
Abstract
Chymotrypsin-like cysteine protease, also known as main protease (3CLpro/Mpro) of SARS-CoV-2, is highly conserved among various coronaviruses. Hence, therapeutics targeting the main protease are likely to show broad-spectrum activity. Peptides are a promising avenue for antiviral therapeutics as they are capable of offering a sustainable strategy to combat infectious diseases. In this work, we screened clinically proven antimicrobial peptides against the cysteine protease of SARS-CoV-2 using state-of-the-art cheminformatics methods including docking and dynamics simulation, statistical analysis, and structure-activity relationship studies. From the molecular docking investigation, three peptides were chosen which showed the high binding affinities [DRAMP18152 (∆G = − 56.56 kcal/mol), DRAMP18160 (∆G = − 59.9 kcal/mol), DRAMP20773 (∆G = − 56.2 kcal/mol)] and active interactions with His41 and Cys145 residues. Molecular dynamics simulation was employed over 250 ns on these three peptide-Mpro complexes. The MD simulation results reflect the high inhibitory potential of DRAMP18152, DRAMP18160, and DRAMP20773 against Mpro. These three peptides were synthesized using standard solid phase peptide synthesis. Purity (> 90%) and identity of the peptides were established by liquid chromatography and electrospray ionization-mass spectrometry. FRET-based protease assay was conducted for these three top candidates in which only DRAMP18160 showed the inhibition efficiency with an estimated 50% inhibitory concentrations of 59 µM with low cytotoxicity. These results suggest that pursuing further development of peptide-based inhibitors for antiviral applications may be a fruitful endeavor and yield novel antiviral therapeutics.
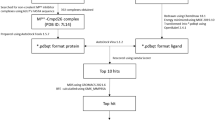
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The outbreak of SARS-CoV-2 originates in Wuhan, Hubei Province of mainland China. Since then, the disease has spread to many other countries and is now considered a global pandemic. SARS-CoV-2, like other similar viruses, is a single-stranded RNA virus that not only infects humans but animals as well. It appears to have transitioned from wildlife to humans in a seafood market in Wuhan. After contracting this disease, most symptomatic patients experience fever, cough, weakness, and other symptoms of upper respiratory tract infections. In more critical cases, the infection can progress to pneumonia and/or death (Velavan and Meyer 2020). As of August 26th, 2023, there have been approximately 769 million cumulative cases of coronavirus infections and globally over 6.95 million deaths (WHO 2023).
Research has quickly identified the crucial proteins of SARS-CoV-2 and their roles in viral infection. The coronavirus main protease (3CLpro/Mpro) has a prominent function in the viral replication in COVID-19. More specifically, it is responsible for the processing of viral polyproteins. This enzymatic structure breaks down those viral polyproteins into functional units by proteolysis. Those functional units can then virally replicate and package within the host cells (Tahir ul Qamar et al. 2020; , Liu et al. 2020). Zhang et al. and Jin et al. demonstrated the x-ray structure of SARS-CoV-2 Mpro, with the crystallographic coordinates available in the Protein Data Bank (Zhang et al. 2020; , Jin et al. 2020). The SARS-CoV-2 Mpro crystal structure shows a protein dimer with three domains for each protomer. After dimerization of two protomers containing three distinctive domains with catalytic dyad His41-Cys145, Mpro becomes activated and cleaves at least 11 specific cleavage sites in the polyprotein1a/1ab derived from ORF1a/ab (Zhang et al. 2020; , Jin et al. 2020). In most cases, the recognition sequence mainly locates in Leu-Gln↓ (Ser, Ala, Gly) (↓ marks the scissile bond). The viral replication process could be interrupted if the activity of this enzyme is inhibited. As no human protein homologs with a comparable cleavage specificity are known, such antiviral agents are suspected not to be toxic, making Mpro an ideal drug target among coronaviruses (Zhang et al. 2020).
Peptide therapeutics have received great attention over the past decade due to multiple reasons. They have seen widespread use in applications pertaining to medicine and biotechnology. Generally, peptides are known for being selective signaling molecules that bind particularly well to cell surface receptors. Not only that, but they have shown exceptional safety, efficacy, and tolerability in humans. Due to these characteristics, it is not unusual for scientists to be intrigued by its potential application in pharmaceutical research and development. Compared to protein-based biopharmaceuticals, peptide therapeutics production is comparatively simple. The manufacturing costs are also minimal when compared to those of small molecules. However, it is important to note that peptides collected from natural sources face some challenges when attempting to apply them as therapeutic agents. They have disadvantageous characteristics such as poor chemical and physical stability. Scientists have been able to solve these weaknesses through what is called “traditional peptide design.” As of 2015, there were nearly 140 therapeutic peptides being assessed in the clinical trials (Magana et al. 2020). Many of these peptides show broad antimicrobial properties and have made it to various stages of FDA approval. Although the antimicrobial effectiveness of these peptides is established, the method by which they achieve their action is still unknown in many cases. Scientists have also turned to antimicrobial peptide therapeutics due to the rapid surge of antibiotic resistance around the world (Mahlapuu et al. 2016). Additionally, antimicrobial peptides are one of the human body’s main components in innate immunity and can also be employed in peptide therapeutics (Mahlapuu et al. 2016).
