
Abstract
Investigations regarding hypertension and dietary sodium, both factors that influence stroke risk, have previously been limited to using genetically disparate treatment and control groups, namely the stroke-prone, spontaneously hypertensive rat and Wistar-Kyoto rat. In this investigation, we have characterized and compared cerebral vasoactive system adaptations following stroke in genetically identical, salt-induced hypertensive, and normotensive control mice. Briefly, ANP+/− (C57BJ/6 × SV129 background) mice were fed chow containing either 0.8 % NaCl (NS) or 8.0 % NaCl (HS) for 7 weeks. Transient cerebral ischemia was induced by middle cerebral artery occlusion (MCAO). Infarct volumes were measured 24-h post-reperfusion and the mRNA expression of five major vasoactive systems was characterized using qPCR. Along with previous publications, our data validate a salt-induced hypertensive state in ANP+/− mice fed HS chow as they displayed left ventricular hypertrophy, increased systolic blood pressure, and increased urinary sodium excretion. Following MCAO, mice fed HS exhibited larger infarct volumes than their dietary counterparts. In addition, significant up-regulation in Et-1 and Nos3 mRNA expression in response to salt and stroke suggests implications with increased cerebral damage in this group. In conclusion, our data demonstrate increased cerebral susceptibility to stroke in salt-induced hypertensive mice. More importantly, however, we have characterized a novel method of investigating hypertension and stroke with the use of genetically identical treatment and control groups. This is the first investigation in which genetic confounding variables have been eliminated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Hypertension is the most important preventable risk factor for premature death worldwide [1], increasing the risk of developing coronary artery disease [2] and stroke [3]. Dietary salt intake is a known contributor to hypertensive disease [4–6].
Approximately, one-third of patients with essential hypertension are sensitive to dietary changes in salt [6]. Increased sodium has been linked to vessel remodeling [7], increased left ventricular (LV) mass, increased blood pressure (BP) [5, 8], and increased urinary sodium excretion [1, 8]. Epidemiological studies have shown a positive correlation between salt intake and increased risk of stroke [6, 9, 10], while others have demonstrated correlations between BP and essential hypertension on stroke [3, 11–13]. No direct relationship between salt-induced hypertension and stroke injury has previously been examined. Vasoactive systems, such as the natriuretic peptide (NP), nitric oxide synthase (NOS), renin–angiotensin (RAS), and endothelin (ET) systems, possess homeostatic roles in normal states to regulate BP. In salt-induced hypertension these systems show adaptive changes that may affect stroke injury.
Atrial natriuretic peptide (ANP) is modulated by volume status and is responsible for the homeostatic control of sodium and water content in the body. Heterozygous ANP gene-disrupted mice (ANP+/−) display normal cardiac mass and BP under normal conditions. Due to the salt-sensitivity of this genotype, ANP+/− mice display left ventricular hypertrophy (LVH) and increased BP when fed high salt (HS) [14, 15]. In the current study, we have used the ANP+/− mouse to investigate the effects of salt-induced hypertension on stroke particularly with interest in cerebral adaptations in the NP, NOS, RAS, ET, and the vascular endothelial growth factor (VEGF) vasoactive systems.
Methods
Animal model and genotyping
Experimental protocols pertaining to the use of mice in this study were approved by the University Animal Care Committee (UACC) of Queen’s University in accordance with the guidelines of the Canadian Council of Animal Care (CCAC). All mice were bred and cared for in the Animal Care Facility at Queen’s University. Mice were housed in cages (up to 4 animals per cage) at room temperature on a 12 h light/dark cycle. Heterozygous (ANP+/−) offspring were obtained by crossing wild-type (ANP+/+, C57BJ/6) females with homozygous ANP gene-disrupted (ANP−/−, C57BJ/6 × SV129 background) [14] males. Mice were weaned at 3 weeks and ear, and tail tissue biopsies were collected for genotyping. The genotype of each mouse was determined using an AccuStartTMII mouse genotyping kit (Quanta Biosciences, Gaithersburg, MD, USA) and previously published PCR methods [16].
