
Abstract
Toll-like receptor 4 (TLR4) signaling requires a number of accessory proteins to initiate a signal. MD-2 is one of the accessory proteins with a relevant role in lipopolysaccharide responses. Although cigarette smoke increases TLR4 expression, TLR4 signaling is altered in smokers and in smokers COPD patients. The main aims of this study were to explore whether MD2 is altered in large and small airways of COPD and of smokers without COPD. The expression of MD2 ex vivo was assessed by immunohistochemistry in surgical specimens from current smokers COPD (s-COPD; n = 14), smokers without COPD (S; n = 7), and from non-smoker non-COPD subjects (C; n = 11. The in vitro effects of cigarette smoke extracts on the MD2 expression in a human bronchial epithelial cell line (16-HBE) were also assessed by flow cytometry. MD2 is reduced in the epithelium and in the submucosa in large airways but not in the epithelium and in the submucosa in small airways of smokers and of s-COPD. The expression of MD2 in the submucosa of the large airways is significantly higher in comparison to the submucosa of the small airways in all the studied groups. In vitro, cigarette smoke is able to increase TLR4 but it reduces MD2 in a dose-dependent manner in bronchial epithelial cells. Cigarette smoke may alter innate immune responses reducing the expression of the MD2, a molecule with an important role in TLR4 signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The respiratory tract is one of the primary routes of entry into the body for invading pathogens. Toll-like receptor 4 (TLR4) is the primary receptor for recognition of lipopolysaccharide (LPS), found in gram-negative bacteria, and was the first mammalian TLR to be characterized [1]. TLR4 signaling requires a number of accessory proteins to initiate a signal [1]. A key component of the TLR4 receptor cluster is Myeloid differentiation-2 (MD-2). MD-2 is a protein that binds to the extracellular domain of TLR4 and is essential for LPS recognition [2]. Under resting conditions, TLR4 cycles between the Golgi and the plasma membrane and translocates to the cell surface upon LPS exposure [3]. The translocation of TLR4 to the cell membrane was reported to depend on glycosylation, for which MD-2 was required [4, 5]. On epithelial cells, soluble MD2 binds to TLR4 and increases TLR4 levels at the cell surface by preventing TLR4 turnover through the endocytic pathway. Thus, sMD2 may prime epithelial cells for enhanced immunoresponsive function prior to LPS exposure [6].
The airway epithelium is active in airway defense mechanisms by releasing cytoprotective mucus and defensins and exerts an important role in coordinating local inflammation and immune responses. The exposure to cigarette smoke, the major risk factor for chronic obstructive pulmonary disease (COPD), alters innate immune responses of the bronchial airway epithelium. In a LPS-stimulated bronchial epithelial cell line, cigarette smoke further increases the expression of TLR4 and orientates the activation of TLR4 toward a reduced release of IP-10 with a preferential activation of the ERK pathway [7, 8]. These events lead to an increased neutrophil chemotaxis and to a reduced lymphocyte chemotaxis thus altering the balance between innate and adaptative responses generated by the epithelium upon microbial stimulation [7, 8]. Although smokers and COPD smokers have an increased expression of TLR4 in the epithelium of both large and small airways [9], COPD smokers have a reduced expression of human beta defensin 2 (HBD2) in large airways. Cigarette smoke reduces HBD2 expression and releases in bronchial epithelial cells [9].
It is unknown whether in smokers and in COPD, the expression of MD2 is altered in large and in small airways and which is the effect of cigarette smoke on MD2 expression.
The aims of the present study were to assess the expression of MD2 in lung tissue samples from smokers and COPD smokers and to evaluate in vitro the effects of CSE on MD2 expression in bronchial epithelial cells.