In our current study, clinically proven antimicrobial peptide sequences were collected from the data repository of antimicrobial peptides (DRAMP) website and screened with the aid computational program targeting the cysteine protease of the SARS-CoV-2. Molecular docking was initially employed to screen the promising peptides targeting the protease of SARS-CoV-2. The selected peptide-protease complexes were assessed by 250 ns molecular dynamics (MD) simulation. Based on the MD results, the three best candidate peptides were synthesized. Standard solid phase peptide synthesis was used for the peptide synthesis and their inhibition efficiency and cell viability were evaluated. The overall aim of this study is to combine a computer aided screening and molecular dynamics approaches to identify potential candidates and then verify the inhibition efficiency with protease assay to provide promising insights into designing and developing of potential peptides for therapeutic intervention against COVID-19.
Methods
Molecular Docking
A total of 13 peptides comprising six antibacterial and eight antimicrobials in the clinical trial were chosen from DRAMP 2.0 database (Kang et al. 2019). Although DRAMP database listed − 78 peptides that are currently in the clinical trials, not all peptides’ sequences are disclosed. Peptides were selected based on their available sequence, small size containing less than 25 amino acids as small peptides are easy to synthesize by solid phase peptide synthesis protocols, and the ability to generate their 3D model. Selected peptides were modelled by CABS-Fold (Blaszczyk et al. 2013). The peptides were docked to the 3D x-ray structure of Mpro (PDB ID: 6Y2G) using PatchDock. The docked pose of 1,000 peptide-protease complexes were generated from PatchDock. The docked complexes were then refined by FireDock (Zhang et al. 2020; , Schneidman-Duhovny et al. 2005; , Andrusier et al. 2007). Molecular dynamics (MD) simulation was implemented on the peptides that exhibited excellent binding affinities and robust interactions with His41 and Cys 145 residues. Binding free energy was also determined for the top 3 peptides by PRODIGY protocol (Xue et al. 2016).
Implementation of the Molecular Dynamics (MD) Simulations
MD simulation was implemented on the best three peptide candidates (DRAMP18152, DRAMP18160, and DRAMP20773) complexed with Mpro retrieved from a molecular docking study for 250 ns. AMBER 14 force field was considered for calculation and the simulation was carried out in YASARA Dynamics software at a simulation speed of 1.25 fs time step and at 7.4 pH, 310 K temperature which was regulated using Berendsen thermostat (Krieger et al. 2012; 10989- Dickson et al. 2014). Water molecules considering 0.998 g/cm3density, and NaCl salt (0.9%) were added to the system for neutralization. The Particle-mesh Ewald method was implemented to detect long-range electrostatic interactions. A cubic simulation cell was built according to the periodic boundary conditions for the simulation where the cell dimensions were 20 Å greater than the protease-peptide complex. Finally, the MD trajectories were collected at every 100 ps interval for the post data processing. PRODIGY protocol, which determines binding free energy (BFE) constructed on intermolecular contacts and properties, was employed on last 100 ns MD snapshots for BFE calculations (Magana et al. 2020).
Principal Components Analysis (PCA)
PCA investigation demonstrates the unseen structural and energy profile within various sets (Islam et al. 2020; , Islam et al. 2019). The structural information such as bond distances, bond angles, dihedral angles, planarity and energy information including Van der Waals energies, and electrostatic energies are included for PCA analysis. MD trajectory data of the last 100 ns for both Apo-form of the protease and peptide-protease complexes were taken into consideration for PCA investigation.
The multivariate elements were placed in the matrix X and cut down into an outcome of two new matrices by employing the equation shown below.
In this equation, matrix X is the result of two new matrices such as Tk and PkT, Tk is the matrix of scores, which indicates the relation of the sample between them, Pk serves as the loading matrix, which conveys the information about the connection of variables to each other. Herein, k is defined as the number of factors into the model. The unmodeled variance are assigned to E.