Dietary treatment and non-invasive tail-cuff BP measurements
Male ANP+/− mice were fed either normal-salt (NS; 0.8 % NaCl) or high-salt chow (HS; 8.0 % NaCl, Lab Diet 5001®, Brentwood, MO, USA) for 7 weeks. A total of 22 NS-treated and 23 HS-treated mice (between 13 and 15 weeks of age) were used. Chow diets and tap water were provided ad libitum. At the beginning of the fourth week of salt treatment, a randomized subset of mice (NS; n = 4, HS; n = 5) underwent a 3-week training period to acquire BP measurements using a CODA non-invasive tail-cuff system (Kent Scientific, Connecticut, USA) [17]. Following training, systolic BP measurements were recorded and the mean value from two separate days at the end of salt treatment was calculated.
To ensure successful salt treatment, a separate randomized subset of mice was used for urine collection (0.8 % NaCl, n = 4; 8.0 % NaCl, n = 4). Urine was collected from each mouse by metabolic cages over a morning, 4-h time period, and analyzed for urinary Na+ excretion. Urinary Na+ concentration was measured using a sodium specific electrode (Model #: K-27504-30, Cole Palmer, Montreal, Canada).
Induction of transient cerebral ischemia
Transient cerebral ischemia was induced by temporary occlusion of the proximal left middle cerebral artery (MCA) using the intraluminal filament method previously described by Longa et al. [18] and later modified by Barber et al. [19]. Mice were anesthetized with isoflurane (3 % initial, 1–1.5 % maintenance), in oxygen to air ratio of 20:80 %. Core body temperature was monitored and maintained at 36.5 °C using a rectal probe with feedback to a heated surgical stage. Transcranial measurements of cerebral blood flow were obtained by laser Doppler flowmetry (Perimed, Periflux System 5010, North Royalton, OH, USA). A reduction of 70 % or more in regional MCA cerebral blood flow was considered adequate to induce ischemia. Occlusion was maintained for 30 min after which reperfusion was achieved. All mice recovered in a cage warmed to 34 °C by a circulating water blanket for 24 h and received 2 mg/kg of bupivacaine subcutaneous (SQ), 0.5 mL of 0.9 % saline SQ and 2 mg/kg of meloxicam SQ for analgesia immediately following surgery. Sham mice were used as controls; all surgical procedures were conducted with exception of MCA occlusion.
Vascular casting
Mice (n = 4) were transcardially perfused with 140 mmol/L NaCl, 10 mmol/L KCl, and 5 mmol/L ethylenediaminetetraacetic acid (EDTA) (pH 7.5). Batson’s #17 polymer (Polysciences Inc., Warrington, PA, USA) (2 mL) was injected (retrograde) through the thoracic aorta and left to polymerize for 24 h. The skull was isolated and the soft and bony tissue digested in 1 mol/L NaOH and 5 % Contrad 70 (Fisher Scientific, Pittsburgh, PA, USA) detergent.
Tissue collection
Mice were anesthetized via an intraperitoneal (IP) injection of euthanyl (100 mg/kg body weight) 24-h post-reperfusion. One millimeter cerebral sections were obtained. The fourth slice (Bregma ± 1 mm) was isolated and snap-frozen for molecular analysis. All other slices were stained in 0.5 % 2,3,5-triphenyltetrazolium chloride (TTC) diluted in 1× phosphate buffered saline (PBS). Infarct volumes were measured using ImageJ software (http://rsbweb.nih.gov/ij/). To account for the overestimation of infarct size due to cerebral swelling, infarct volume measurements were calculated according to Swanson et al. [20]. The heart was excised and dissected into individual chambers. Organ tissue weights were normalized to tibia length.
RNA isolation
Total RNA was isolated using a combination of Trizol (Tri Reagent, Molecular Research Centre, Inc. Burlington, ON, Canada) and a high pure RNA isolation kit (Roche Scientific Co., Laval, QC, Canada). All wash and elution steps were carried out according to the manufacturer’s instructions. RNA was measured using Nano-Drop 2000 spectrometer (Thermo Scientific, Wilmington, DE, USA).