Materials and methods
Study population
The study population of this study was recruited at Istituto Mediterraneo per i Trapianti e Terapie ad Alta Specializzazione (ISMETT) in Palermo, Italy. All recruited patients underwent surgery for lung cancer. The study protocol was approved by the ISMETT Ethic Committee (#149311-29/05/2006) and informed written consent was obtained from each patient. The patients were recruited from September 2006 to January 2008 and classified into the following groups as previously reported [9]: (1) Non-smoking patients without chronic pulmonary underlying events (Controls) (n = 11); (2) current smoking patients (>15 pack/year) without COPD (S) (n = −7); and (3) current smoking patients (>15 pack/year) with stable moderate COPD (s-COPD) (n = 14). COPD patients were treated with bronchodilators. The patients were not under corticosteroid therapy (neither inhaled nor systemic) and not under theophylline or antibiotics and did not have exacerbations during the month preceding the study. All the COPD patients included in the study were classified on the basis of preoperative lung function test: FEV1/FVC < 70 %; 50 % ≤ FEV1 < 80 % predicted and broncho-dilatation reversibility less than 12 %. The following variables at admission were recorded: age, gender, and smoking habits (Table 1).
Histology
Tissue specimens from central bronchi or sub-pleural parenchyma samples were taken away from the tumor site, fixed with 10 % Neutral buffer formalin, and embedded in paraffin wax. A pathologist examined the lung parenchyma and confirmed that they were tumor free and surrounded by tumor-free tissue. Deparaffinised sequential sections (4-µm thick) were stained with hematoxylin and eosin (HE) and used for immunohistochemistry [10]. MD-2 was assessed in large (internal perimeter > 6 mm) and small (internal perimeter < or = 6 mm) airways.
The following antibody was used: rabbit anti-human polyclonal anti-TLR4 (Santa Cruz; sc-10741) and anti-MD2 antibody (Santa Cruz; sc-20668) (1:10 overnight). The reaction was revealed by using the Avidin–Biotin technique (AP-LSAB2 KIT; DAKO). An antibody diluent (DAKO) and irrelevant antibody (Santa Cruz Biotechnology) of the same isotype were used as negative controls.
The cell nuclei were stained for 1 min with Hematoxylin (DAKO). For distal airways examination, at least four non-cartilaginous peripheral airways with an internal perimeter less than 6 mm were selected for each patient. To avoid measurements in tangentially cut airways, bronchioles with a short/long diameter ratio less than one-third were excluded from the study. In each airway, we measured the internal perimeter along the subepithelial basement membrane and the luminal diameter as the greater distance in a plane perpendicular to the long axis of the lumen [11, 12].
The immunoreactivity in the epithelium of large and small airways [9, 13–15] was evaluated blindly by two independent investigators (M.F; G. C.) and was assessed as positive epithelial cells/mm basement membrane. The immunoreactivity in the submucosa was evaluated as previously described [16]. Results are expressed as medians and 25–75 percentiles [9, 14, 15].
Preparation of cigarette smoke extracts (CSE)
Commercial cigarettes (Marlboro) were used in this study. Cigarette smoke solution was prepared as described previously [7]. Each cigarette was smoked for 5 min and one cigarette was used per 10 ml of PBS to generate a CSE-PBS solution. The CSE solution was filtered through a 0.22-μm pore filter to remove bacteria and large particles. The smoke solution was then adjusted to pH 7.4 and used within 30 min of preparation. This solution was considered to be 100 % CSE and diluted to obtain the desired concentration in each experiments. The concentration of CSE was calculated spectrophotometrically measuring the OD as previously described [17] at the wavelength of 320 nm. The pattern of absorbance, among different batches, showed very little differences and the mean OD of the different batches was 1.37 ± 0.16. The presence of contaminating LPS on undiluted CSE was assessed by a commercially available kit (Cambrex Corporation, East Rutherford, New Jersey, USA) and was below the detection limit of 0.1 EU/ml.
Bronchial epithelial cell cultures
16-HBE, an immortalized normal bronchial epithelial cell line [18], was used in this study. Bronchial epithelial cells were maintained in MEM (Gibco, BRL, Germany), supplemented with 10 % fetal calf serum (Gibco). Cell cultures were maintained in a humidified atmosphere of 5 % CO2 in air at 37 °C. Cells were cultured in the presence and absence of CSE (2.5, 5, and 10 %) for 24 h. In some experiments, bronchial epithelial cells were cultured with both CSE 10 % and LPS (1 μg/ml) (Sigma-Aldrich Corporate, St Louis, MO) for 24 h. At the end of stimulation, cells were collected for further flow cytometry evaluations.
