
Abstract
B cell translocation gene 3 (BTG3) is a tumor suppressor by inhibiting cell proliferation, migration, and regulating cell cycle progression in several tumors. However, its role in esophageal adenocarcinoma (EAC) remains unknown. Here, we detected the expression of BTG3 in EAC tissues and subsequent progression. BTG3 expression was significant decreased in EAC tissues and cell lines detected by real-time RT–PCR and Western blot. Relationships of BTG3 with EAC clinicopathology were analyzed statistically. The decrease expression of BTG3 is associated with lymph node metastases. In vitro assay demonstrated that overexpression of BTG3 significantly suppressed colony formation and proliferation of EAC cells. The suppressed migration and invasion abilities found in BTG3-overexpressing EAC cells. Our findings suggested that BTG3 is suppressor in the progression of EAC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Esophageal carcinoma is one of the most common malignancies in the world [1]. The predominant histological type is esophageal squamous cell carcinoma (ESCC); however, the incidence of esophageal adenocarcinoma (EAC) has increased over recent decades [2, 3], and the reasons underlying this trend are incompletely understood. EAC has one of the poorest outcomes of all solid tumors, with only 14 % of patients surviving 5 years [4]. Surgery remains the main curative treatment but it is only suitable for ~50 % of patients due to the majority of EAC patients being diagnosed at an advanced stage [5]. A better understanding of EAC underpins efforts to improve early detection and treatment outcomes.
The protein encoded by B-cell translocation gene 3 (BTG3) belongs to an antiproliferative BTG/transducer of ErbB2 (Tob) family, which also includes BTG1, BTG2, TOB, TOB2, and PC3b in human cells [6]. These protein are characterized by the conserved N-terminal domain spanning 104–106 amino acids and regulate cell cycle progression in a variety of cell types [6, 7]. BTG/Tob proteins can be activated or repressed by transcriptional mechanisms, as well as via post-translational modifications. Several studies have suggested the direct involvement of BTG/Tob proteins in cancer. In lung and thyroid cancer patients, the presence of the phosphorylated inactive form of Tob1 or reduced Tob1 expression is frequently observed [8, 9]. Downregulation or impaired expression of BTG2 is observed in prostate and breast cancer [10, 11]. BTG3 interacts with E2F1, a transcription factor important for S-phase entry and cell cycle progression [12]. As for Tob1, reduced expression of BTG3 is also observed in a high frequency of lung cancer specimens [13]. Mice lacking BTG3 also display an increased incidence of lung tumors [13]. BTG3 expression is reduced by promoter inactivation through DNA methylation in renal cancer [14]. However, the expression pattern and function of BTG3 in EAC remain unknown.
In this study, we detected the expression of BTG3 in EAC clinical samples and examined the effect of BTG3 on EAC cell proliferation, migration, and invasion.
Materials and methods
Patients and tissue specimens
Thirty-nine EAC samples and paired adjacent normal esophageal tissues were collected from patients treated surgically at the First Affiliated Hospital of Zhengzhou University and the Tumor Hospital of Linzhou City Henan, China, between 2012 and 2013. Samples were frozen immediately and stored in liquid nitrogen. No patients received chemotherapy or radiotherapy prior to surgery. Clinical and pathological characteristics were determined. Informed consent was obtained from all patients. The procedures were approved by the Zhengzhou University Ethics Committee.
Cell lines and stable transfection
The EAC cell lines OE-33 and OE-19 were purchased from the Cell Bank of the Chinese Academy of Medical Sciences. Cells were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) with 10 % fetal calf serum and supplemented with penicillin/streptomycin. The human esophageal squamous epithelial cell line HET-1A was obtained from the Cell Bank of the Chinese Academy of Medical Sciences. HET-1A cells were cultured in bronchial epithelial cell basal medium (Lonza Group, Basel, Switzerland) supplemented with triiodothyronine, insulin, transferrin, retinoic acid, hydrocortisone, human recombinant epidermal growth factor, epinephrine, and bovine pituitary extract.
