
Abstract
This study was designed to assess the potential effect of vitamin D3 (VD3) in avoiding atherothrombosis by modulation of lipid metabolism and platelet activation in type 1 diabetic rats. Male wistar rats were divided into eight groups (n = 5–10): Control/Saline (Sal); Control/Metformin 500 mg/kg (Metf); Control/Vitamin D3 90 µg/kg (VD3); Control/Metformin 500 mg/kg + VD3 90 µg/kg (Metf + VD3); Diabetic/Saline (Sal); Diabetic/Metformin 500 mg/kg (Metf); Diabetic/Vitamin D3 90 µg/kg (VD3); Diabetic/Metformin 500 mg/kg + VD3 90 µg/kg (Metf + VD3). Treatments were administered during 30 days after diabetes induction with streptozotocin (STZ). After 31 days, the rats were euthanized and blood was collected and separated into serum and platelets, both used for lipid profile and ectonucleotidase activity assays, respectively. Ectonucleoside triphosphate phosphohydrolase (E-NTPDase), ectonucleotide pyrophosphatase/phosphodiesterase (E-NPP), and 5′-nucleotidase and adenosine deaminase (E-ADA) were significantly higher in the Diabetic than in Control group. Treatment with Metf and/or VD3 prevented the increase in NTPDase and E-NPP activities in diabetic rats. Only Metf + VD3 significantly prevented the increase in 5′-nucleotidase. VD3 alone, but not Metf, prevented the increase in ADA activity when compared to saline-treated diabetic rats. Treatment of rats with VD3, Metf, and Metf + VD3 was also effective in the prevention of lipid metabolism disorder in diabetic and was able to ameliorate lipid metabolism in non-diabetic rats. These results provide evidence for the potential of Metf and VD3 in the treatment of platelet dysfunction and lipid metabolism impairment in T1D, which may be important in the control and prevention of atherothrombosis in diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperglycemia plays a causal role in the platelet hyper-reactivity in diabetic patients; it is responsible for impairment of Ca2+ homeostasis in platelets, which is consistent with enhanced sensitivity to aggregating agents, platelet adhesion, activation, and aggregation [1, 2]. These factors are critically important for inflammatory processes in the vascular wall, the major pathophysiological mechanism underlying myocardial infarction and ischemic stroke [3].
In this context, adenosine nucleotides are also involved in modulating platelet activity: ATP has been suggested to regulate platelet aggregation; ADP promotes platelet aggregation and the nucleoside adenosine (Ado) is an endogenous inhibitor of platelet aggregation and limits the degree of vascular injury by reducing the release of reactive oxygen species [4].
The family of ectonucleotidases, whose enzymes are responsible for extracellular nucleotide breakdown, plays an important role in hemostasis and platelet aggregation mainly by regulating ADP catabolism and adenosine production. The enzymes nucleoside triphosphate diphosphohydrolase (NTPDase1 or CD39) and ecto-5′-nucleotidase (CD73) are located in the platelet membrane and complete the hydrolysis of ATP to adenosine. Alterations in NTPDase and 5′-nucleotidase activities in human and animal blood platelets suggest that ectonucleotidases are involved in the thromboregulation process in diabetic conditions [5–8].
The association between hyperlipidemia and platelet hyper-reactivity may cause heart disease, stroke, and death in about 50 % of people with diabetes. Diabetic dyslipidemia is characterized by hypertriglyceridemia, a shift from large low-density lipoprotein (LDL) to smaller and denser particles, and reduced high-density lipoprotein (HDL) cholesterol [9]. In this sense, avoiding the establishment of dyslipidemia is important to prevent atherosclerosis and thrombus developments in diabetes.
Metformin (N1, N1-dimethylbiguanide) is classified as an antihyperglycemic drug and has been shown to be effective in reducing platelet hypersensitivity [10] and ameliorating vascular and endothelial functions [11] and the lipid profile in diabetes [12, 13]. Although vitamin D is not used as a medication in the treatment of diabetes, its deficiency [25(OH)D3 < 20 ng/mL] has been indicated as an environmental factor related with insulin-dependent diabetes and cardiovascular risk [14]. This hypothesis has attracted the interest of researchers to discover whether vitamin D can act in the treatment of diabetes. Vitamin D3 administration appears to be involved in the amelioration of glycemic conditions from diabetic rats induced with STZ [15, 16]; insulin secretion, glucose tolerance [17, 18]; and insulin sensitivity [19, 20] in individuals with diabetes. Studies have demonstrated the benefic effects of vitamin D against a number of vascular disorders such as atherosclerosis and myocardial infarction [30, 49–53]. Therefore, vitamin D appears to play an important role in maintaining normal antithrombotic homeostasis.
In the present study, we aim to evaluate whether vitamin D3 affects the nucleotides ATP, ADP, AMP, and adenosine hydrolysis in platelets from diabetic and non-diabetic wistar rats as well as to verify whether vitamin D3 has potential as an antithrombotic compound, preventing alterations in the lipid profile caused by the diabetic condition.
Materials and methods
Chemicals
The substrates ATP, ADP, AMP, p-nitrophenyl thymidine 5′-monophosphate (p-Nph-5′-TMP), adenosine, as well as Trizma base, sodium azide HEPES, cholecalciferol (vitamin D3), and Coomassie Brilliant Blue G were obtained from Sigma Chemical Co (St. Louis, MO, USA) and bovine serum albumin, K2HPO4, from Reagen (Colombo, PR, Brazil). All the other chemicals used in this experiment were of the highest purity. Vitamin D3 and metformin were dissolved in corn oil and saline, respectively.