Reverse transcription
Total RNA (1 μg) was reverse transcribed into cDNA using a high capacity cDNA reverse transcription kit (LifeTechnologies, Carlsbad, CA). Protocol was carried out according to the manufacturer’s instructions. Samples were incubated for 1 h with oligo-dT18 and an additional hour with random hexamers.
Real-time quantitative PCR (qPCR)
Oligonucleotide primers were designed using Primer Design 2.0 software (Scientific & Educational Software, Cary, NC, USA) from published mRNA sequences from NIH GenBank (www.ncbi.nlm.nih.gov/Genbank) (National Center for Biotechnical Information, Bethesda, MD, USA) (Table 1). Primer sets yielding a standard curve with efficiency within 1.7–2.0 were deemed adequate. Levels of mRNA expression were measured using the standard curve method for each gene with hypoxanthine–guanine phosphoribosyltransferase (HPRT) as a reference gene. All qPCR was performed using the LightCycler® 480 system II (Roche Scientific, Laval, QU, USA) and all samples were run in triplicates.
Data and statistical analysis
All statistical analyses were performed using Prism 6.0 Software (GraphPad Software Inc., La Jolla, CA, USA). Organ tissue weights to tibia length ratios, systolic blood pressure (SBP) measurements for NS and HS treatment groups, and infarct volumes were compared by unpaired, two-tailed Student’s t test and plotted as mean ± SD. Target gene mRNA expressional data were compared by two-way ANOVA with multiple comparisons and Tukey post hoc test. Data are presented as mean ± SEM. P ≤ 0.05 was deemed statistically significant. Potential outliers were determined for all statistical analyses using Grubb’s test.
Results
ANP+/− mice exhibit salt-induced hypertension, LVH and a complete circle of Willis
Body weight and cardiac mass to tibia length ratios are presented in Table 2. Previously, we have demonstrated that ANP+/− mice are salt sensitive [14, 15]. When fed HS chow over a 7 week period, ANP+/− mice exhibited significant LVH, a clear indicator for cardiac changes associated with the development of hypertension. Tibia lengths were used as a non-confounding variable to normalize all physical masses. As compared to ANP+/− mice fed NS chow, those fed a HS diet, demonstrated a 12 % increase in cardiac mass by the end of the dietary treatment period. Furthermore, consistent cerebrovascular casting demonstrated a complete circle of Willis in all mice with the presence of posterior communicating arteries (PCommAs) in both right and left cerebral hemispheres (Fig. 1).

Representative cerebrovascular cast. Arrows demonstrate presence of posterior communicating arteries forming a complete circle of Willis (n = 4)
Salt-induced hypertensive ANP+/− mice exhibit larger infarct volumes
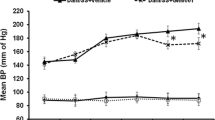
A subset of ANP+/− mice underwent non-invasive tail-cuff BP measurements to ensure the development of hypertension. As expected, a significant increase (22 %) in SBP was shown in ANP+/− mice fed HS chow (159.5 ± 4.5 mmHg) as compared to the NS (130.8 ± 10.2 mmHg) group (Fig. 2a). Only SBP is being reported as other investigators have demonstrated inaccuracies in measuring diastolic blood pressure (DBP) using non-invasive tail-cuff methods [21]. In addition, mice fed HS chow excrete more urinary Na+ as compared to those fed NS (Fig. 2b).

Systolic blood pressure (a) and urinary sodium excretion (b) measurements in NS and HS treatment groups. a Data represented as mean ± SEM, n = 4–5; b data represented as mean ± SD, n = 4. *P ≤ 0.05 using unpaired, two-tailed Student’s t test
Following middle cerebral artery occlusion (MCAO), mice treated with 8.0 % NaCl exhibited an approximate threefold increase in infarct size as compared to the 0.8 % NaCl group (P = 0.0002) (Figs. 2c, 3). Thus, the development of salt-induced hypertension in ANP+/− mice significantly impacted cerebral ischemic infarct size.