OE-33 or OE-19 cells were plated into 6-well plates 24 h before transfection. The cells were transfected with pcDNA3.1-BTG using Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA). For 48 h after transfection, the cells were cultured in medium supplemented with G418 at 500 µg/ml for 4 weeks. Clones overexpressing BTG3 (Ex-BTG3) were selected for further study. Cells stably transfected with pcDNA3.1 were also generated (NC).
Real-time polymerase chain reaction (PCR)
Total RNA was extracted from EAC and normal tissues, and control and transfected EAC cell lines (OE-33 and OE-19) using TRIzol Reagent (Invitrogen). The concentration and purity of RNA were determined using spectrophotometry. cDNA was synthesized using the PrimeScript RT Reagent Kit (Takara, Dalian China). Quantitative real-time PCR analysis was done using the Premix Ex Taq (Probe qPCR), ROX Plus Kit (Takara). The primers used were BTG3 Forward: 5′-CTCCTCCTGTTCCATTTGGT-3′, Reverse: 5′-TAATCCAGTGATTCCGGTCA-3′, Probe: 5′-Fam-CGCCCAATTCCAGTGACATGG-Tamra-3′; β-actin Forward: 5′-CACTCTTCCAGCCTTCCTTC-3′, Reverse: 5′-GGATGTCCACGTCACACTTC-3′, Probe: 5′-Fam-TGCCACAGGACTCCATGCCC-Tamra-3′.
Western blotting
Frozen tissues were homogenized on ice in cell lysis buffer (Beyotime, Jiangsu, China). Protein lysates were collected following centrifugation at speed of 12,000g. For BTG3 transfected EAC cell lines or controls, cells were harvested by trypsinization, washed in ice-cold PBS, and then lysed in cell lysis buffer. Total protein concentrations were estimated using the Bradford method. 15 μg proteins were used for Western blotting. The protein blots were incubated with primary goat anti-human BTG, Cyclin A, Cyclin D1, CDK2, and β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4 °C. Secondary antibodies (donkey anti-goat IgG) conjugated with horseradish peroxidase (Santa Cruz Biotechnology) were used and the blots were visualized by enhanced chemiluminescence (GE Healthcare, Chalfont St. Giles, UK).
Cell proliferation assay
Cells were seeded at 5 × 103 per well in 96-well plates. Zero, 1, 2, and 3 days after seeding, 0.5 mg/ml MTT (Sigma–Aldrich, St Louis, MO, USA) was added and the plates were incubated for 4 h at 37 °C in the dark. The medium containing MTT was removed. The blue-colored formazan that formed was dissolved in 150 μl DMSO per well. The absorbance was measured at 490 nm using an ELISA plate reader.
Anchorage-independent growth
EAC cell lines transfected with BTG3 construct or control cells (1 × 103 per well) were suspended in RPMI 1640 medium with 10 % FBS and were added to 0.3 % agar over a base layer of 0.6 % agar culture medium. After 2 weeks incubation, the cells were stained with 0.1 % crystal violet for 15 min. Clones with >50 cells were manually counted.
Flow cytometry
Cells were harvested by trypsinization, washed once with PBS, and fixed in 70 % chilled ethanol for a minimum of 1 h. After fixation, cells were suspended in PBS containing 50 µg/ml propidium iodide (PI) and 0.5 mg/ml DNase-free ribonuclease. The cells were stained for 1 h at 4 °C. Percentages of cells within each of the cell cycle compartments (G0/G1, S, or G2/M) and apoptosis were determined using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Transwell invasion assay
BTG3-overexpressing EAC cells lines or control cells (1 × 105 per well) suspended in RPMI 1640 medium with 10 % FBS were seeded on the upper chamber of the Transwell filter with 8 μm pores (6.5 mm). The bottom chambers were filled with RPMI-1640 with 10 % FBS, and the cells were incubated for 24 h at 37 °C. Cells in the upper chamber that had not migrated were gently scraped away. Cells present in the coated membrane were fixed with 10 % formaldehyde in PBS, and stained with methylene blue (Beyotime) for 30 min. The membrane was examined under a microscope at ×100 magnification.