Flow cytometry
The expression of MD2 in 16-HBE cells was evaluated as previously described [19] by flow cytometry using a FACS Calibur (Becton–Dickinson, Mountain View, CA). To evaluate the expression of MD-2, cells were fixed with PBS containing 4 % paraformaldehyde for 20 min at room temperature. Fixed cells were washed twice in permeabilization buffer (PBS containing 1 % FBS, 0.3 % saponin, and 0.1 % Na azide) for 5 min at 4 °C and then incubated with a rabbit polyclonal anti-MD2 antibody (Santa Cruz; sc-20668) (1:100) for 30’, followed by a FITC conjugated anti-rabbit IgG (Dako, Glostrup, Denmark). Negative controls were performed using rabbit immunoglobulins negative control (Dako). Data are expressed as percentage of positive cells.
Statistics
Data are expressed as medians and inter-quartile range or as mean counts ± SD. A non-parametric Mann–Whitney test was applied for comparisons between the two subject groups. For in vitro experiments, the differences between different experimental conditions were evaluated by non-parametric Wilcoxon test. P < 0.05 was accepted as statistically significant.
Results
Demographic characteristics of the subjects
The demographic characteristics and the functional evaluations of the studied groups are shown in Table 1. All recruited patient groups were similar with regard to age. The number of packs/year was similar between S and s-COPD (Table 1).
Expression of MD2 in the epithelium and in submucosa of large and small airways
TLR4 signaling requires a number of accessory proteins to initiate a signal. One of the accessory proteins with a crucial role for LPS recognition is represented by MD2 [2].
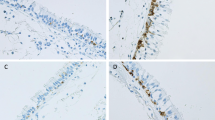
Since TLR4 expression is increased in the epithelium of both large and small airways in S and in s-COPD [9], in the present study, we assessed the expression of MD2 in large and small airways from controls, S and s-COPD. MD2 expression is reduced in the epithelium (Fig. 1a, b) and in the submucosa (Fig. 2a, b) in large airways of smokers and of s-COPD in comparison to controls. MD2 expression is not reduced in both the epithelium (Fig. 3a, b) and the submucosa (Fig. 4a, b) in small airways of smokers and of s-COPD in comparison to the controls. The expression of MD2 in the submucosa of the large airways was significantly higher than in the submucosa of the small airways in all the studied groups (large airways: median 17.5; small airways: median 0.1; P < 0.002). The expression of MD2 in the submucosa was mainly concentrated on inflammatory cells. TLR4 expression is increased in the epithelium in large airways in smokers and s-COPD in comparison to controls [Controls = 14(8–19); Smokers = 30(26–45); s-COPD 28(17–47)].

MD2 expression is reduced in the epithelium of large airways. Immunohistochemistry for MD2 in large airways from surgical samples of Controls (n = 11), S (n = 7), and s-COPD (n = 14) subjects. Cells were stained with an anti-MD2 antibody. Negative controls were performed using rabbit immunoglobulins negative control (see Materials and Methods for details). a Individual counts for the number of positive epithelial cells/mm basement membrane in large airways. Horizontal bars represent median values. *P < 0.05 values in figure represent Mann–Whitney U test analyses. b Representative MD2 immunostaining (red stain) in large airways of a Control, of a S, and of a s-COPD. ×400 magnification

MD2 expression is reduced in the submucosa of large airways. Immunohistochemistry for MD2 in large airways from surgical samples of Controls (n = 11), S (n = 7), and s-COPD (n = 14) subjects. Cells were stained with an anti-MD2 antibody. Negative controls were performed using rabbit immunoglobulins negative control (see Materials and Methods for details). a Individual counts for the number of positive cells/mm2 area. Horizontal bars represent median values. *P < 0.05 values in figure represent Mann–Whitney U test analyses. b Representative MD2 immunostaining (red stain) in submucosa of large airways of a Control, of a S, and of a s-COPD. ×400 magnification

MD2 expression is not reduced in the epithelium of small airways. Immunohistochemistry for MD2 in small airways from surgical samples of Controls (n = 11), S (n = 7), and s-COPD (n = 14) subjects. Cells were stained with an anti-MD2 antibody. a Individual counts for the number of positive epithelial cells/mm basement membrane in small airways. Horizontal bars represent median values. b Representative MD2 immunostaining (red stain) in small airways of a Control, of a S, and of a s-COPD. ×400 magnification