Animals
Adult male Wistar rats (300–400 g) from the Central Animal House of the University Federal of Santa Maria (UFSM) were used in the experiment. Animals were maintained at a constant temperature (23 ± 1 °C) on a 12 h light/dark cycle with free access to food and water. During the 30 days of experiment, all rats consumed a commercial diet (Supralab, São Leopoldo, RS) containing a minimum of 2000 IU of vitamin D3 for each kilo of food. All animal procedures were approved by the Animal Ethics Committee from the Federal University of Santa Maria (protocol under number: 23/2012).
Induction of experimental diabetes
Diabetes was induced by a single i.p. injection of streptozotocin (STZ, Sigma, St. Louis, MO) in fasted rats at dose of 55 mg/kg. STZ was freshly dissolved in 0.1 M cold sodium citrate buffer, pH 4.5. Development of diabetes was confirmed 72 h after injection of STZ by measuring fasted blood glucose levels. Only rats with fasting blood glucose levels greater than 250 mg/dL were considered diabetic and then included in the experiment. Treatments were started on the eighth day after STZ injection and continued for 30 days.
Experimental design
Rats were randomly distributed into two groups (Control and Diabetic rats; n = 5–10), both groups divided in subgroups
-
Saline (Sal): normal control rats, received 2 mL/kg/day of saline solution.
-
Metformin (Metf): rats received 500 mg/kg/day of metformin chloride dissolved in 2 mL of saline.
-
Vitamin D3 (VD3): rats received 90 µg/kg/day of cholecalciferol dissolved in one mL of corn oil.
-
Metformin plus Vitamin D3 (Metf + VD3): rats received 500 mg/kg/day of metformin chloride plus 90 µg/kg/day of cholecalciferol.
On 31th day of treatment, the rats were submitted to euthanasia and the blood was collected by cardiac puncture for platelet preparation and serum.
Platelet preparation
Platelet-Rich Plasma (PRP) was prepared by the method of Lunkes et al. [5] with the following minor modifications: total blood was collected by cardiac puncture with 0.120 M sodium citrate as anticoagulant. The total blood–citrate system was centrifuged at 160×g during 15 min. PRP was centrifuged at 1400×g for 30 min and washed twice with 3.5 mM HEPES buffer, pH 7.0, containing 142 mM NaCl, 2.5 mM KCl, and 5.5 mM glucose. The washed platelets were suspended in HEPES isosmolar buffer and adjusted to 0.4–0.6 mg of protein per milliliter for enzymatic activities.
Platelet count and coagulation time
The preparation of Platelet-Rich Plasma (PRP) was obtained as previously described. The platelet count was performed in PRP and expressed as number of platelets per mL of blood.
We also evaluated the time for whole blood visual coagulation in all the groups of this experiment. Blood was incubated at 30 °C and the whole blood coagulation was monitored carefully. Results are expressed as time (s) spent in coagulations.
E-NTPDase and E-5′-nucleotidase activity assay
As described by Lunkes et al. [5], the NTPDase measure was performed in a medium containing 5 mM CaCl2, 100 mM NaCl, 4 mM KCl, 5 mM glucose, and 50 mM Tris–HCl buffer, pH 7.4, at a final volume of 200 μL. The ecto-5′-nucleotidase activity was also carried out as previously described by Lunkes et al. [5]. to measure AMP hydrolysis. However, the 5 mM CaCl2 was replaced by 10 mM MgCl2 to perform the assay. The enzyme activities were expressed as nmol Pi released/min/mg of protein. Briefly, 20 µL of the enzyme preparation was added to the reaction mixture and the preincubation proceeded for 10 min at 37 °C. The reaction was initiated by the addition of ATP or ADP at a final concentration of 1.0 mM, and AMP at a final concentration of 2 mM. The time of incubation was 60 min. Both enzyme assays were stopped by the addition of 200 μl of 10 % trichloroacetic acid to provide a final concentration of 5 %. Subsequently, the tubes were chilled on ice for 10 min. Released inorganic phosphate (Pi) was assayed by the method of Chan et al. [21] using malachite green as the colorimetric reagent and KH2PO4 as standard.
E-NPP activity assay
As previously described by Fürstenau et al. [22], the ecto-nucleotide pyrophosphatase/phosphodiesterase (E-NPP) determination was measured using p-nitrophenyl 5′-thymidine monophosphate (p-Nph-5′-TMP) as substrate and was expressed as nmol of p-nitrophenol released per minute per milligram of protein (nmol p-nitrophenol released/min/mg protein). The reaction medium containing 50 mM Tris–HCl buffer, 120 mM NaCl, 5.0 mM KCl, 60 mM glucose, and 5.0 mM CaCl2, pH 8.9, was preincubated with approximately 20 mg per tube of platelet protein for 10 min at 37 °C in a final volume of 200 mL. The enzyme reaction was started by the addition of p-Nph-5′-TMP at a final concentration of 0.5 mM. After 80 min of incubation, 200 mL NaOH 0.2 N was added to the medium to stop the reaction. The amount of p-nitrophenol released from the substrate was measured at 400 nm using a molar extinction coefficient of 18.8 × 10−3/M/cm. Enzyme activities have been expressed as nmol of p-nitrophenol released per minute per milligram of protein (nmol p-nitrophenol/min/mg protein).
E-ADA activity assay
Ecto-adenosine deaminase (E-ADA) activity determination was performed as described by Guisti and Galanti [23] which is based on the direct measurement of the formation of ammonia, produced when adenosine deaminase acts in excess of adenosine. Briefly, 50 μL of platelets reacted with 21 mmol/L of adenosine, pH 6.5, and was incubated at 37 °C for 60 min. The protein content used for the platelet experiment was adjusted between 0.7 and 0.9 mg/mL. Results have been expressed in units per milligram of protein (U/mg protein). One unit (1 U) of E-ADA is the amount of enzyme required to release 1 mmol of ammonia per minute from adenosine at standard assay conditions.