Peptides Structure Activity Relationship (SAR) Analysis
All peptides were chosen for the structure-activity relationship (SAR) studies. ProtParam tool was utilized to determine related properties of the peptides including acidic residues, basic residues, aromatic residues, polar residues, nonpolar residues, molecular weight, theoretical pI and number of amino acids (Wilkins et al. 1999). Stepwise multiple linear regression was executed at first, taking these peptide properties to estimate the computed binding affinity between the protease of SARS-CoV-2 and the test peptides. To further investigate the structural variance, a PCA was conducted considering the 5 most significant properties of peptides to aggregate them in a biplot.
Solid Phase Peptides Synthesis and LC–MS Characterization
Peptides were synthesized on Fmoc-Rink amide resin via automated solid phase synthesis employing Fmoc-protected strategy. A Liberty Blue Microwave Synthesizer (CEM) was employed to perform the synthesis and yield the nascent resin-bound peptide. DMF was used as the main solvent. For performing deprotection, 20% piperidine solution in DMF was used. N,N′-Diisopropylcarbodiimide was used as an activator with Oxyma which served as an activator-base. After synthesis, the resin-bound peptide was washed several times with DCM (3 × 15mL) and allowed to dry for 1 h on a vacuum filter. The cleavage cocktail was prepared by combining TFA, water, and triisopropylsilane in proportions of 95:2.5:2.5 v/v/v for a total volume of 10 mL. The freshly prepared cocktail was combined with the dry product in a reaction vessel and heated to 42 °C in a water bath. This reaction was allowed to progress for 45 min with occasional shaking to ensure proper mixing of components. The suspension was then filtered using a frit syringe and the filtrate was evaporated to a reduced volume using nitrogen gas. Once the volume was reduced to 5 mL the peptide product was precipitated by using 45 mL of cold diethyl ether. Centrifugation at 7500 rpm for 10 min at 0 °C yielded the peptide pellet. After decantation, the peptide was dissolved using 10 mL of 10% acetic acid solution. Subsequently, the solution was frozen in an − 80 °C freezer and later lyophilized using Labconco FreeZone 4.5 Freeze Dryer Lyophilizer for 24 h to yield the final dry peptide product.
The HPLC system (Agilent 1290 Infinity II) was used for all LC experiments. Peptides were dissolved in 0.1% formic acid containing 10–40% acetonitrile. ZORBAX C18 column was utilized, and UV detector excitation was set at 214 nm. Water (A) and acetonitrile (B) with 0.1% FA were used for the mobile phase. A short gradient of 12 min was developed with 20% B in 2 min, 20–40% B from 2 to 6.6 min, 40–90% B from 6.6 to 9 min. Mass spectrometer was operated in positive mode on an LTQ XL mass spectrometer (Thermo Scientific). For ionization source, the heated electrospray ionization (HESI) was employed. The HESI spray voltage was approximately 4 kV and the heater temperature was fixed at 50 °C. Relatively higher sheath gas flow rate is set at 40 with an Auxiliary gas flow being kept at 7. The capillary temperature, capillary voltage and capillary tube lens were fixed at 350 °C, 10 V, and 100 V, respectively. All mass spectrometry experiments were conducted with 3 microscan and a maximal injection time of 50ms. Mass spectra were obtained using a mass range of 200–2000 m/z.
Protease FRET Activity and Cell Viability Assay
The natural cleavage target (KTSAVLQ↓SGFRKME) of Mpro was purchased from CPC Scientific already modified with the chromophore-fluorophore pair DABCYL-EDANS. The fluorogenic substrate peptide DABCYL-KTSAVLQSGFRKME-EDANS was reconstituted using DMSO to produce a stock solution (2.5 mM). The main protease (purchased from Sigma Aldrich) assay was performed on an opaque 96 well plate with a total volume of 100 µL comprised of buffer (LC–MS grade water, Tris-HCl 20 mM, pH 7.41), Main protease enzyme (5 µg/mL), fluorogenic substrate (5 µM), and inhibitor. All components other than substrate were plated first with thorough mixing and allowed to incubate for 15 min. Fluorogenic substrate was added in each well and the plate was allowed to develop for 45 min at room temperature. Fluorescent intensity of each well was determined using a SpectraMax iD5 multiplate reader at an excitation wavelength of 340 nm and an emission wavelength of 490 nm. Graphpad Prism v9 software was used to compute the non-linear regression curve and determine IC50 from that function.