MD2 expression is not reduced in the submucosa of small airways. Immunohistochemistry for MD2 in small airways from surgical samples of Controls (n = 11), S (n = 7), and s-COPD (n = 14) subjects. Cells were stained with an anti-MD2 antibody. a Individual counts for the number of positive cells/mm2 area. Horizontal bars represent median values. b Representative MD2 immunostaining (red stain) in submucosa of small airways of a Control, of a S, and of a s-COPD. ×400 magnification
Effect of cigarette smoke exposure on bronchial epithelial cells
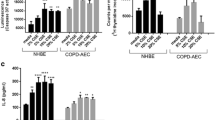
To assess the specific role of cigarette smoke on the MD2 expression, the effect of CSE on a bronchial epithelial cell line was tested. In in vitro dose response experiments, cigarette smoke was able to reduce MD2 expression in a dose-dependent manner (Fig. 5), and CSE 10 % was able to significantly reduce MD2 expression on bronchial epithelial cells (Figs. 6, 7) and was able to increase, in parallel, TLR4 while it reducing MD2 (Fig. 7), suggesting that cigarette smoke exposure promotes the expression of a not fully functional TLR4 in bronchial epithelial cells. Finally, we explored the effects of the agonist of TLR4, the LPS, on MD2 expression in bronchial epithelial cells exposed to CSE. LPS alone did not significantly modify both the constitutive expression and the CSE-modified MD2 expression in bronchial epithelial cells (Baseline = 71 ± 15; CSE = 63 ± 14; LPS = 66 ± 14; LPS + CSE 10 % = 66 ± 13).

CSE is able to reduce MD2 expression in a dose-dependent manner in bronchial epithelial cells. 16-HBE cells were cultured in the presence and in the absence of different concentrations of CSE (2.5, 5, and 10 %) for 24 h and were used for assessing MD2 expression. The expression of MD2 was evaluated by flow cytometry. Representative histogram plots of the MD2 expression from two different experiments are shown

CSE 10 % is able to significantly reduce MD2 expression in bronchial epithelial cells. 16-HBE cells were cultured in the presence and in the absence of CSE 10 % for 18 h and were used for assessing MD2 expression (n = 7). The expression of MD2 was evaluated by flow cytometry and the results are expressed as Percentage of positive cells (means ± SD). *P < 0.05

CSE 10 % is able to reduce MD2 expression while it increases TLR4 expression in bronchial epithelial cells. 16-HBE cells were cultured in the presence and in the absence of CSE 10 % for 18 h and were used for assessing MD2 and TLR4 expression. Representative histogram plots are shown
Discussion
Cell surface and endosomal TLRs constitute one of the major cell receptors involved in innate immunity of the lung mediating immediate immune responses against microbial pathogens.
TLR4, one of the most extensively studied TLRs, is a type I transmembrane protein with an extracellular domain of leucine-rich repeats that provides, via the activation of MD2, the recognition of LPS, the invariant virulence factor of Gram-negative bacteria [1]. LPS-induced cell responses are tightly regulated via distinct pathways including subcellular TLR4 localization and thus ligand sensing [20]. The role of lung cell-specific micro-environmental factors in regulating expression and intracellular TLR4 localization in response to LPS is largely unknown.
The present study investigated for the first time the expression of MD2 in smokers and in s-COPD and the specific role of cigarette smoke exposure on MD2 expression in a model of airway epithelial cells.
MD-2 was first described by Shimazu et al. [2], documenting the need for assembling of MD-2 and TLR4 for cells to be LPS sensitive. Studies of the receptor structure have demonstrated that MD-2 binds directly to LPS causing an allosteric change that facilitates its binding to a second TLR4 protein thus forming a receptor dimer [21, 22]. These studies also suggest that LPS does not directly bind to the TLR4 receptor, thus explaining the obligate requirement for MD-2. In contrast to CD14, MD-2 is selectively associated with TLR4 and, in contrast to CD14, does not transmit signals from danger-associated molecular patterns [23].