25(OH)D3 assay
The serum 25(OH)D3 levels were detected using a RIA kit from DiaSorin (Stillwater, MN). The results have been expressed as ng/mL.
Lipid profile assay
Whole blood was separated by cardiac puncture and in tubes without an anticoagulant system and centrifuged at 1800×g for 10 min. The precipitate was discarded and the serum was used to determine the lipid profile. The levels of serum total cholesterol and triglyceride concentrations were measured using standard enzymatic methods by use of Ortho-ClinicalDiagnostics® reagents on the fully automated analyzer (Vitros 950® dry chemistry system; Johnson & Johnson, Rochester, NY, USA). High-density lipoprotein cholesterol was measured in the supernatant plasma after the precipitation of polipoprotein B-containing lipoproteins with dextran sulfate and magnesium chloride as previously described Bachorik and Albers [24].
Protein determination
Protein content was measured by the Coomassie Blue method according to Bradford [25], using bovine serum albumin as the standard. This assay is based on the binding of the dye Coomassie Blue G-250 to protein, and this binding is accompanied by measuring the absorbance maximum of the solution at 595 nm.
Statistical analysis
Data are presented as mean ± S.E.M. and were analyzed statistically by Two-Way ANOVA, followed by Duncan’s test. Differences between groups were considered to be significant when P < 0.05.
Results
Table 1 shows the effect of VD3 and Metf treatments on final body weights, net food and water consumption, and blood glucose levels. Blood glucose and water and food intakes in the diabetic group were significantly increased when compared to the control group, besides a significant decrease in body weight of diabetic rats (P < 0.0001). At the end of 30 days, however, no differences were observed between the diabetic non-treated and treated rats. Treatment with compounds VD3, Metf, and VD3 + Metf per se for 30 days did not produce any effect on glucose levels.
The number of platelets (Table 2) significantly decreased in diabetic rats when compared with control ones. Treatment with VD3, but not with Metf or Metf + VD3, partially prevented this decrease. At the same time, whole blood visual coagulation time (Table 2) was not altered in any of the groups or treatments.
The findings of the present study demonstrate that vitamin D3 prevented the increase in ectonucleotidase activities from platelets of diabetic rats after treatment for 30 days. As can be observed in Fig. 1, STZ-induced diabetes increased ATP hydrolysis by 70 % in saline-treated diabetic when compared with saline-treated control rats (P < 0.001, Fig. 1a). On the other hand, treatment with metformin and/or VD3 prevented the increase in ATP hydrolysis, keeping the enzyme activity at same level as the Control group.

Effects of treatment with metformin and vitamin D3 on a E-NTPDase (ATP), b E-NTPDase (ADP) and c E-5′-nucleotidase activities in platelets of diabetic rats
The Diabetic group showed a significant increase in ADP hydrolysis when compared with the Control group [(1,46) = 100.22, P < 0.0001]. This increase was about 160 % higher in Diabetic/Sal than Control/Sal rats [P < 0.001, Fig. 1b]. There was a partial but significant prevention of this increase in the treatments with metformin and/or VD3, although this prevention was not great enough to maintain enzyme activity at control levels.
AMP hydrolysis by ecto-5′-nucleotidase was significantly higher in the diabetic group when compared to that in the control group [(1,52) = 9.58, P = 0.003] and 65 % higher in Diabetic/Sal-treated rats compared to Control/Sal-treated rats (P < 0.01, Fig. 1c). Only the combined treatment with Metf and VD3 in diabetic rats was able to prevent this increase in AMP hydrolysis.
In relation to E-ADA activity (Fig. 2), there was a significant increase (42 %) in its activity in Diabetic/Sal when compared to Control/Sal group (P < 0.05). Metf alone was not able to prevent the increase. However, Metf with VD3, and VD3 alone prevented the enzyme activity increase.

Effects of treatment with metformin and vitamin D3 on E-ADA activity in platelets of diabetic rats
Figure 3 shows the effects of diabetes and treatments on E-NPP activity in platelets. Results demonstrate that the diabetic state significantly increased E-NPP, by 37 %, and treatments prevented this increase. Treatment per se with Metf and/or VD3 did not cause any alterations in the enzymes analyzed in this work.

Effects of treatment with metformin and vitamin D3 on E-NPP activity in platelets of diabetic rats
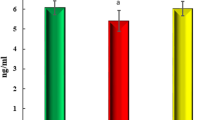
The lipid profile and 25(OH)D3 content are shown in Table 3. VD3 levels were higher in rats treated with VD3 or Metf + VD3 when compared to rats treated with Metf alone or Sal, regardless of the group (Diabetic or Non-diabetic). Diabetes had no effect on the decreased VD3 level in saline-treated rats when compared to non-diabetic saline-treated rats. On the other hand, lipid metabolism was impaired, presenting a significant increase in LDL-c (P < 0.0001) and triglycerides (P = 0.001), by 140 and 74 %, respectively, and a significant decrease in HDL-c (P = 0.00002), by about 18 % in saline-treated diabetic rats. Treatments with Metf + VD3, Metf, and VD3 per se had a significant effect on decreased LDL-c and triglyceride levels in the control group, whereas these same treatments had a preventive effect on the increase of these parameters in diabetic rats. In relation to HDL-c levels, treatments with Metf, VD3 and Metf + VD3 totally prevented its decrease in diabetic rats, which increased by about 95, 95, and 118 %, respectively, when compared to the Diabetic/Sal group. Treatment with VD3 per se, but not with Metf, also had an effect on HDL-c level, increasing it to 52 % (P = 0.00005) in relation to Non-diabetic/Sal-treated rats.