Cell viability assay of the DRAMP18160 was performed with Vero E6 cells (ATCC, American Tissue Culture Type). These cell lines are maintained in Dulbecco’s modified eagle’s medium (DMEM) containing phenol red. Moreover, fetal bovine serum (FBS) at 10% and L glutamine at 1% were added as supplements. The temperature was retained at 37 °C and maintained atmosphere with 5% CO2. Utilizing 0.25% trypsin-EDTA, cells were harvested from flasks. Beckman Coulter Epics XL MCL Flow Cytometry Analyzer (Beckman, CA, USA) was used for cell viability test by staining with propidium iodide. The DRAMP18160 peptide was solubilized at 1 mM concentration using 10% DMSO. The final concentrations of DMSO were kept close to 0.5%. Vero E6 cells were added into a 96-well plate. Cells were added until the density was 10 K cells/well in 50 µL of assay medium. Incubation followed for 24 h at 37 °C and 5% CO2 with high humidity. After incubation, 5 µL of DRAMP18160 ranging from concentrations of 33 to 0.5 µM was added to incubated cells for 72 h. Luminescence measurements were then conducted utilizing a PerkinElmer plate reader (PerkinElmer, MA, USA).
Results and Discussion
Molecular Docking
The cysteine protease of SARS-CoV-2 catalyzes the proteolytic cleavage of viral polypeptides. SARS-CoV-2 infection cycle and viral pathogenesis can thus be halted by blocking the main protease. The binding groove of SARS-CoV-2 Mpro has a catalytic dyad consisting of Cys145 and His41 amino acids; peptides must bind to these crucial amino acids to interrupt the enzyme’s activity. The rigid body docking approach of PatchDock-FireDock was executed for peptides docking against 3CLpro. The peptide sequence, length (collected from DRAMP 2.0 database), charge, hydrophobicity (calculated from Peptide2.0), estimated solubility (calculated from PepCalc) and binding affinity (predicted by FireDock) of 13 peptides are summarized in Table 1. DRAMP18152, DRAMP18160, and DRAMP20773 were the highest ranked peptides based on the stronger binding affinity scores of − 56.56, − 59.9, − 56.2 respectively, to the target protein. Among these three peptides, DRAMP18160 contained four charge and showed good water solubility. The intermolecular contacts with the nearby residues belonging to Mpro’s active site are represented in the docking scores (Fig. 1). DRAMP18152, DRAMP18160 shows interactions with both the His-Cys catalytic dyad (His41and Cys145) along with the other active site residues but DRAMP20773 exhibits binding to only one of the catalytic amino acids (Cys145) of the cysteine protease. Both hydrogen bond and hydrophobic interaction were predominantly observed in the non-bonding interactions of the selected peptides bound to the target protein. DRAMP18152, DRAMP18160, and DRAMP20773 have greater interactions with the catalytic amino acid Cys145, with bond distances of 3.95, 2.59, and 2.72, respectively. (Table 2). Prodigy was used to measure the binding energy of the three complexes, which measures the binding free energy of the peptide-protease complex based on intermolecular interactions. The calculated binding free energy of DRAMP18152, DRAMP18160, and DRAMP20773 peptides with Mpro are – 10.3, − 10.5, and – 8.5 kcal/mol (Table 2). Thus, we observed that the binding affinities calculated by FireDock agreed with the score of PRODIGY. To further ascribe the stability of the peptides revealed from docking, 250 ns MD simulation was performed.

A Molecular surface representation of SARS-CoV-2 Mpro with the covalently docked peptide substrate in the protein binding site. B–D Nonbonding interactions of three selected peptide candidates with the main protease of SARS-CoV-2. B DRAMP18152 C DRAMP18160 D DRAMP20773
Molecular Dynamics (MD) Simulation
The best three peptide candidates (DRAMP18152, DRAMP18160, and DRAMP20773) as well as for Mpro-apo were considered for 250 ns MD simulation for further validation of the docking results. Root-mean-square-deviation (RMSD), solvent-accessible-surface-area (SASA), Radius of gyration (Rg), and root-mean-square-fluctuation (RMSF) were investigated for MD simulation trajectory analysis. DRAMP18152, DRAMP18160, and DRAMP20773 complexes maintained the most stable RMSD profile, as evidenced by Fig. 2A. A marginal hike in RMSD trajectory was observed for DRAMP18152 towards 240ns, but it does not alter peptides binding to the 3CLpro’s active site. The mean RMSD recorded for the