Representative brain images stained with TTC 24 h post reperfusion (a) and infarct volume measurements (b) demonstrating the significant infarct volume differences across ANP+/− treatment groups; a control sham (I), 0.8 % NaCl (II) and 8.0 % NaCl (III). All brain sections (1 mm) were stained with TTC to measure infarct volume. Arrows locate infarcted area. b Data are represented as mean ± SD, n = 6. *P ≤ 0.05 using unpaired, two-tailed Student’s t test
Cerebral vasoactive gene expression in response to salt-induced hypertension and transient cerebral ischemia
The mRNA expression levels of various vasoactive system genes were assessed by qPCR methods. Sham operated mice were used as controls to better assess gene expression differences in response to stroke and/or dietary treatment.
Natriuretic peptide (NP) system
Gene mRNA expression levels of C-type natriuretic peptide (Nppc) and its specific receptor, natriuretic peptide receptor 2 (Npr2) demonstrated no significant differences across dietary treatment or across surgical intervention (refer to Fig. S-1 in the Supplemental Figures). All components of the NP system, including ANP (Nppa), B-type natriuretic peptide (Nppb) and natriuretic peptide receptor 1 (Npr1) were assessed; however, expression levels of these peptides and receptor were undetectable by qPCR methods.
Nitric oxide synthase (NOS) system
Figure 4 summarizes the changes in mRNA expression of neuronal nitric oxide synthase (Nos1) (Fig. 4a), endothelial-derived nitric oxide synthase (Nos3) (Fig. 4b) and receptors soluble guanylyl cyclase alpha 1 (sGC 1α1 ) (Fig. 4c) and beta-1 (sGC 1β1 ) (Fig. 4d). A general, non-significant trend toward increased expression in response to HS was observed in both receptor genes as well as Nos1 with no influence from stroke. The expression of Nos3 significantly increased in stroked, HS-treated (HS-MCAO) mice as compared to the NS, stroke (NS-MCAO), and HS-treated sham (HS-Sham) groups. Nos3 levels appear to change in response to the combined effect of HS treatment and stroke.

Changes in mRNA expression of Nos1 (a), Nos3 (b), sGC a1 (c), and sGC b1 (d) in left hemispheric brain tissue of sham and left MCAO mice fed either NS or HS diet. Data are presented as mean ± SEM. *P ≤ 0.05, using two-way ANOVA with Tukey’s post hoc test. n = 6
Renin–angiotensin (RAS) system
The resultant mRNA expression of angiotensinogen (Agt), angiotensin-converting enzyme (Ace), angiotensin II receptor type 1a (Agtr1a), and angiotensin II receptor type 2 (Agtr2) did not show significant changes although some trends were observed. Both Agt and Agtr1 target genes demonstrated a pattern of decreased gene expression in response to stroke in both dietary groups equally, thus increased salt did not influence these genetic changes. Agtr2 mRNA expression increased in response to high salt alone. (refer to Fig. S-2 in the Supplemental Figures).
Endothelin (ET) system
Endothelin-1 (Et-1) mRNA levels demonstrated a trend toward increased expression following stroke in normotensive mice (Fig. 5a). A significant increase in Et-1 expression was observed in the HS-MCAO group as compared to all other treatment groups (Fig. 5a). This pattern of expression suggests that the combined presence of increased sodium, hypertension and stroke stimulates this system, above all, to respond. Although, no significant changes were observed, a similar trend of increased endothelin receptor A (Et-A) mRNA expression was seen in the HS-MCAO group compared to all others, which corresponds to Et-1 levels (Fig. 5b).

Changes in mRNA expression of Et-1 (a) and Et-A (b) in left hemispheric brain tissue of sham and left MCAO mice fed either NS or HS diet. Data are presented as mean ± SEM. *P ≤ 0.05, using two-way ANOVA with Tukey’s post hoc test. n = 6
Vascular endothelial growth factor (VEGF) system
Vascular endothelial growth factor type A (VEGFA), vascular endothelial growth factor receptor 1 (Flt-1), and vascular endothelial growth factor receptor 2 (Flk-1) were assessed by qPCR to determine whether cerebral ischemia and/or hypertension stimulates this physiological response. A general trend to increased Vegfa expression (Fig. 6a) in response to salt was seen. Receptors Flt-1 (Fig. 6b) and Flk-2 (Fig. 6c) exhibited similar expression patterns. Both receptors showed an increase in mRNA expression in the HS-MCAO group; however, only the Flk-1 expression was significantly increased. As a result, these data demonstrate angiogenic responses to a combined salt-induced hypertensive and cerebral ischemic event.