Discussion
The present study was carried out to evaluate the effects of VD3 treatment on ectonucleotidase activities in rat platelets. To the best of our knowledge, there are no studies in the literature evaluating the effects of VD3 or Metf on purinergic signaling. The vitamin D3 concentration (90 µg/kg/day) used to perform the experiment was based on the previous Oral Glucose Tolerance Test (OGTT) as mentioned in Calgaroto et al. [26]. We performed the 25(OH)D3 assay (see Table 2) in order to assess the relationship between vitamin D3 administered and vitamin D3 absorbed and to verify whether diabetes and treatments induced alterations in vitamin D status as well as to confirm the association between the effects observed on the enzyme activities and vitamin D3 levels. As expected, 25(OH)D3 levels significantly increased in VD3- or Metf + VD3-treated rats in both control and diabetic groups. In addition, saline-treated diabetic rats presented 25(OH)D3 levels statistically equal to VD3 saline-treated control rats. Therefore, we suggest that VD3 supplementation, and not endogenous VD3, is involved in changes in enzyme activities from platelets.
Seven days after diabetes induction, diabetic rats showed polyphagia and polydipsia symptoms associated with weight loss (Table 1). Blood glucose levels in the Diabetic/Sal group were found to be significantly increased, by about five-fold, when compared to the Control/Sal group. On the other hand, and in contradiction to recent reports [38–42], VD3 and/or Metf had no effect on glucose levels in either diabetic or non-diabetic rats. The absence of an effect on glucose levels in diabetic rats might be due to the severity of the STZ-induced diabetes, which can promote serious damage on tissues and organs, thus incapacitating them to respond efficiently to treatments that could revert or ameliorate the diabetic condition.
We assumed that an irreversible increase in blood glucose level occurred because STZ scathes specifically the pancreatic β-cells, which are responsible for insulin secretion. In view of this, there was not a great enough number of β-cells remaining after damage induced by STZ to release the amount insulin needed to reduce glucose levels. In an opposite situation, Del Pino-Montes et al. [27] showed that when β-cells are not impaired, VD3 is able to exert its (indirect) effects on glycaemia, by converting VD3 to 1.25-(OH)2D3, which acts on the pancreatic β-cell through vitamin D-receptor activation, increasing insulin synthesis, and secretion in both the vitamin D-depleted and vitamin D-repleted states.
Plasma and cellular components of the homeostatic system are often abnormal in diabetic patients, and some of these abnormalities may play a role in the high risk of thrombosis [28]. In fact, our results showed that hydrolysis of ATP, ADP, and AMP in platelets from the diabetic group was increased after 30 days of diabetes induction while in the control group the treatment with Metf, VD3 or both had no effect on enzyme activities analyzed. In addition, adenosine deamination was also increased in the platelets from diabetic animals.
Corroborating with these results, previous studies with animals as well as in patients with diabetes type 2 carried out in our laboratory [5, 7, 8] demonstrated that the diabetic condition caused an increase in E-NTPDase and E-5′-nucleotidase activities in platelets. Taken together, these findings indicate that the up-regulation of ectoenzymes may play an important role in the control of cellular responses induced by diabetic pathology [8].
Circulating or locally released nucleotides ATP and ADP are rapidly metabolized by sequential activity of ecto-enzymes on the cell surface, which is known as the purinergic cascade [29]. These enzymes have an important role in the thromboregulation process and alterations in their activities have been observed in several diseases, suggesting that they are an important physiological and pathological parameter [8]. The increase in E-NTPDases, E-NPP and E-5′-nucleotidase activities in diabetic rats could be related to an attempt of the organism to compensate for organic alterations induced by diabetes, since ATP and ADP hydrolysis favors the production of adenosine, a molecule that induces vasodilatation and inhibits platelet aggregation [4]. Thus, the organism could be avoiding thrombotic processes by ADP depletion and enhancing adenosine synthesis.
Despite the compensatory response observed in most of the enzymes analyzed, E-ADA activity was increased in diabetic saline-treated rats. The rapid deamination of adenosine by E-ADA and the resulting fall in adenosine levels may be associated with the development of vascular complications observed in the diabetic state, since adenosine plays an important role in preventing the thrombotic process.
The elevation in E-ADA activity in tissues of diabetic rats induced with STZ was also observed by Rutkiewicz and Górski [30] and Schmatz et al. [8]. Insulin appears to be involved in the regulation of E-ADA activity in diabetes, and insulin administration is capable of decreasing the elevated activity of this enzyme in several tissues [30, 31]. Thus, we suggest that the increased E-ADA activity observed in this study with diabetic rats could be explained, in part, by the insulin deficits caused by STZ administration due to its destructive effects on pancreatic β-cells.
When VD3 treatment was associated with diabetes, a decrease in E-NTPDases, E-5′-nucleotidase, E-NPP, and E-ADA platelet activities was found (Figs. 1, 2, 3). These findings suggest a modulation of the purinergic system by VD3 administration in diabetic rats, driving the activities to normal levels similar to those found in the control group. While untreated rats responded to diabetes by producing the antiaggregant Ado, the VD3-treated diabetic rats responded in a different way. VD3 supplementation acted to avoid ADP synthesis, through inhibition of E-NTPDases and an increase in Ado levels by decreasing E-ADA activity. Consequently, in VD3-treated rats, the organism was able to avoid thrombotic processes by ADP depletion and by enhancing adenosine production.