DRAMP18152, DRAMP18160, and DRAMP20773 complexes were 1.99 ± 0.36 Å, 2.09 ± 0.24 Å, 1.74 ± 0.30 Å respectively, which is lower than apo-Mpro’s RMSD (2.39 ± 0.46 Å) (Table 3). A similar trend was documented in Rg trajectory analysis of DRAMP18152, DRAMP18160, and DRAMP20773 complexes, where all three showed stable Rg profiles during the simulation time span (Fig. 2B). DRAMP20773 reported lower-most Rg scores, implying that the peptide caused greater compactness and rigidity in Mpro upon binding (Table 3). Moreover, the lowest solvent accessible surface area (SASA) was noticed for DRAMP20773 (Fig. 2C) (Table 3). Root-mean-square-fluctuation (RMSF) based on the single residue for all protease-peptide complexes were quite similar for all three simulated peptides-protein complexes (Fig. 2D). As shown in Fig. 3, it is confirmed that all three peptides were stable in their initial binding position, exhibiting minimal deviation throughout the 250 ns MD simulation. The overall count of intermolecular H-bonds in peptide-protein complexes is examined to further understand conformational stability. The integrity of protein structure is strongly influenced by hydrogen bonds. The mean count of H-bonds registered in apo-protein, DRAMP18152, DRAMP18160, and DRAMP20773 complexes are 509, 541, 535, and 523, respectively (Fig. 2E). The DRAMP18152-Mpro complex shows the highest average number of hydrogen bonds, whereas apo-Mpro form records the lowest average number of hydrogen bonds. The average binding free energy of DRAMP18152, DRAMP18160, and DRAMP20773 complexes with Mpro are − 7.03 ± 0.59, − 8.90 ± 0.24, − 0.7.74 ± 0.33 kcal/mol, respectively (Table 3). DRAMP18160 has shown the highest binding free energy calculated from MD snapshots, which is similar to the result determined for the docked complexes. Overall, the MD simulation analysis shows the peptides are very stable when bound to the protein Mpro’s active site.

Molecular dynamics simulation. A Root mean square deviation (RMSD); B Radius of gyration (Rg); C Solvent accessible surface area (SASA); D Root mean square fluctuation (RMSF); E Total hydrogen bond count of top three peptides-Mpro complexes over 250 ns MD simulation

Representative snapshots during 250 ns MD simulation. A Mpro-DRAMP18152 (red) B Mpro-DRAMP18160 (magenta) C Mpro-DRAMP20773 (purple)
Principle Component Analysis (PCA)
For revealing the structural and energy profiles variation among peptide-protein complexes with Apo protein, a PCA model containing of 4 training sets (Mpro-Apo and three Mpro-peptide complexes) was established during MD simulation. Here, PC1 and PC2 illustrated 66.6 and 17% of the total 83.6% of variances. Although, the PC1 and PC2 plot (Fig. 4A) revealed that all three Mpro-peptide complexes resided almost near to each other moving from Apo. The loading plot (Fig. 4B) indicates that the bond distance, bond angle, van der Waals energy, and coulomb played an important role in this clustering pattern. The DRAMP18152 complex is at a great distance from the apo-protein, reflecting large transformation in its Coulomb interactions. The downward shift of DRAMP18152, DRAMP18160 and DRAMP20773 complexes in Fig. 4B indicates a large alteration in the dihedral angles and planarity factors during the simulation.

Principle component analysis (PCA). A Score plot of peptide-Mpro complexes including Apo-Mpro; B Loading plot of peptide-Mpro complexes including Apo- Mpro
Structure-Activity Relationship (SAR) Analysis
Based on the relevant properties of the peptides, multiple linear regression (MLR) assessment was implemented to investigate the critical predictors of the binding and interaction of the test peptides (Table 4). MLR analysis suggests that aromatic, non-polar, basic residues, length, and molecular weight of the peptides were the major predictors which illustrated the predominant feature of hydrogen bonding and hydrophobic interactions observed in the protease-peptide complexes. This regression model is also valid for peptides-protein dynamic evolution throughout 250 ns MD simulation. In the clustering pattern of the top-performing peptides (DRAMP18152, DRAMP18160 and DRAMP20773), nonpolar residues are major contributors to PC1 and the basic residues to PC2, which combinedly represent 92.5% structural variance (Fig. 5). The SAR analysis suggests that by combining basic and non-polar residues, effective peptides for SARS-CoV-2 Mpro can be designed.