Wound healing assay
Cells were seeded in six-well plates and were cultured for 24 and 48 h, until completely confluent. The cell monolayer was scratched with a 200 μl pipette tip and incubated for 24 and 48 h. The distance between the scratched line and the growing edge of the cells was measured. The relative percent wounded area filled through cell migration was then calculated as follows: [(mean wound width−mean remaining width)/mean wound width] × 100 (%).
Statistical analysis
Results were expressed as mean ± SD. Statistical analysis was performed by Student’s t test or one-way analysis of variance for normally distributed data. P < 0.05 was considered to be significant.
Results
BTG3 is downregulated in human EAC
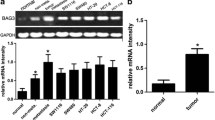
BTG3 mRNA level in 39 patients were detected by real-time PCR. We found that the mRNA levels of BTG3 in EAC were downregulated compared to adjacent normal esophageal tissue (Fig. 1a, P < 0.05). Western blotting also revealed significant downregulation of BTG3 in EAC compared with the adjacent normal esophageal tissue (Fig. 1b, P < 0.05).

Analysis of BTG3 expression in EAC patients and cell lines. a BTG3 mRNA level in EAC tissues and paired adjacent normal tissues analyzed by real-time PCR. b Western blot analysis of BTG3 protein in EAC tissues and paired adjacent normal tissues. c, d BTG3 mRNA and protein level in OE-33 and OE-19 EAC cell lines compared with human esophageal squamous epithelial cell line HET-1A. e Correlation between BTG3 mRNA level and EAC cell differentiation. Correlation between BTG3 expression and tumor staging (f) and lymph node metastases (g) (*P < 0.05)
A similar expression profile of BTG3 was also observed in EAC cell lines. The mRNA and protein levels of BTG3 in two OE-33 and OE-19 EAC cell lines were downregulated compared to human esophageal squamous epithelial cell line HET-1A (Fig. 1c, d, P < 0.05).
We characterized the correlation between BTG3 mRNA level and clinical features of EAC including cell differentiation, tumor staging, and lymph node metastases (Table 1). Expression of BTG3 was not correlated with age and sex. As shown in Fig. 1e, BTG3 mRNA level was not associated with cell differentiation of EAC (P > 0.05). BTG mRNA level in TNM-I of EAC was higher compared with TNM-III (P < 0.05, Fig. 1f). There was a significant difference in BTG3 between EAC with and without lymph node metastases (P < 0.05, Fig. 1g).
Overexpression of BTG3 downregulated the proliferation of EAC cell lines
To test the ability of BTG3 to suppress tumor formation, we performed an MTT assay and colony formation. OE-33 and OE-19 EAC cells were transfected with a BTG3 expression or empty control construct. Overexpression of BTG3 in Ex-BTG3 cells was validated by real-time PCR and Western blotting (Fig. 2a–c). Proliferation was significantly inhibited in Ex-BTG cells from the first day after transfection (P < 0.0.05; Fig. 2d, e). Colonies that formed were Giemsa stained (Fig. 2f, g). In both tumor cell lines, overexpression of BTG3 reduced the number of colonies. These data suggest that BTG3 exhibits a tumor suppressive function in EAC cells.

Overexpression of BTG3 reduces colony formation and proliferation in EAC cell lines. a–c Expression of BTG3 was measured by PCR and Western blotting after transfection with BTG3 expression construct in OE-33 and OE-19 EAC cell lines. d, e Proliferation of OE-33 and OE-19 cells transfected with BTG3 construct was evaluated by MTT assay. f, g Cells were stained with Giemsa after transfection with BTG3 expression construct in OE-33 and OE-19 cells; mean colonies numbers are shown according to three repeated experiments (*P < 0.05)
BTG3 effects cell proliferation through S-phase arrest and apoptosis
To understand the mechanisms by which cell proliferation is affected, flow cytometry was performed to analyze changes in apoptosis and the cell cycle profile of EAC cell lines. Compared to OE-33 and OE-19 cells, their corresponding BTG3- overexpressing counterparts displayed a strongly decreased S-phase fraction. A marked increase in G1 fraction was detectable in BTG3-overexpressing EAC cell lines (Fig. 3a, b). Overexpress BTG3 downregulate the protein levels of Cyclin A and Cyclin D1 in OE-19 and OE-33 cell lines (Fig. 3c, d). Flow cytometry showed that the proportion of cells undergoing apoptosis was significantly higher in BTG3-overexpressing OE-33 cells compared with negative controls (Fig. 4a). Overexpression of BTG3 increased apoptosis in OE-19 cells (Fig. 4b). Thus, downregulation in proliferation of EAC cells was mediated through S-phase arrest and consequently through apoptosis.