It has also been suggested that microaggregates of platelets via P2Y12 receptors could play a key role in the hyper-reactivity of platelets in diabetic patients [32]. Higher extracellular ATP concentrations are involved in the modulation of platelet aggregation by binding to P2 receptors to antagonize various agonist-induced platelet responses through a competitive mechanism with ADP. Recent evidence indicates that while competition is the major mechanism of inhibition of ADP-induced platelet responses, an additional mechanism antagonizes other agonist responses via ATP binding to a second platelet P receptor [33]. This latter mechanism appears to involve the G protein coupled to induction of cAMP and inhibition of internal calcium mobilization, thereby limiting further recruitment of platelets [34]; it induces dilatation of the vascular wall and at the same time promotes its own stimulated release from endothelial cells [35]. In addition, the activity of ectonucleoside diphosphokinase (E-NDPK) can interconvert extracellular nucleotides, and ATP can be used as a co-substrate of ecto-protein kinase in the phosphorylation of surface-located proteins. Thus, the inhibition of ATP hydrolysis by VD3 is important, since more free ATP in the circulation (due to the inhibition of the ecto-enzyme of platelets) will allow phosphorylation of surface proteins by a platelet E-NDPK, which protects platelets from spontaneous aggregation and thus can play an important role in homeostatic mechanisms that maintain circulating platelets in a resting, inactivated state [36].
Conversely, the inhibition of platelet ADPase activity by VD3 could lead to an increase in ADP extracellular levels. ADP is recognized to induce changes in platelet shape and aggregation. However, if, on one hand VD3 and Metf promote an increase in extracellular ADP levels, favoring platelet aggregation and thrombotic processes, on the other hand, the inhibition of ATP hydrolysis (a competitive inhibitor of ADP-induced platelet aggregation) by these same compounds should be sighted as a compensatory mechanism in thromboregulation, as suggested by Fürstenau et al. [37] in a study about the effect of ebselen on nucleotidase enzymes.
Experimental studies suggest that metformin exhibits antiatherogenic properties through several mechanisms, including a reduction in lipid accumulation in the arterial wall, inhibition of leukocyte–endothelial interaction, foam cell formation, smooth muscle cell proliferation, and platelet aggregation [38]. In the present study, we found similar effects of VD3 treatment on nucleotide breakdown. Both Metf and Metf + VD3 treatments caused inhibition of E-NTPDase and E-5′-nucleotidase activities. Metf, however, had no effect in decreasing E-ADA activity in diabetic rats, as did VD3 or Metf + VD3. Thus, Metf showed lower efficiency than VD3 in modulating nucleotide breakdown. At same time, Metf treatment acted as a purinergic modulator in platelets because it avoided ADP production by decreasing activity of E-NTPDases and 5′-nucleotidase enzymes.
The number of platelets was significantly decreased in diabetic rats when compared to controls (Table 2). While platelet number differed among treatments, the coagulation time remained the same for all treatments analyzed, which resulted in differences between treatments and groups with regard to platelet count/time of blood coagulation ratio. This might indicate platelet hyper-reactivity in diabetic rats, since the significant decrease in platelet count did not result in lower blood coagulation time.
In our study, the possible high platelet reactivity in diabetic rats observed by coagulation time/platelet count ratio may be due to STZ effects on general cells. Our hypothesis is that STZ may have interfered in platelet reactivity via hyperglycemia and insulin deficiency as a consequence of β-cell destruction. These findings suggest that improvement of glycemic control may have a beneficial effect on platelet adhesion and subsequent activation.
Treatment with VD3, but not with Metf or Metf + VD3, partially prevented this decrease in platelet count. Studies such as that conducted by Winocour et al. [39] also observed that STZ-induced diabetes induced decreased platelet count in animals. According to them, the significant decrease in platelet number in the diabetic rats was due to the shortened platelet survival, which was caused mainly by non-platelet factors and platelet defects, 28 days after diabetes induction. In agreement with our findings, other researchers have reported significant drops in platelet count in STZ-induced diabetic rats [40] and diabetic patients [41, 42].
Unexpectedly, the Metf + VD3 group induced a significant decrease in platelet count in control rats and had no effect in restoring platelet count in diabetic rats (Table 2). This may demonstrate an interaction between compounds and it would be interesting to perform future studies to elucidate the mechanisms involved. In fact, during this study, 4 out of 20 non-diabetic rats treated with Metf + VD3 died by convulsion as soon as metformin was administered. We are investigating the possible neurologic factors involved in this situation.
VD3 treatment, on the other hand, restored the platelet count in diabetic rats when compared to the Control/Saline group (Table 2). We suggest that VD3 administration exercised its function in restoring the number of platelets through VDR activity. As modulator of calcium fluxes, VDR could play an essential role in megakaryocytopoiesis, platelet activation, and apoptosis, which are calcium-dependent events [43]. The control of calcium homeostasis is the most probable non-genomic function of platelet VDR, given the important role of calcium fluxes in platelet formation, aggregation, and granule content release. This possibility is supported by observations on non-genomic effects of sex hormones in platelets [44], such as the observation that in vitro testosterone appears to enhance platelet aggregation [45]. Together, these effects may potentially explain the restoration of platelet levels upon VD3 treatment. Platelet restoration was also obtained in patients who have autoimmune thrombocytopenia and treated with high-dose vitamin D supplementation [46]. Moreover, the megakaryocyte differentiation process is known to be triggered by estrogen and vitamin D [47, 48].