Changes in mRNA expression of Vegfa (a), Flt-1 (b), and Flk-2 (c) in left hemispheric brain tissue of sham and left MCAO mice fed either NS or HS diet. Data are presented as mean ± SEM. *P ≤ 0.05, using two-way ANOVA with Tukey’s post hoc test. n = 6
Discussion
We have used a novel approach to study the effects of salt-induced hypertension on adaptations in vasoactive systems in acute stroke. The genotype expressed by the ANP+/− mouse allowed us to examine the effects of salt-sensitivity without confounding from genetic influences. Experiments using the spontaneously hypertensive rat (SHR) and spontaneously hypertensive stroke-prone (SHR-SP) rat developed by Okamoto et al. [22], often use the Wistar-Kyoto (WKY) rat as the control group, a genetically disparate animal from SHR and SHR-SPs [23]. The genetic factors by which these rats develop hypertension or become more vulnerable to stroke is still poorly understood. This is also the case with Dahl salt-sensitive and Dahl salt-resistant rat models [24, 25] for which the genetic basis of salt-sensitivity has also not been elucidated. This is the first study in which the influence of salt-induced hypertension on stroke tissue injury has been examined using treatment and control groups with well-defined and identical genotypes. Through evaluation of urinary sodium excretion, SBP and cardiac hypertrophy, we have validated the establishment of salt-induced hypertension in the ANP+/− mouse. This is in accordance with previously published work [14, 15, 26].
Vascular casting in the ANP+/− mouse demonstrated an intact circle of Willis without anastomotic variations, thus allowing comparison of cerebral responses across treatment groups following stroke. As a result, we can be confident that the increased infarct size we have observed in the HS group is due to mechanistic changes that have been influenced by the acquired salt-induced hypertensive state.
Cerebrovascular adaptations, resulting from increased salt and high BP, could contribute to the large infarct volumes experienced by hypertensive ANP+/− mice. The NP and RAS systems did not show significant changes in mRNA expression of the peptides or receptors investigated implying that these systems are not responsible for cerebrovascular changes in this state. However, changes involving NOS1, NOS3, ET-1, and VEGF were seen.
Nitric oxide (NO) production is known to play a pathophysiological role for various vascular diseases, in particular, those related to aberrant blood flow and inflammatory response. Increases in NOS1 have been implicated with larger infarct volumes due to its neurotoxic effects, while increases in NOS3 play a neuroprotective and angiogenic role [27, 28]. In response to the HS dietary treatment, cerebral mRNA expression of NOS1 increased in ANP+/− mice, suggesting that NOS1-associated neurotoxicity may be stimulated in the presence of high sodium. Cerebral NOS3 expression increased markedly in response to the combined presence of high sodium and stroke, perhaps reflecting an induced endogenous protective response.
The effect of NO on vascular smooth muscle cells is calcium dependent. Binding of NO to sGC receptors catalyzes the conversion of GTP to cGMP causing further alterations in calcium availability leading to vasodilation. Gene mapping of sGC receptor isoforms has revealed a connection between expression of these receptors, salt-sensitivity, and BP [29]. Our molecular analysis demonstrates a similar role for the α-1 and β-1 sGC isoforms as trending increased mRNA expression was seen in the hypertensive animal.
The ET system is responsible for vasoconstriction of blood vessels, in particular, small arterioles, and controlling myogenic tone of capillary beds. The plasma concentration of ET-1 is increased in acute stroke patients [30, 31] and salt-sensitive/hypertensive individuals [32]. In addition, ET-1 has been shown to disrupt microcirculation in the brain rendering the cerebrovasculature at risk for damage [33]. Although not significant, increased Et-1 mRNA expression was seen in response to stroke. In contrast, there was a significant rise in cerebral mRNA expression of Et-1 in the HS-MCAO group alluding to a potential role for Et-1 in contributing to the large infarct volumes in this subset of mice. Future experimentation is required to better understand the mechanism of these alterations and the role of ET-1.