TLRs establish the inflammatory setting in response to infections or tissue damage and provide a low-grade activation of the innate immune system for day-to-day lung structure stability [24, 25]. High grade activation of TLR signaling, leading to increased production of cytokines and reactive oxidant, contributes to experimental emphysema [26].
MD-2 is found both as a membrane bound and soluble protein and binds to TLR4 to assemble the functional receptor.
Under resting conditions, TLR4 cycles between the Golgi and the plasma membrane and translocates to the cell surface upon LPS exposure [3]. Trafficking of TLR4 from the Golgi to the plasma membrane is regulated by the small TLR4-associated glycoprotein MD2 [4, 5]. However, in the present study, LPS exposure is not able to modify both the constitutive expression and the CSE-modified MD2 expression in bronchial epithelial cells.
In this regard, it has been demonstrated that abnormal TLR4 trafficking leads to decreased TLR4 degradation, which affects down-regulation of the proinflammatory state [27]. Here it has been demonstrated that MD2 expression is reduced in the epithelium and in the submucosa in large airways but not in small airways in smokers and in s-COPD in comparison to controls. Differently, in large airways, TLR4 is increased in the epithelium in smokers and in s-COPD in comparison to controls. Although the predominant pathology is present in small airways and lung parenchyma in COPD [28], the contribution of the alterations within the large airways is becoming increasingly important. In large airways, ciliated cells, goblet cells, and submucosal glands produce mucus that traps inhaled pathogens and other particulate material. Then coordinated beating of cilia sweeps the trapped material from more distally located airways toward proximal, a defense mechanism known as mucociliary escalator [29]. Cigarette smoke interferes with the innate host defense system by increasing mucus production, reducing mucociliary clearance, disrupting the epithelial barrier, and stimulating the migration of inflammatory and immune cells. The data of the present study further support the concept that a defect in the innate immune responses in the large airways of smokers may predispose to the development of COPD. Cigarette smoke exposure increases TLR4 expression but reduces MD-2 expression in bronchial epithelial cells. In this regard, it has been previously demonstrated that although TLR4 expression is increased, HBD2 expression is reduced in the epithelium of large airways and its expression positively correlates with FEV1/FVC ratio and inversely correlates with the cigarette smoke exposure [9] in smokers with COPD.
Cigarette smoke extracts reduce the expression of HBD2 in primary bronchial epithelial cells from smokers and from COPD patients [30] and, in bronchial epithelial cells, block the induction of HBD2 mRNA generated by exposure to IL-1 beta, a cytokine with a crucial role in the inflammation of COPD [9]. On the other hand, cigarette smoke extracts increase the expression of TLR4 in a bronchial epithelial cell line [7, 8] and orientate the activation of TLR4 toward an increased IL-8 release and a reduced IP-10 release leading to an increased neutrophil chemotaxis and to a reduced lymphocyte chemotaxis, thus altering the balance between innate and adaptative responses [7, 8]. This unbalance may amplify lung inflammation since lung inflammation can be excessive when the adaptive pulmonary immune responses are inappropriate [31]. The MD2 expression in the submucosa was significantly higher in large airways than (P < 0.002) in small airways in all the studied groups. Taken together, these findings suggest that physiologically the innate immune responses are highly activated in the upper airways to block pathogens at this level, thus protecting lower airways and lung parenchyma from infections and from excessive inflammation.
As consequence, the defect in large airways may activate an excessive activation of the innate immune responses in the small airways and in the lung parenchyma, thus amplifying the inflammatory responses at this level. Physiologically, the distal airways are sterile while the airways of COPD patients are chronically colonized by potential respiratory pathogens [32]. Pneumonia caused by Gram-negative bacteria is a major cause of morbidity and mortality in humans with an increasing prevalence of community-acquired and early-onset ventilator-associated pneumonia [33]. Chronic bacterial colonization together to an oxidant/antioxidant unbalance can stimulate the host immune system and cause a chronic airway inflammation [34] that in turn may promote the tissue damage observed in distal airways and lung parenchyma of COPD patients. Bronchiolar inflammation correlates with functional impairment and temporally precedes emphysema [35]. The inflammatory processes promote the structural and functional changes associated with chronic bronchitis in the larger bronchi [36] while in the smaller bronchi and bronchioles, they cause the occlusion of the lumen by mucus, thickening of the walls, and narrowing of the lumen [28].