Although there was no glycemic control with treatments in this study, there were alterations of other factors related with platelet activation, such as the ectonucleotidase activities observed in this work. Indeed, high glucose concentrations, when chronic, promote alteration in ATP/ADP levels and may be an important factor involved in platelet hyperactivity in the course of diabetes [49]. Extracellular ATP regulates platelet reactivity by way of direct action on platelet purinergic receptors or by hydrolysis to ADP [4]. Extracellular ATP antagonized various agonist-induced platelet responses solely by a competitive mechanism with ADP. More recent evidence indicates that while competition is the major mechanism of inhibition of ADP-induced platelet responses an additional mechanism antagonizes other agonist responses via ATP binding to a second platelet P receptor. This latter mechanism appears to involve the G protein coupled induction of cAMP and inhibition of internal calcium mobilization, inhibiting collagen–thrombin- and thromboxane analog-induced platelet aggregations. The ATP response is synergistic with a second potent physiological platelet aggregation inhibitor, prostacyclin. The simultaneous presentation of prostacyclin and ATP could play a significant role in limiting clot formation to the site of vascular injury and excluding clot formation beyond the margin of disrupted endothelium [34].
The presence of diabetes was shown to confer an enhanced CVD risk when compared with other traditional risk factors, in particular association with dyslipidemia [50]. Although atherosclerotic vascular disease has a multi-factorial etiology, disorders of lipid metabolism play a central role. The impairment of the lipid profile is due to underutilization of glucose, leading to excessive mobilization of fat from adipose tissue. Patients with poorly controlled T1D present elevated levels of triglyceride (TG)-rich lipoproteins [very low-density lipoproteins (VLDL) and chylomicrons] due to a reduction in the activity of lipoprotein lipase (LPL) in the muscle and adipocytes [51]. This increase in TG-rich lipoproteins promotes an increased exchange of high-density lipoprotein (HDL) and low-density lipoprotein (LDL) cholesterol esters with TG in chylomicrons and VLDL, which in turn reduces HDL-c levels and generates small, dense LDL [51]. Insulin deficiency is also associated with an increase in the absolute levels of LDL-c, LDL particle number, and apolipoprotein B-100, because LDL receptor expression is regulated, in part, by insulin [52].
Corroborating with several studies in humans [53, 54] and rodents [55–58] with uncontrolled hyperglycemia, we found worse lipid profile parameters, such as total cholesterol, HDL-c, and triglycerides, in diabetic saline-treated rats, when compared with non-diabetic rats (Table 3). This impairment in lipid metabolism was partially prevented with Metf and VD3 treatments, such that the association between Metf and VD3 (Metf + VD3 group) presented the greatest prevention of lipid impairment, because there were larger increases in HDL-c and greater decreases in total cholesterol and triglycerides when compared with Metf or VD3 administered alone. On the other hand, treatments with Metf, VD3, and Metf + VD3 ameliorated the lipid metabolism in non-diabetic rats, through the decrease in LDL-c and triglycerides, and increase in HDL-c. Our results corroborate to prove that the diabetic condition is caused by lipid metabolism impairment. Because it is the origin of atherosclerotic plaque development, Metf, VD3, and mainly supplementation with Metf + VD3, may be prevent lipid metabolism dysfunction and consequently play a role in avoiding plaque initiation and all other diseases following this event.
With this paper, we report, for the first time, the effects of the drug VD3 on nucleotide hydrolysis by platelets. The knowledge of the exact mechanism of ATP and ADP hydrolysis inhibition by VD3 in platelets from rats may represent an important approach for the use of this inhibitor as an anti-inflammatory drug. Thus, the commercial use of this drug is expected to improve the possibilities of treating inflammatory diseases, but additional studies are necessary to confirm this premise.
The alterations in enzymes observed in this work may indicate some beneficial effects of treatment with VD3 on platelet function and lipid profile resulting in an important development in the prevention of platelet dysfunction and atherothrombosis observed in saline-treated diabetic rats. In summary, our findings suggest that beneficial effects of VD3 on cardiovascular diseases could be associated with the modulation of adenine nucleotide hydrolysis in platelets.
Abbreviations
- 25(OH)D3 :
-
25-Hydroxyvitamin D3
- T1D:
-
Type 1 Diabetes
- VD3 :
-
Vitamin D3
- Metf:
-
Metformin
- Metf + VD3 :
-
Metformin plus Vitamin D3
- CVD:
-
Cardiovascular Disease
- TG:
-
Triglycerides
- LDL-c:
-
Low Density Lipid Cholesterol
- VLDL-c:
-
Very Low Density Lipid Cholesterol
- HDL-c:
-
High Density Lipid Cholesterol
- VDR:
-
Vitamin D Receptor
- PGI2:
-
Prostacyclin
- ADA:
-
Adenosine desaminase
- NTPDase:
-
Ectonucleoside Triphosphate Phosphoydrolase
- E-NPP:
-
Ectonucleotide pyrophosphatase/phosphodiesterase