The biplot of the selected 13 high binding affinity peptides clustered based on five peptide properties
Peptide Synthesis and Characterization
These peptides were synthesized by standard Fmoc-based synthesis protocols. The synthesis and purification of these peptides were verified by liquid chromatography and electrospray ionization-mass spectrometry. The acquired liquid chromatograms and mass spectra of DRAMP18152, DRAMP18160 and DRAMP20773 are shown in Fig. 6. The overall purity of these peptides is over 90.0%. The chromatogram of DRAMP152 showed a single intense peak. This peptide is eluted early at 1.9 min. In the mass spectrum, three peaks are detected. The peak of low intensity noticed at m/z 1392.08 can be attributed to the singly protonated [M + H]+ ion of the peptide. A second notable peak of the highest intensity is observed at m/z 696.67 and can be attributed to the [M + 2 H]2+ ion of this peptide. A third peak attributable to the [M + 3 H]3+ ion detected at m/z 464.92. Similarly, a single strong chromatogram peak was noticed for DRAMP18160 peptide. As this peptide contains several basic residues (ILRWPWWPWRRK), two representative peaks of [M + 3 H]3+ and [M + 4 H]4+ are found at m/z 594.17 and 445.92 respectively. These peaks are agreed with the theoretical masses (594.01 and 445.76). The DRAMP20773 was eluted at 1.87 min and showed two peaks at m/z 277.83 and 554.58 for [M + H]2+ and [M + H]+ ion, respectively. The theoretical mass (555.36 Da) of [M + H]+ ion matched with the experimental one (554.58 Da).

Liquid chromatographs (A–C) and mass spectra (D–F) of DRAMP20773, DRAMP18160 and DRAMP18152 obtained by Agilent 1290 UPLC coupled with Thermo Scientific LTQ XL mass spectrometer
Main Protease Inhibition Assay and Cell Viability of DRAMP18160
FRET based protease activity assays were utilized to determine the effect each of the inhibitor peptides had on the Main protease’s function. Peptides DRAMP20773 and DRAMP18152 showed no indication that they were inhibiting significantly at concentrations approaching 1 mM and thus were not investigated further. Only DRAMP18160 showed a clear dose dependent response. DRAMP18160 is a derivative of Indolicidin which is a known antimicrobial peptide generated by bovine neutrophils. The virucidal effects of Indolicidin have been documented previously and have included activity against HIV and Herpes virus (Yasin et al. 2000). DRAMP18160 differs from Indolicidin in several transpositions and substitutions of the amino acid composition but retains the tryptophan rich-proline interspersed motif. When tested against the Main Protease, DRAMP18160 displayed an IC50 value of 59 ± 3.16 µM (Fig. 7A).

A Dose response curve showing inhibition of Main Protease activity by DRAMP18160 peptides. The activity of the Main Protease was measured with DABCYL-KTSAVLQSGFRKME-EDANS Protease Activity Assay. B Cell viability of DRAMP18160
Peptide based inhibitors have been explored using linear, cyclic, and enantiomeric sequences. Previously Ullrich et al (2021) produced linear peptides that were able to achieve IC50 values of 71 µM but were unable to produce any cyclic peptides achieving activity below 100 µM. Kreutzer et al. (2021) developed a novel cyclic peptide that was able to achieve an IC50 of 160 µM, likewise failing to come in under the acceptable value of 100 µM which would indicate high affinity binding (Kreutzer et al. 2021). Eberle et al., chose a different method and instead developed a sequence based on Crotamine which is a small cationic venom peptide isolated from the rattlesnake Crotalus durissus terrificus which is known to be an active antimicrobial compound (Falcao and Radis-Baptista 2020). They produced several D-enantiomeric analogues of this peptide and showed that IC50 values ranging in the 1–10 µM range was possible for non-warhead-based peptide inhibitors of lengths not exceeding 15 amino acids (Eberle et al. 2022). Chan et al., thoroughly investigated the interactions between the natural substrate of the Main Protease and the enzyme itself to try and create an inhibitor that would bind tightly and competitively. They were successful in creating potent peptide inhibitors with IC50 values ranging from 3 to 5 µM but discovered that their peptides were undergoing proteolysis by the enzyme. Chan et al. uncovered a key finding which was that the N-terminal downstream site on the inhibitors, specifically the P2 position, if substituted with a Tryp residue, resisted proteolysis (Chan et al. 2021). DRAMP18160 is a cationic peptide with a linear chain not exceeding 12 amino acids and also has an abundance of tryptophan residues. These factors combined suggest that DRAMP18160 is a viable peptide for further development. The cell viability was assessed by utilizing Vero E6 cells with varying the concentration of the DRAMP18160 from 33 to 0.5 µM (Fig.7B). At the concentration of 4 µM and lower, the peptide did not show any cytotoxicity. The cellular toxicity data suggests that DRAMP18160 has an off-target effect that may have lethal consequences for human cells. This alternative target may be a protein or a membrane. More experiments will have to be conducted to either expose the vulnerability being targeted or to determine a more innocuous peptide structure/modification that proves to be safer for cells in-vitro.