In conclusions, cigarette smoke exposure in smokers as well as in COPD patients alters the physiological activation of innate immune responses in large airways increasing TLR4 but not the accessory protein MD2 with a crucial role in LPS responses. The altered immune competence of large airways may increase inflammation in small airways and lung parenchyma. All these events may contribute to increase the risk of infections and exacerbations in COPD patients, thus promoting disease progression.
Abbreviations
- TLR4:
-
Toll-like receptor 4
- COPD:
-
Chronic obstructive pulmonary disease
- CSE:
-
Cigarette smoke extracts
- LPS:
-
Lipopolysaccharide
- MD-2:
-
Myeloid differentiation-2
References
Sender V, Stamme C (2014) Lung cell-specific modulation of LPS-induced TLR4 receptor and adaptor localization. Commun Integr Biol 7:e29053
Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782
Latz E, Visintin A, Lien E, Fitzgerald KA, Monks BG, Kurt-Jones EA, Golenbock DT, Espevik T (2002) Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem 277:47834–47843
Ohnishi T, Muroi M, Tanamoto K (2001) N-linked glycosylations at Asn(26) and Asn(114) of human MD-2 are required for toll-like receptor 4-mediated activation of NF-κB by lipopolysaccharide. J Immunol 167:3354–3359
Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K (2002) Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol 3:667–672
Lauer S, Kunde YA, Apodaca TA, Goldstein B, Hong-Geller E (2009) Soluble MD2 increases TLR4 levels on the epithelial cell surface. Cell Immunol 255:8–16
Pace E, Ferraro M, Siena L, Melis M, Montalbano AM, Johnson M, Bonsignore MR, Bonsignore G, Gjomarkaj M (2008) Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology 124:401–411
Pace E, Ferraro M, Uasuf CG, La Grutta S, Liotta G, Giarratano A, Johnson M, Gjomarkaj M (2011) Cilomilast counteracts the effects of cigarette smoke in innate responses of airway epithelial cells. Cell Immunol 268:47–53
Pace E, Ferraro M, Minervini MI, Vitulo P, Pipitone L, Vitulo G, Chiappara P, Siena L, Montalbano AM, Johnson M, Gjomarkaj M (2012) Beta defensin-2 is reduced in central but not in distal airways of smoker COPD patients. PLoS ONE 7:e33601
Chiappara G, Chanez P, Bruno A, Pace E, Pompeo F, Bousquet J, Bonsignore G, Gjomarkaj M, Bousquet J (2007) p-CREB expression depicts different asthma phenotype. Allergy 62:787–794
Saetta M, Turato G, Baraldo S, Zanin A, Braccioni F, Mapp EC, Maestrelli P, Cavallesco G, Papi A, Fabbri ML (2000) Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with both symptoms of chronic bronchitis and chronic airflow limitation. Am J Respir Crit Care Med 161:1016–1021
Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L et al (1998) CD8 + T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 157:822–826
Miotto D, Hollenberg MD, Bunnett NW, Papi A, Braccioni F et al (2002) Expression of protease activated receptor-2 (PAR-2) in central airways of smokers and non-smokers. Thorax 57:146–151
Chiappara G, Gjomarkaj M, Virzì A, Sciarrino S, Ferraro M, Bruno A, Montalbano AM, Vitulo P, Minervini MI, Pipitone L, Pace E (2013) The role of p21 Waf1/Cip1 in large airway epithelium in smokers with and without COPD. Biochim Biophys Acta 1832:1473–1481
Chiappara G, Gjomarkaj M, Sciarrino S, Vitulo P, Pipitone L, Pace E (2014) Altered expression of p21, activated caspase-3, and PCNA in bronchiolar epithelium of smokers with and without chronic obstructive pulmonary disease. Exp Lung Res 40:343–353
Siena L, Gjomarkaj M, Elliot J, Pace E, Bruno A, Baraldo S, Saetta M, Bonsignore MR, James A (2011) Reduced apoptosis of CD8+ T-lymphocytes in the airways of smokers with mild/moderate COPD. Respir Med 105:1491–1500
Luppi F, Aarbiou J, van Wetering S, Rahman I, de Boer WI, Rabe KF, Hiemstra PS (2005) Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respir Res 6:140