BTG3 arrested Ex-BTG3 EAC cells at G1 phase. Flow cytometry was performed to analyze changes in the cell cycle profile. The effect of BTG3 on the distribution (%) of OE-33 cells (a) and OE-19 cells (b) in the G1, S, and G2 phases of the cell cycle is shown. Compared to NC group, Cyclin A and Cyclin D1 were low expressed in Ex-BTG3 group both in OE-19 and OE-33, and the expression of CDK2 has no significant difference. (c, d) (*P < 0.05)

BTG3 induced apoptosis of EAC cell lines. Apoptotic cells were identified by flow cytometric analysis. Representative histograms are shown. a left OE-33 cells, b left OE-19 cells. The percentages of apoptotic OE-33 (a right) and OE-19 (b right) cells are shown (*P < 0.05)
BTG3 suppresses migration and invasion of EAC cells
The effect of BTG3 on tumor cell migration and invasion was assessed by wound healing assay. Figure 5a shows that OE-33 cells overexpressing BTG3 had markedly decreased migration. Similar results were observed in OE-19 cells (Fig. 5b). The Transwell invasion assay showed that BTG3-overexpressing OE-33 and OE-19 EAC cell lines had significantly reduced invasiveness (P < 0.05, Fig. 5c, d).

BTG3 inhibited the migration and invasion ability of EAC cells. Wound healing assay (a OE-33 cells, b OE-19 cells) showed migration of cells overexpressing BTG3 was suppressed compared with control cells. (c OE-33 cells, d OE-19 cells). Cell invasion was measured using the Transwell invasion assay. The numbers of migrating and invading cells were counted (*P < 0.05)
Discussion
Many studies have suggested that BTG3 plays an important role in tumor suppression. Downregulation of BTG3 is observed in renal, breast, and prostate cancer, and it is a candidate tumor suppressor gene in several cancers [14–19]. However, the expression pattern and function of BTG3 in the progression of EAC remain unknown. In this study, we observed that BTG3 expression was significantly downregulated in EAC tissues compared with adjacent normal tissues by real-time PCR and Western blotting. In addition, the relationship between differentiation, tumor staging, and distant metastasis with BTG3 expression was evaluated. Our data demonstrated that the decreased expression of BTG3 was associated with lymph node metastases.
Inactivation or impaired expression of BTG/Tob proteins is frequently observed in the clinical samples of cancer patients. Boiko et al. [20] identified BTG2 as an important transcriptional target gene of p53. BTG4 inactivation by promoter silencing contributes to colon cancer [21]. By contrast, several models show that enforced expression of BTG/Tob genes can result in inhibition of tumor growth [22, 23]. These findings suggest that BTG/Tob proteins have important roles in preventing tumourigenesis and cancer.
Here, we showed that BTG3 strongly inhibited proliferation of EAC cells, by colony formation and proliferation assays. Inhibition of cell proliferation by BTG3 is thought to result from cell cycle arrest [14]. Control of cell cycle progression in cancer cells is a potentially effective strategy for the control of tumor growth because molecular analysis of human cancers has revealed that cell cycle regulators are frequently mutated in many common malignancies [24]. Our in vitro data indicated that overexpression of BTG3 in EAC cell lines resulted in significant S-phase arrest and may be associated with apoptosis. Several studies have demonstrated that BTG3 inhibits E2F1 transcription factor, suggesting that it has a negative regulatory influence consistent with its role in inhibiting progression into S-phase [14]. BTG3 is also a direct transcriptional target gene of p53 [12], which affects its role in tumor suppression and DNA damage response. BTG3 interacts with ChHK1, which is a key effector kinase in the cell cycle checkpoint response, and regulates its phosphorylation and activation [25].