VEGF is involved in angiogenesis [34] and enhanced vascular permeability [35–37]. Ischemic conditions induce the production of VEGF to promote new vessel formation [38], but it remains unclear if this is beneficial or detrimental to stroke injury. Increased Vegfa, Flt-1, and Flk-2 in response to HS may reflect a propensity for increased vascularization and possibly contribute to larger infarct size. Further experimentation would be needed to clarify this.
Conclusion
We have successfully examined potential influences of salt-induced hypertension on stroke with genetically identical treatment and control groups using the ANP+/− mouse. Our data indicate that a salt-induced state of hypertension renders the brain more susceptible to tissue damage following an ischemic event. We provide evidence for the notion that the large infarct volumes observed are due to alterations in vasoactive systems, in particular the NOS, ET, and VEGF systems, which change the response and activity of the cerebrovasculature. Further investigation is required to identify the mechanistic role the proposed vasoactive systems play with respect to salt-induced hypertension and stroke.
References
World Health Organization (2013) GlobalHealthRisks_report_full, pp 1–70
Lawes CM, Bennett DA, Lewington S, Rodgers A (2002) Blood pressure and coronary heart disease: a review of the evidence. Semin Vasc Med 02:355–368. doi:10.1055/s-2002-36765
Lawes CM, Bennett DA, Feigin VL, Rodgers A (2004) Blood pressure and stroke: an overview of published reviews. Stroke 35:1024. doi:10.1161/01.STR.0000126208.14181.DD
Dahl LK (1961) Possible role of chronic excess salt consumption in the pathogenesis of essential hypertension. Am J Cardiol 8:571–575. doi:10.1016/0002-9149(61)90137-0
Stamler J, Rose G, Elliott P, et al (1989) The INTERSALT study. Hypertension
Strazzullo P, D’Elia L, Kandala N-B, Cappuccio FP (2009) Salt intake, stroke, and cardiovascular disease: meta-analysis of prospective studies. BMJ 339:b4567–b4567. doi:10.1136/bmj.b4567
Yu HCM, Burrell LM, Black MJ et al (1998) Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation 98:2621–2628. doi:10.1161/01.CIR.98.23.2621
Elliott P, Stamler J, Nichols R et al (1996) Intersalt revisited: further analyses of 24 hour sodium excretion and blood pressure within and across populations. Intersalt Cooperative Research Group. BMJ 312:1249–1253
Nagata C, Takatsuka N, Shimizu N, Shimizu H (2004) Sodium intake and risk of death from stroke in Japanese men and women. Stroke 35:1543–1547. doi:10.1161/01.STR.0000130425.50441.b0
Gardener H, Rundek T, Wright CB et al (2012) Dietary sodium and risk of stroke in the Northern Manhattan study. Stroke 43:1200–1205
Kannel WB, Dawber TR, Sorlie P, Wolf PA (1976) Components of blood pressure and risk of atherothrombotic brain infarction: the Framingham study. Stroke 7:327–331
Kannel WB, Wolf PA, Verter J, McNamara PM (1996) Epidemiologic assessment of the role of blood pressure in stroke the Framingham study. JAMA 276:1269. doi:10.1001/jama.1996.03540150071040
Fujii K, Weno BL, Baumbach GL, Heistad DD (1992) Effect of antihypertensive treatment on focal cerebral infarction. Hypertension 19:713–716
John SW, Krege JH, Oliver PM et al (1995) Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science 267:679–681
Sangaralingham SJ, Tse MY, Pang SC (2007) Estrogen protects against the development of salt-induced cardiac hypertrophy in heterozygous proANP gene-disrupted mice. J Endocrinol 194:143–152. doi:10.1677/JOE-07-0130
Angelis E, Tse MY, Pang SC (2005) Interactions between atrial natriuretic peptide and the renin–angiotensin system during salt-sensitivity exhibited by the proANP gene-disrupted mouse. Mol Cell Biochem 276:121–131