Conclusion
The COVID-19 pandemic continues to infect and kill hundreds every day; therefore, there is a critical need for an effective treatment against SARS-CoV-2. Peptide therapeutics received considerable attention over the past decade due to their attractive pharmacological characteristics. In this molecular modeling study, we have selected peptides in clinical trials to investigate their potential as therapeutics against the main protease (Mpro) of SARS-CoV-2. Docking and binding free energy data reveal high binding affinities between DRAMP18152, DRAMP18160, and DRAMP20773 against Mpro. All three peptides showed interactions with either Cys145 or His41 catalytic residues. DRAMP18160 and DRAMP18152 showed strong interactions with both residues. MD simulations of Mpro crystal structure and complexes of DRAMP18152, DRAMP18160, and DRAMP20773 were performed for 250 ns. Analysis of MD simulations reveal that all three peptides maintained relative stability throughout the simulation time. These peptide candidates were synthesized using the standard Fmoc synthesis protocols by CEM Liberty Blue peptide synthesizer. Peptides’ characterizations were then conducted by liquid chromatography and mass spectrometry. A strong peptide candidate (DRAMP18160) was chosen and subjected to a series of in-vitro analyses to determine its effectiveness at inhibiting the function of Mpro. By utilizing Fӧrster resonance energy transfer-based assays it was found that DRAMP18160 exhibited an IC50 value of 59 µM which further establishes the viability of natural product repurposing for widespread antiviral applications. Although these are promising results, there are challenges inherent to clinically applying peptide therapeutics. Low aqueous solubility, poor oral bioavailability, and short in-vivo half-life are some challenges which need to be addressed for peptide-based drugs so that they can compete with small molecules. Peptide stapling offers a potential solution to biological instability as well as a promising avenue for improving binding affinity. In the future, various potent analogous and their corresponding staple peptides of DRAMP18160 will be designed to improve the inhibition effectiveness against the SARS-CoV-2 main protease.
References
Andrusier N, Nussinov R, Wolfson HJ (2007) FireDock: fast interaction refinement in molecular docking. Prot: Struct Funct Genet 69:139–159. https://doi.org/10.1002/prot.21495
Blaszczyk M, Jamroz M, Kmiecik S, Kolinski A (2013) CABS-fold: server for the de novo and consensus-based prediction of protein structure. Nucl Acids Res. https://doi.org/10.1093/nar/gkt462
Chan HTH, Moesser MA, Walters RK, Malla TR, Twidale RM, John T, Deeks HM, Johnston-Wood T, Mikhailov V, Sessions RB, Dawson W, Salah E, Lukacik P, Strain-Damerell C, Owen CD, Nakajima T, Świderek K, Lodola A, Moliner V, Glowacki DR, Spencer J, Walsh MA, Schofield CJ, Genovese L, Shoemark DK, Mulholland AJ, Duarte F, Morris GM (2021) Discovery of SARS-CoV-2 Mpro peptide inhibitors from modelling substrate and ligand binding. Chem Sci 12:13686–13703. https://doi.org/10.1039/D1SC03628A
Dickson CJ, Madej BD, Skjevik ÃA, Betz RM, Teigen K, Gould IR, Walker RC (2014) Lipid14: the amber lipid force field. J Chem Theory Comput 10:865–879. https://doi.org/10.1021/ct4010307
Eberle RJ, Gering I, Tusche M, Ostermann PN, Müller L, Adams O, Schaal H, Olivier DS, Amaral MS, Arni RK, Willbold D, Coronado MA (2022) Design of D-Amino acids SARS-CoV-2 main protease inhibitors using the cationic peptide from rattlesnake venom as a scaffold. Pharmaceuticals. https://doi.org/10.3390/ph15050540
Falcao CB, Radis-Baptista G (2020) Crotamine and crotalicidin, membrane active peptides from Crotalus durissus terrificus rattlesnake venom, and their structurally minimized fragments for applications in medicine and biotechnology. Peptides 126:170234. https://doi.org/10.1016/j.peptides.2019.170234
Islam MJ, Khan AM, Parves MR, Hossain MN, Halim MA (2019) Prediction of deleterious non-synonymous SNPs of human STK11 gene by combining algorithms, molecular docking, and molecular dynamics simulation. Sci Rep. https://doi.org/10.1038/s41598-019-52308-0
Islam R, Parves MR, Paul AS, Uddin N, Rahman MS, Al Mamun A, Hossain MN, Ali MA, Halim MA (2020) A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1761883
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293. https://doi.org/10.1038/s41586-020-2223-y
