
Yan A, Avraham T, Zampell JC, Haviv YS, Weitman E, Mehrara BJ (2011) Adipose-derived stem cells promote lymphangiogenesis in response to VEGF-C stimulation or TGF-β1 inhibition. Future Oncol 7:1457–1473
Shoji T, Ii M, Mifune Y, Matsumoto T, Kawamoto A, Kwon SM, Kuroda T, Kuroda R, Kurosaka M, Asahara T (2010) Local transplantation of human multipotent adipose-derived stem cells accelerates fracture healing via enhanced osteogenesis and angiogenesis. Lab Invest 90:637–649
Zhu M, Feng Y, Dangelmajer S, Guerrero-Cázares H, Chaichana KL, Smith CL, Levchenko A, Lei T, Quiñones-Hinojosa A (2015) Human cerebrospinal fluid regulates proliferation and migration of stem cells through insulin-like growth factor-1. Stem Cells Dev 24:160–171
Sobral LM, Bufalino A, Lopes MA, Graner E, Salo T, Coletta RD (2011) Myofibroblasts in the stroma of oral cancer promote tumorigenesis via secretion of activin A. Oral Oncol 47:840–846
Licona-Limón P, Alemán-Muench G, Chimal-Monroy J, Macías-Silva M, García-Zepeda EA, Matzuk MM, Fortoul TI, Soldevila G (2009) Activins and inhibins: novel regulators of thymocyte development. Biochem Biophys Res Commun 381:229–235
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Drago D, Cossetti C, Iraci N, Gaude E, Musco G, Bachi A, Pluchino S (2013) The stem cell secretome and its role in brain repair. Biochimie 95:2271–2285
Gazdhar A, Grad I, Tamò L, Gugger M, Feki A, Geiser T (2014) The secretome of induced pluripotent stem cells reduces lung fibrosis in part by hepatocyte growth factor. Stem Cell Res Ther 5:123
Goldschmidt E, Hem S, Ajler P, Ielpi M, Loresi M, Giunta D, Carrizo A, Yampolsky C, Argibay P (2013) A new model for dura mater healing: human dural fibroblast culture. Neurol Res 35:300–307
Varodayan FP, Zhu XJ, Cui XN, Porter BE (2009) Seizures increase cell proliferation in the dentate gyrus by shortening progenitor cell-cycle length. Epilepsia 50:2638–2647
Auld CA, Morrison RF (2006) Evidence for cytosolic p27(Kip1) ubiquitylation and degradation during adipocyte hyperplasia. Obesity (Silver Spring) 14:2136–2144
Nagata S (1997) Apoptosis by death factor. Cell 88:3553–3565
Feng R, Han J, Ziegler J, Yang M, Castranova V (2012) Apaf-1 deficiency confers resistance to ultraviolet-induced apoptosis in mouse embryonic fibroblasts by disrupting reactive oxygen species amplification production and mitochondrial pathway. Free Radic Biol Med 52:889–897
Rotem-Dai N, Oberkovitz G, Abu-Ghanem S, Livneh E (2009) PKCeta confers protection against apoptosis by inhibiting the pro-apoptotic JNK activity in MCF-7 cells. Exp Cell Res 315:2616–2623
Chiou HL, Hsieh YS, Hsieh MR, Chen TY (2006) HCV E2 may induce apoptosis of Huh-7 cells via a mitochondrial-related caspase pathway. Biochem Biophys Res Commun 345:453–458
Sobral LM, Bufalino A, Lopes MA, Graner E, Salo T, Coletta RD (2011) Myofibroblasts in the stroma of oral cancer promote tumorigenesis via secretion of activin A. Oral Oncol 47(9):840–846
Yu J, Ye J, Liu X, Han Y, Wang C (2011) Protective effect of l-carnitine against H2O2-induced neurotoxicity in neuroblastoma (SH-SY5Y) cells. Neurol Res 33:708–716
Glover KP, Markell LK, Donner EM, Han X (2014) Protein kinase C-activating tumor promoters modulate the DNA damage response in UVC-irradiated TK6 cells. Toxicol Lett 229:210–219
Kim HS, Lee JH, Park HS, Lee GS, Kim HW, Ha KT, Kim BJ (2015) Schizandra chinensis extracts induce apoptosis in human gastric cancer cells via JNK/p38 MAPK activation and the ROS-mediated/mitochondria-dependent pathway. Pharm Biol 53:212–219
Chathoth S, Thayyullathil F, Hago A, Shahin A, Patel M, Galadari S (2009) UVC-induced apoptosis in Dubca cells is independent of JNK activation and p53(Ser-15) phosphorylation. Biochem Biophys Res Commun 383:426–432
Fritsch RM, Schneider G, Saur D, Scheibel M, Schmid RM (2007) Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J Biol Chem 282:22551–22562
Kostic I, Toffoletto B, Toller M, Beltrami CA, Ambesi-Impiombato FS, Curcio F (2010) UVC radiation-induced effect on human primary thyroid cell proliferation and HLA-DR expression. Horm Metab Res 42:846–853
Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 68:383–424
Li Y, Zhang JF, Zhang YM, Ma XB (2009) The protective effect of genistein postconditioning on hypoxia/reoxygenation-induced injury in human gastric epithelial cells. Acta Pharmacol Sin 30:576–581