- Sal:
-
Saline
- STZ:
-
Streptozotocin
References
Lambert P, Bingley PJ (2002) What is type 1 diabetes? Medicine 30:1–5. doi:10.1383/medc.30.1.1.28264
Junod A, Lambert AE, Stauffacher W et al (1969) Diabetogenic action of streptozotocin: relationship of dose to metabolic response. J Clin Invest 48:2129–2139. doi:10.1172/JCI106180
Gawaz M, Langer H, May AE (2005) Platelets in inflammation and atherogenesis. J Clin Invest 115:3378–3384. doi:10.1172/JCI27196
Birk AV, Broekman MJ, Gladek EM et al (2002) Role of extracellular ATP metabolism in regulation of platelet reactivity. J Lab Clin Med 140:166–175
Lunkes GI, Lunkes DS, Morsch VM et al (2004) NTPDase and 5′-nucleotidase activities in rats with alloxan-induced diabetes. Diabetes Res Clin Pract 65:1–6. doi:10.1016/j.diabres.2003.11.016S0168822703003164
Lunkes GI, Lunkes DS, Leal D et al (2008) Effect of high glucose levels in human platelet NTPDase and 5′-nucleotidase activities. Diabetes Res Clin Pract 81:351–357. doi:10.1016/j.diabres.2008.06.001
Lunkes GI, Lunkes D, Stefanello F et al (2003) Enzymes that hydrolyze adenine nucleotides in diabetes and associated pathologies. Thromb Res 109:189–194
Schmatz R, Schetinger MR, Spanevello RM et al (2009) Effects of resveratrol on nucleotide degrading enzymes in streptozotocin-induced diabetic rats. Life Sci 84:345–350. doi:10.1016/j.lfs.2008.12.019
Berthezène F (2002) Diabetic dyslipidaemia. Br J Diabetes Vasc Dis 2:S12–S17
Gargiulo P, Caccese D, Pignatelli P et al (2002) Metformin decreases platelet superoxide anion production in diabetic patients. Diabetes Metab Res Rev 18:156–159. doi:10.1002/dmrr.282
Chan NN (2001) Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol 38:2131–2132
El-Batran SA, Abdel-Salam OM, Nofal SM et al (2006) Effect of rosiglitazone and nateglinide on serum glucose and lipid profile alone or in combination with the biguanide metformin in diabetic rats. Pharmacol Res 53:69–74. doi:10.1016/j.phrs.2005.08.008
Ghatak SB, Dhamecha PS, Bhadada SV et al (2011) Investigation of the potential effects of metformin on atherothrombotic risk factors in hyperlipidemic rats. Eur J Pharmacol 659:213–223. doi:10.1016/j.ejphar.2011.03.029
Kassi E, Adamopoulos C, Basdra EK et al (2013) Role of vitamin D in atherosclerosis. Circulation 128:2517–2531. doi:10.1161/CIRCULATIONAHA.113.002654
Peeyush KT, Savitha B, Sherin A et al (2010) Cholinergic, dopaminergic and insulin receptors gene expression in the cerebellum of streptozotocin-induced diabetic rats: functional regulation with Vitamin D3 supplementation. Pharmacol Biochem Behav 95:216–222. doi:10.1016/j.pbb.2010.01.008
George N, Kumar TP, Antony S et al (2012) Effect of vitamin D3 in reducing metabolic and oxidative stress in the liver of streptozotocin-induced diabetic rats. Br J Nutr 108:1410–1418. doi:10.1017/S0007114511006830
Rudnicki PM, Molsted-Pedersen L (1997) Effect of 1,25-dihydroxycholecalciferol on glucose metabolism in gestational diabetes mellitus. Diabetologia 40:40–44. doi:10.1007/s001250050640
Borissova AM, Tankova T, Kirilov G et al (2003) The effect of vitamin D3 on insulin secretion and peripheral insulin sensitivity in type 2 diabetic patients. Int J Clin Pract 57:258–261
Clark SA, Stumpf WE, Sar M (1981) Effect of 1,25 dihydroxyvitamin D3 on insulin secretion. Diabetes 30:382–386
Chiu KC, Chu A, Go VL et al (2004) Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr 79:820–825
Chan KM, Delfert D, Junger KD (1986) A direct colorimetric assay for Ca2+-stimulated ATPase activity. Anal Biochem 157:375–380
Furstenau CR, Trentin Dda S, Barreto-Chaves ML et al (2006) Ecto-nucleotide pyrophosphatase/phosphodiesterase as part of a multiple system for nucleotide hydrolysis by platelets from rats: kinetic characterization and biochemical properties. Platelets 17:84–91. doi:10.1080/09537100500246641
Guisti G, Galanti B (1984) Colorimetric method. Bergmeyer HU. Verlag Chemie, Weinheim, pp 315–323
Bachorik PS, Albers JJ (1986) Precipitation methods for quantification of lipoproteins. Methods Enzymol 129:78–100
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Calgaroto NS, Thome GR, da Costa P et al (2014) Effect of vitamin D3 on behavioural and biochemical parameters in diabetes type 1-induced rats. Cell Biochem Funct 32:502–510. doi:10.1002/cbf.3044
Del Pino-Montes J, Benito GE, Fernandez-Salazar MP et al (2004) Calcitriol improves streptozotocin-induced diabetes and recovers bone mineral density in diabetic rats. Calcif Tissue Int 75:526–532. doi:10.1007/s00223-004-0118-9
Carr ME (2001) Diabetes mellitus: a hypercoagulable state. J Diabetes Complicat 15:44–54
Bours MJ, Swennen EL, Di Virgilio F et al (2006) Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112:358–404. doi:10.1016/j.pharmthera.2005.04.013
Rutkiewicz J, Gorski J (1990) On the role of insulin in regulation of adenosine deaminase activity in rat tissues. FEBS Lett 271:79–80
Kurtul N, Pence S, Akarsu E et al (2004) Adenosine deaminase activity in the serum of type 2 diabetic patients. Acta Medica (Hradec Kralove) 47:33–35
Matsuno H, Tokuda H, Ishisaki A et al (2005) P2Y12 receptors play a significant role in the development of platelet microaggregation in patients with diabetes. J Clin Endocrinol Metab 90:920–927. doi:10.1210/jc.2004-0137