Kang X, Dong F, Shi C, Liu S, Sun J, Chen J, Li H, Xu H, Lao X, Zheng H (2019) DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci Data. https://doi.org/10.1038/s41597-019-0154-y
Kreutzer AG, Krumberger M, Diessner EM, Parrocha CMT, Morris MA, Guaglianone G, Butts CT, Nowick JS (2021) A cyclic peptide inhibitor of the SARS-CoV-2 main protease. Eur J Med Chem 221:113530. https://doi.org/10.1016/j.ejmech.2021.113530
Krieger E, Dunbrack RL, Hooft RWW, Krieger B (2012) Assignment of protonation states in proteins and ligands: combining pK a prediction with hydrogen bonding network optimization. Methods Mol Biol 819:405–421. https://doi.org/10.1007/978-1-61779-465-0_25
Liu C, Zhou Q, Li Y, Garner LV, Watkins SP, Carter LJ, Smoot J, Gregg AC, Daniels AD, Jervey S, Albaiu D (2020) Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent Sci 6:315–331. https://doi.org/10.1021/acscentsci.0c00272
Magana M, Pushpanathan M, Santos AL, Leanse L, Fernandez M, Ioannidis A, Giulianotti MA, Apidianakis Y, Bradfute S, Ferguson AL, Cherkasov A, Seleem MN, Pinilla C, de la Fuente-Nunez C, Lazaridis T, Dai T, Houghten RA, Hancock REW, Tegos GP (2020) The value of antimicrobial peptides in the age of resistance. Lancet Infect Dis 20:e216–e230. https://doi.org/10.1016/S1473-3099(20)30327-3
Mahlapuu M, Håkansson J, Ringstad L, Björn C (2016) Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2016.00194
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucl Acids Res. https://doi.org/10.1093/nar/gki481
Tahir ul Qamar M, Alqahtani SM, Alamri MA, Chen LL (2020) Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal 10:313–319. https://doi.org/10.1016/j.jpha.2020.03.009
Ullrich S, Sasi VM, Mahawaththa MC, Ekanayake KB, Morewood R, George J, Shuttleworth L, Zhang X, Whitefield C, Otting G, Jackson C, Nitsche C (2021) Challenges of short substrate analogues as SARS-CoV-2 main protease inhibitors. Bioorg Med Chem Lett 50:128333. https://doi.org/10.1016/j.bmcl.2021.128333
Velavan TP, Meyer CG (2020) The COVID-19 epidemic. Trop Med Int Health 25:278–280. https://doi.org/10.1111/tmi.13383
WHO (2023) WHO Coronavirus (Covid-19) Dashboard. https://covid19.who.int/
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552. https://doi.org/10.1385/1-59259-584-7:531
Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A (2016) A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 32:3676–3678. https://doi.org/10.1093/bioinformatics/btw514
Yasin B, Pang M, Turner JS, Cho Y, Dinh N-N, Waring AJ, Lehrer RI, Wagar EA (2000) Evaluation of the inactivation of infectious herpes simplex virus by host-defense peptides. Eur J Clin Microbiol Infect Dis 19:187–194. https://doi.org/10.1007/s100960050457
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 368:409–412. https://doi.org/10.1126/science.abb3405
Acknowledgements
This project was supported by Funds to Sustain Research Excellence and Mentor Protégé Program at College of Science and Mathematics, Kennesaw State University (KSU). Noam Lewit would like to thank First-Year and Sophomore Scholar Program at Office of Undergraduate Research, KSU for providing stipend and funding. This project was also partially supported by the Research Technology Core of the Arkansas INBRE program, supported by a grant from the National Institute of General Medical Sciences, (NIGMS), P20 GM103429 from the National Institutes of Health. Authors like to thank Cordell Gilreath and Dr. Shanzhi Wang for performing the initial protease assay in this project and Rimon Parves, Md. Abu Sufian, Dr. Md. Nayeem Hossain for providing PCA and SAR codes and assisting with some statistical analysis related to this project. Authors would like to thank UTHSC Regional Biocontainment Lab (Memphis, Tennessee) for performing cell viability assay.
Author information
Authors and Affiliations
Contributions
MAH conceived the idea. SD, MMH, MAA, CBW performed molecular docking, dynamics and statistical analysis. RF, JS, NL and PO did the peptide synthesis, characterization, and protease assay. RF, MMH, SD wrote the main manuscript text and prepared the figures. ST and MAH supervised and revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Faddis, R., Du, S., Stewart, J. et al. Molecular Modelling, Synthesis, and In-Vitro Assay to Identify Potential Antiviral Peptides Targeting the 3-Chymotrypsin-Like Protease of SARS-CoV-2. Int J Pept Res Ther 29, 89 (2023). https://doi.org/10.1007/s10989-023-10563-w
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-023-10563-w