Yuan Y, Peng C, Li K, Hussain M, Sikharam C, Guthikonda M, Ding Y (2012) Ethanol reduces expression of apoptotic proteins after hypoxia/reoxygenation in a brain slice model. Neurol Res 34(4):373–378
Wang Z, An LJ, Duan YL, Li YC, Jiang B (2008) Catalpol protects rat pheochromocytoma cells against oxygen and glucose deprivation-induced injury. Neurol Res 30:106–112
Qiu J, Zhou XY, Zhou XG, Cheng R, Liu HY, Li Y (2013) Neuroprotective effects of microRNA-210 on hypoxic-ischemic encephalopathy. Biomed Res Int 2013:350419
Pyle AD, Lock LF, Donovan PJ (2006) Neurotrophins mediate human embryonic stem cell survival. Nat Biotechnol 24:344–350
Ogawa T, Yogo K, Ishida N, Takeya T (2003) Synergistic effects of activin and FSH on hyperphosphorylation of Rb and G1/S transition in rat primary granulosa cells. Mol Cell Endocrinol 210:31–38
Yasuda M, Yamamoto M, Ochiai H, Eguchi Y, Arishima K (2007) Effects of growth factors on development of fetal islet B-cells in vitro. J Vet Med Sci 69:807–811
Krieglstein K, Suter-Crazzolara C, Fischer WH, Unsicker K (1995) TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP + toxicity. EMBO J 14:736–742
Komura M, Komura H, Konishi K, Ishimaru T, Hoshi K, Takato T, Tabata Y, Iwanaka T (2014) Promotion of tracheal cartilage growth by intra-tracheal injection of basic fibroblast growth factor (b-FGF). J Pediatr Surg 49:296–300
Feng Y, Dai X, Li X, Wang H, Liu J, Zhang J, Du Y, Xia L (2012) EGF signalling pathway regulates colon cancer stem cell proliferation and apoptosis. Cell Prolif 45:413–419
Carvalho KA, Guarita-Souza LC, Rebelatto CL, Senegaglia AC, Hansen P, Mendonca JG, Cury CC, Francisco JC, Brofman PR (2004) Aneural culture of rat myoblasts for myocardial transplant. Transpl Proc 36:1023–1024
Cetin N, Ball K, Gokden M, Cruz NF, Dienel GA (2003) Effect of reactive cell density on net [2-14C] acetate uptake into rat brain: labeling of clusters containing GFAP + - and lectin + -immunoreactive cells. Neurochem Int 42:359–374
Poon VK, Huang L, Burd A (2005) Biostimulation of dermal fibroblast by sublethal Q-switched Nd:YAG 532 nm laser: collagen remodeling and pigmentation. J Photochem Photobiol, B 81:1–8
Sakai T, Inoue S, Otsuka T, Matsuyama TA, Saito T, Murakami M, Ota H, Katagiri T (2004) Cell cycle regulator expression after coronary stenting in humans. Jpn Heart J 45:133–145
Menetrey J, Kasemkijwattana C, Day CS, Bosch P, Vogt M, Fu FH, Moreland MS, Huard J (2000) Growth factors improve muscle healing in vivo. J Bone Joint Surg Br 82:131–137
Sumino Y, Hirata Y, Sato F, Mimata H (2007) Growth mechanism of satellite cells in human urethral rhabdosphincter. Neurourol Urodyn 26:552–561
Grant MB, Wargovich TJ, Ellis EA, Caballero S, Mansour M, Pepine CJ (1994) Localization of insulin-like growth factor I and inhibition of coronary smooth muscle cell growth by somatostatin analogues in human coronary smooth muscle cells. A potential treatment for restenosis? Circulation 89:1511–1517
Martins P, Schmitt F, Almeida H, Frazão JM (2008) Evaluation of parathyroid gland angiogenesis in chronic kidney disease associated with secondary hyperparathyroidism. Nephrol Dial Transpl 23:2889–2894
Taléns-Visconti R, Bonora A, Jover R, Mirabet V, Carbonell F, Castell JV, Gómez-Lechón MJ (2007) Human mesenchymal stem cells from adipose tissue: differentiation into hepatic lineage. Toxicol In Vitro 21:324–329
Villageois P, Wdziekonski B, Zaragosi LE, Plaisant M, Mohsen-Kanson T, Lay N, Ladoux A, Peraldi P, Dani C (2011) Regulators of human adipose-derived stem cell self-renewal. Am J Stem Cells 1:42–47
Acknowledgments
This study was supported by National Natural Science Foundation of China (No. 81371689), collaborated grant for HK-Macao-TW of Ministry of Science and Technology (2012DFH30060) and Special Funds for Major Science and Technology Projects of Guangdong Province (2015B010125007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Rui-Ling Lian and Xiao-Ling Guo have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Lian, RL., Guo, XL., Chen, JS. et al. Effects of induced pluripotent stem cells-derived conditioned medium on the proliferation and anti-apoptosis of human adipose-derived stem cells. Mol Cell Biochem 413, 69–85 (2016). https://doi.org/10.1007/s11010-015-2640-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2640-7