Feng M, DiPetrillo K (2009) Non-invasive blood pressure measurement in mice. Methods Mol Biol 573:45–55. doi:10.1007/978-1-60761-247-6_3
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Barber PA, Hoyte L, Colbourne F, Buchan AM (2004) Temperature-regulated model of focal ischemia in the mouse: a study with histopathological and behavioral outcomes. Stroke 35:1720–1725. doi:10.1161/01.STR.0000129653.22241.d7
Swanson RAR, Morton MTM, Tsao-Wu GG et al (1990) A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab 10:290–293. doi:10.1038/jcbfm.1990.47
Feng M, Whitesall S, Zhang Y et al (2008) Validation of volume–pressure recording tail-cuff blood pressure measurements. Am J Hypertens 21:1288–1291. doi:10.1038/ajh.2008.301
Okamoto K, Aoki K (1963) Development of a strain of spontaneously hypertensive rats. Jpn Circ J 27:282–293
H’Doubler PB, Peterson M, Shek W et al (1991) Spontaneously hypertensive and Wistar Kyoto rats are genetically disparate. Lab Anim Sci 41:471–473
Rapp JP (1982) Dahl salt-susceptible and salt-resistant rats. A review. Hypertension 4:753–763
Dahl LK, Heine M, Tassinari L (1962) Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature 194:480–482. doi:10.1038/194480b0
Armstrong DWJ, Tse MY, O’Tierney-Ginn PF et al (2013) Gestational hypertension in atrial natriuretic peptide knockout mice and the developmental origins of salt-sensitivity and cardiac hypertrophy. Regul Pept 186C:108–115. doi:10.1016/j.regpep.2013.08.006
Huang Z, Huang PL, Panahian N et al (1994) Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 265:1883–1885
Huang Z, Huang PL, Ma J et al (1996) Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J Cereb Blood Flow Metab 16:981–987. doi:10.1097/00004647-199609000-00023
Azam M, Gupta G, Chen W et al (1998) Genetic mapping of soluble guanylyl cyclase genes : implications for linkage to blood pressure in the Dahl rat. Hypertension 32:149–154. doi:10.1161/01.HYP.32.1.149
Alioglu Z, Orem A, Bulbul I et al (2013) Evaluation of plasma endothelin-1 levels in patients with cerebral infarction. Angiology 53:77–82
Ziv II, Fleminger GG, Djaldetti RR et al (1992) Increased plasma endothelin-1 in acute ischemic stroke. Stroke 23:1014–1016
Ergul A (2000) Hypertension in black patients: an emerging role of the endothelin system in salt-sensitive hypertension. Hypertension 36:62–67
Faraco G, Moraga A, Moore J et al (2013) Circulating endothelin-1 alters critical mechanisms regulating cerebral microcirculation. Hypertension 62:759–766. doi:10.1161/HYPERTENSIONAHA.113.01761/-/DC1
Leung DW, Cachianes G, Kuang WJ et al (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246:1306–1309
Hayashi T, Abe K, Suzuki H, Itoyama Y (1997) Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke 28:2039–2044. doi:10.1161/01.STR.28.10.2039
Lennmyr F, Ata KA, Funa K et al (1998) Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol 57:874–882
Sun Y, Jin K, Xie L et al (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest 111:1843–1851. doi:10.1172/JCI17977
Shweiki D, Itin A, Soffer D, Keshet E (1992) Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359:843–845. doi:10.1038/359843a0
Acknowledgments
The authors would like to thank Dr. Alastair Ferguson, Department of Biomedical and Molecular Sciences, Queen’s University, for the use of the CODA non-invasive tail-cuff BP system. NMV is a recipient of the Franklin Bracken Student Fellowship. Research equipment funding (real-time PCR) was provided by the Canadian Foundation of Innovation (CFI).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ventura, N.M., Peterson, N.T., Tse, M.Y. et al. Molecular adaptations in vasoactive systems during acute stroke in salt-induced hypertension. Mol Cell Biochem 399, 39–47 (2015). https://doi.org/10.1007/s11010-014-2230-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-014-2230-0