Soslau G, McKenzie RJ, Brodsky I et al (1995) Extracellular ATP inhibits agonist-induced mobilization of internal calcium in human platelets. Biochim Biophys Acta 1268:73–80
Soslau G, Youngprapakorn D (1997) A possible dual physiological role of extracellular ATP in the modulation of platelet aggregation. Biochim Biophys Acta 1355:131–140
Bodin P, Burnstock G (1996) ATP-stimulated release of ATP by human endothelial cells. J Cardiovasc Pharmacol 27:872–875
Redegeld FA, Caldwell CC, Sitkovsky MV (1999) Ecto-protein kinases: ecto-domain phosphorylation as a novel target for pharmacological manipulation? Trends Pharmacol Sci 20:453–459
Furstenau CR, Spier AP, Rucker B et al (2004) The effect of ebselen on adenine nucleotide hydrolysis by platelets from adult rats. Chem Biol Interact 148:93–99. doi:10.1016/j.cbi.2004.04.003
Mamputu JC, Wiernsperger NF, Renier G (2003) Antiatherogenic properties of metformin: the experimental evidence. Diabetes Metab 29:6S71–6S76
Winocour PD, Laimins M, Colwell JA (1984) Platelet survival in streptozotocin-induced diabetic rats. Thromb Haemost 51:307–312
Martin FJ, Miguez JM, Aldegunde M et al (1995) Platelet serotonin transport is altered in streptozotocin-induced diabetic rats. Life Sci 56:1807–1815
James J, Padayatti P, Paul T et al (1997) Platelet monoamine changes in diabetic patients and streptozotocin-induced diabetic rats. Curr Sci 72:137–139
Paton RC (1979) Platelet survival in diabetes mellitus using an aspirin-labelling technique. Thromb Res 15:793–802
Leytin V, Freedman J (2003) Platelet apoptosis in stored platelet concentrates and other models. Transfus Apher Sci 28:285–295. doi:10.1016/S1473-0502(03)00048-X
Pilo R, Aharony D, Raz A (1981) Testosterone potentiation of ionophore and ADP induced platelet aggregation: relationship to arachidonic acid metabolism. Thromb Haemost 46:538–542
Khetawat G, Faraday N, Nealen ML et al (2000) Human megakaryocytes and platelets contain the estrogen receptor beta and androgen receptor (AR): testosterone regulates AR expression. Blood 95:2289–2296
Bockow B, Kaplan TB (2013) Refractory immune thrombocytopenia successfully treated with high-dose vitamin D supplementation and hydroxychloroquine: two case reports. J Med Case Rep. doi:10.1186/1752-1947-7-91
Silvagno F, De Vivo E, Attanasio A et al (2010) Mitochondrial localization of vitamin D receptor in human platelets and differentiated megakaryocytes. PLoS One 5:e8670. doi:10.1371/journal.pone.0008670
Song LN, Cheng T (1993) Glucocorticoid-induced growth inhibition and differentiation of a human megakaryoblastic leukemia cell line: involvement of glucocorticoid receptor. Stem Cells 11:312–318. doi:10.1002/stem.5530110409
Michno A, Bielarczyk H, Pawelczyk T et al (2007) Alterations of adenine nucleotide metabolism and function of blood platelets in patients with diabetes. Diabetes 56:462–467. doi:10.2337/db06-0390
Grundy SM (2006) Diabetes and coronary risk equivalency: what does it mean? Diabetes Care 29:457–460
Chahil TJ, Ginsberg HN (2006) Diabetic dyslipidemia. Endocrinol Metab Clin North Am 35(491–510):vii–viii. doi:10.1016/j.ecl.2006.06.002
Jaiswal M, Schinske A, Pop-Busui R (2014) Lipids and lipid management in diabetes. Best Pract Res Clin Endocrinol Metab 28:325–338. doi:10.1016/j.beem.2013.12.001
Maahs DM, Dabelea D, D’Agostino RB Jr et al (2013) Glucose control predicts 2-year change in lipid profile in youth with type 1 diabetes. J Pediatr 162(101–107):e101. doi:10.1016/j.jpeds.2012.06.006
Valdivielso P, Sanchez-Chaparro MA, Calvo-Bonacho E et al (2009) Association of moderate and severe hypertriglyceridemia with obesity, diabetes mellitus and vascular disease in the Spanish working population: results of the ICARIA study. Atherosclerosis 207:573–578. doi:10.1016/j.atherosclerosis.2009.05.024
Hamzah RU, Odetola AA, Erukainure OL et al (2012) Peperomia pellucida in diets modulates hyperglyceamia, oxidative stress and dyslipidemia in diabetic rats. J Acute Dis 1(2):135–140
Gonçalves AESS, Lellis-Santos C, Curi R et al (2014) Frozen pulp extracts of camu-camu(Myrciaria dubia McVaugh) attenuate the hyperlipidemia and lipid peroxidation of Type 1 diabetic rats. Food Res Int 64:1–8
Pillai SI, Subramanian SP, Kandaswamy M (2014) Antidyslipidemic effect of a novel vanadium-3-hydroxy flavone complex in streptozotocin-induced experimental diabetes in rats. Biomed Prev Nut 4:189–193
Ugochukwu NH, Figgers CL (2007) Attenuation of plasma dyslipidemia and oxidative damage by dietary caloric restriction in streptozotocin-induced diabetic rats. Chem Biol Interact 169:32–41. doi:10.1016/j.cbi.2007.05.002
Conflict of interests
The authors declare that they have no Conflict of interests.
Compliance with ethical standards
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Federal University of Santa Maria (protocol under number: 23/2012). All efforts were made to minimize suffering.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Calgaroto, N.S., da Costa, P., Cardoso, A.M. et al. Vitamin D3 prevents the increase in ectonucleotidase activities and ameliorates lipid profile in type 1 diabetic rats. Mol Cell Biochem 405, 11–21 (2015). https://doi.org/10.1007/s11010-015-2390-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2390-6