Cozens AL, Yezzi MJ, Yamaya M, Steiger D, Wagner JA, Garber SS, Chin L, Simon EM, Cutting GR, Gardner P (1992) A transformed human epithelial cell line that retains tight junctions post crisis. In Vitro Cell Dev Biol 28:735–744
Pace E, Di Sano C, Sciarrino S, Scafidi V, Ferraro M, Chiappara G, Siena L, Gangemi S, Vitulo P, Giarratano A, Gjomarkaj M (2014) Cigarette smoke alters IL-33 expression and release in airway epithelial cells. Biochim Biophys Acta 1842:1630–1637
Sender V, Stamme C (2014) Lung-cell specific modulation of LPS-induced TLR4 receptor and adaptor localization. Commun Integr Biol 7:e29053
Kim HM, Park BS, Kim JI, Kim SE, Lee J et al (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130:906–917
Ohto U, Fukase K, Miyake K, Satow Y (2007) Crystal structures of human MD-2 and its complex with anti endotoxic lipid IVa. Science 316:1632–1634
Chun KH, Seong SY (2010) CD14 but not MD2 transmit signals from DAMP. Int Immunopharmacol 10:98–106
Droemann D, Goldmann T, Tiedje T, Zabel P, Dalhoff K et al (2005) Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir Res 6:68
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW (2005) Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11:1173–1179
Zhang X, Shan P, Jiang G, Cohn L, Lee PJ (2006) Toll-like receptor 4 deficiency causes pulmonary emphysema. J Clin Investig 116:3050–3059
Bruscia EM, Zhang PX, Satoh A, Caputo C, Medzhitov R, Shenoy A, Egan ME, Krause DS (2011) Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J Immunol 186:6990–6998
Hogg JC (2004) Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 364:709–721
Ganesan S, Comstock AT, Sajjan US (2013) Barrier function of airway tract epithelium. Tissue Barriers 1:e24997
Zhang W, Case S, Bowler RP, Martin RJ, Jiang D et al (2011) () Cigarette smoke modulates PGE(2) and host defence against Moraxella catarrhalis infection in human airway epithelial cells. Respirology 16:508–516
Curtis JL, Freeman CM, Hogg JC (2007) The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc 4:512–521
Sethi S, Murphy TF (2001) Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin Microbiol Rev 14:336–363
Koulenti D, Rello J (2006) Gram-negative bacterial pneumonia: aetiology and management. Curr Opin Pulm Med 12:198–204
Pace E, Giarratano A, Ferraro M, Bruno A, Siena L et al (2011) TLR4 upregulation underpins airway neutrophilia in smokers with chronic obstructive pulmonary disease and acute respiratory failure. Hum Immunol 72:54–62
O’Donnell R, Breen D, Wilson S, Djukanovic R (2006) Inflammatory cells in the airways in COPD. Thorax 61:448–454
Kim V, Rogers TJ, Criner GJ (2008) New concepts in the pathobiology of chronic obstructive pulmonary disease. Proc Am Thorac Soc 5:478–485
Acknowledgments
This work was supported by the Italian National Research Council. Elisabetta Pace and Maria Ferraro designed the study. Elisabetta Pace performed the statistical analysis of the data, wrote the manuscript, and declares that she has had access to and takes responsibility for the integrity of the data and the accuracy of the data analysis. Maria Ferraro, Giuseppina Chiappara, and Serena Di Vincenzo performed the experiments of the study and participated to the interpretation of the data. Patrizio Vitulo contributed to the patient selection. Loredana Pipitone collected and managed biological samples. Mark Gjomarkaj contributed to the interpretation of the data and to writing out the manuscript.
Conflict of interest
The Authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Elisabetta Pace and Maria Ferraro have equally contributed to the work.
Rights and permissions
About this article

Cite this article
Pace, E., Ferraro, M., Chiappara, G. et al. MD2 expression is reduced in large airways of smokers and COPD smokers. Mol Cell Biochem 407, 289–297 (2015). https://doi.org/10.1007/s11010-015-2476-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2476-1