To verify the ability of BTG3 to suppress EAC tumorigenesis, we performed Transwell invasion and wound healing assays. BTG3 significantly suppressed the invasive ability of EAC cell lines. BTG3 is also linked with aggressiveness of ovarian and hepatocellular carcinoma [26, 27]. The mechanisms underlying the inhibition of EAC cell metastasis needs further investigation.
The observations reported here present a significant association between downregulation of BTG3 and EAC tumorigenesis. BTG3 inhibits proliferation through inducing cell cycle arrest and invasion of EAC cells. BTG3 may act as a suppressor in the progression of EAC.
References
Pennathur A, Gibson MK, Jobe BA, Luketich JD (2013) Oesophageal carcinoma. Lancet 381:400–412
Pohl H, Welch HG (2005) The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 97:142–146
Francis AM, Sepesi B, Correa AM, Blum MA, Erasmus JJ, Lee JH, Maru DM, Mehran RJ, Rice DC, Roth JA, Vaporciyan AA, Walsh GL, Welsh JW, Swisher SG, Hofstetter WL, University of Texas MD Anderson Esophageal Cancer Group (2013) The influence of histopathologic tumor viability on long-term survival and recurrence rates following neoadjuvant therapy for esophageal adenocarcinoma. Ann Surg 258:500–507
Eloubeidi MA, Mason AC, Desmond RA, El-Serag HB (2003) Temporal trends (1973–1997) in survival of patients with esophageal adenocarcinoma in the United States: a glimmer of hope? Am J Gastroenterol 98:1627–1633
Li X, Galipeau PC, Paulson TG, Sanchez CA, Arnaudo J, Liu K, Sather CL, Kostadinov RL, Odze RD, Kuhner MK, Maley CC, Self SG, Vaughan TL, Blount PL, Reid BJ (2014) Temporal and spatial evolution of somatic chromosomal alterations: a case-cohort study of Barrett’s esophagus. Cancer Prev Res 7:114–127
Matsuda S, Rouault J, Magaud J, Berthet C (2001) In search of a function for the TIS21/PC3/BTG1/TOB family. FEBS Lett 497:67–72
Winkler GS (2010) The mammalian anti-proliferative BTG/Tob protein family. J Cell Physiol 222:66–72
Iwanaga K, Sueoka N, Sato A, Sakuragi T, Sakao Y, Tominaga M, Suzuki T, Yoshida Y, Tsuzuku JK, Yamamoto T, Hayashi S, Nagasawa K, Sueoka E (2003) Alteration of expression or phosphorylation status of tob, a novel tumor suppressor gene product, is an early event in lung cancer. Cancer Lett 202:71–79
Ito Y, Suzuki T, Yoshida H, Tomoda C, Uruno T, Takamura Y, Miya A, Kobayashi K, Matsuzuka F, Kuma K, Yamamoto T, Miyauchi A (2005) Phosphorylation and inactivation tob contributes to the progression of papillary carcinoma of the thyroid. Cancer Lett 220:237–242
Ficazzola MA, Fraiman M, Gitlin J, Woo K, Melamed J, Rubin MA, Walden PD (2001) Antiproliferative B cell translocation gene 2 protein is down-regulated post transcriptionally as an early event in prostate carcinogenesis. Carcinogenesis 22:1271–1279
Kawakubo H, Brachtel E, Hayashida T, Yeo G, Kish J, Muzikansky A, Walden PD, Maheswaran S (2006) Loss of B-cell translocation gene-2 in estrogen receptor-positive breast carcinoma is associated with tumor grade and overexpression of cyclin d1 protein. Cancer Res 66:7075–7082
Ou YH, Chung PH, Hsu FF, Sun TP, Chang WY, Shieh SY (2007) The candidate tumor suppressor BTG3 is a transcriptional target of p53 that inhibits E2F1. EMBO J 26:3968–3980
Yoneda M, Suzuki T, Nakamura T, Ajima R, Yoshida Y, Kakuta S, Sudo K, Iwakura Y, Shibutani M, Mitsumori K, Yokota J, Yamamoto T (2009) Deficiency of antiproliferative family protein Ana correlates with development of lung adenocarcinoma. Cancer Sci 100:225–232
Majid S, Dar AA, Ahmad A, Hirata H, Kawakami K, Shahryari V, Saini S, Tanaka Y, Dahiya AV, Khatri G, Dahiya R (2009) BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 30:662–667
Lin TY, Cheng YC, Yang HC, Lin WC, Wang CC, Lai PL, Shieh SY (2012) Loss of the candidate tumor suppressor BTG3 triggers acute cellular senescence via the ERK-JMJD3-p16(INK4a) signaling axis. Oncogene 31:3287–3297
Majid S, Dar AA, Shahryari V, Hirata H, Ahmad A, Saini S, Tanaka Y, Dahiya AV, Dahiya R (2010) Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer 116:66–76
Chen XB, Chen GY, Cao XG, Zhou YD, Yang TJ, Wei SD (2013) Downregulation of BTG3 in non-small cell lung cancer. Biochem Biophys Res Commun 437:173–178
Deng BY, Zhao Y, Gou WF, Chen S, Mao XY, Takano Y, Zheng HC (2013) Decreased expression of BTG3 was linked to carcinogenesis, aggressiveness, and prognosis of ovarian carcinoma. Tumor Biol 34:2617–2624
Ren XL, Zhu XH, Li XM, Li YL, Wang JM, Wu PX, Lv ZB, Ma WH, Liao WT, Wang W, Ding YQ, Liang L (2015) Down-regulation of BTG3 promotes cell proliferation, migration and invasion and predicts survival in gastric cancer. J Cancer Res Clin Oncol 141:397–405
Boiko AD, Porteous S, Razorenova OV, Krivokrysenko VI, Williams BR, Gudkov AV (2006) A systematic search for downstream mediators of tumor suppressor function of p53 reveals a major role of BTG2 in suppression of Ras-induced transformation. Genes Dev 20:236–252
Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T (2008) Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 68:4123–4132
Yanagie H, Tanabe T, Sumimoto H, Sugiyama H, Matsuda S, Nonaka Y, Ogiwara N, Sasaki K, Tani K, Takamoto S, Takahashi H, Eriguchi M (2009) Tumor growth suppression by adenovirus-mediated introduction of a cell growth suppressing gene tob in a pancreatic cancer model. Biomed Pharmacother 63:275–286
Farioli-Vecchioli S, Tanori M, Micheli L, Mancuso M, Leonardi L, Saran A, Ciotti MT, Ferretti E, Gulino A, Pazzaglia S, Tirone F (2007) Inhibition of medulloblastoma tumorigenesis by the antiproliferative and pro-differentiative gene PC3. FASEB J 21:2215–2225
Molinari M (2000) Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif 33:261–274
Cheng YC, Lin TY, Shieh SY (2013) Candidate tumor suppressor BTG3 maintains genomic stability by promoting Lys63-linked ubiquitination and activation of the checkpoint kinase CHK1. Proc Natl Acad Sci USA 110:5993–5998
Deng B, Zhao Y, Gou W, Chen S, Mao X, Takano Y, Zheng H (2013) Decreased expression of BTG3 was linked to carcinogenesis, aggressiveness, and prognosis of ovarian carcinoma. Tumour Biol 34:2617–2624
Lv Z, Zou H, Peng K, Wang J, Ding Y, Li Y, Ren X, Wang F, Chang R, Liang L, Ding Y (2013) The suppressive role and aberrant promoter methylation of BTG3 in the progression of hepatocellular carcinoma. PLoS ONE 8:e77473
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Du, Y., Liu, P., Zang, W. et al. BTG3 upregulation induces cell apoptosis and suppresses invasion in esophageal adenocarcinoma. Mol Cell Biochem 404, 31–38 (2015). https://doi.org/10.1007/s11010-015-2363-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2363-9