
Abstract
Visceral leishmaniasis (VL) infection is mostly caused by Leishmania donovani and affects countries worldwide. Despite the need for a safe and effective vaccine against leishmaniasis due to the increased drug resistance, however, no vaccine has yet been licensed for clinical use. This study revolves around the immunoinformatics approach to design a multi-epitope vaccine against VL infection. In this case, the proteome of L. donovani has been investigated, and three host non-homologous and antigenic extracellular secretory proteins have been identified as potential vaccine candidates with low transmembrane helices (≤ 1). The multi-epitope subunit vaccine construct consists of T-cell (cytotoxic T-lymphocyte (CTL) and helper T-lymphocyte (HTL)) epitopes accompanied by appropriate adjuvant and linkers. A 372-amino acid vaccine construct has been established with specific characteristics, such as soluble, stable, antigenic, non-allergenic, non-toxic, and non-host homologous. Besides, the tertiary structure of the designed vaccine was modeled and validated. Also, the stability and affinity of the vaccine- TLR4 complex were confirmed by using molecular docking and molecular dynamics (MD) simulation. In addition, in silico immunization assay showed the efficiency of this candidate vaccine to stimulate an effective immune response. Furthermore, the refined vaccine was optimized and cloned in the pET28a (+) vector, and its successful expression was confirmed virtually. However, the experimental validation is required to verify the multi-epitope vaccine efficacy against VL infection.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Leishmaniasis is a vector-borne disease caused by infection with Leishmania parasites that predominantly affects the world’s most vulnerable and poorest populations. Leishmania parasites are transmitted to humans by the bite of infected female sandflies (Alvar et al. 2006). The clinical manifestations of leishmaniasis depend on the causative Leishmania species. There are mainly three forms of leishmaniasis, including cutaneous, mucocutaneous, and visceral. The latest estimate of the World Health Organization (WHO) in 2019 demonstrated that more than 90% of the new cases of visceral leishmaniasis (VL) were reported in ten countries, including Brazil, Ethiopia, Eritrea, India, Iraq, Kenya, Nepal, Somalia, South Sudan, and Sudan (https://www.who.int/news-room/fact-sheets/detail/leishmaniasis). The VL, caused by Leishmania donovani and Leishmania infantum, is the most severe form of the disease. Also, it can lead to death if not treated. The mortality rate in VL is higher than in other types of disease. This issue is related to the emergence of resistance in the Leishmania strains against most antibiotics (Moore et al. 2010).
Various VL vaccines have been tested in animal models, such as the fucose mannose ligand (FML)-saponin (Santos et al. 2003), Leishmania excreted/secreted antigens (Lemesre et al. 2005), and liposome-based vaccines (Afrin et al. 1997; Sharma et al. 2006). Although vaccines are the most effective way to prevent diseases, there is still no approved vaccine against human VL (Kedzierski et al. 2006). This situation is due to the pathogen antigenic variation and gene polymorphism (Ghorbani et al. 2018; Kumar et al. 2014). One of the computational methods for detecting protective antigens by screening the proteome of a pathogen is reverse vaccination (RV), which is highly recommended for vaccine development (Rappuoli 2000). Therefore, researchers’ have focused on selecting of the appropriate pathogenic antigen in designing vaccines. Immunoinformatics approaches are cost-effective methods used for designing a peptide-based vaccine. This vaccine contains the antigen part of pathogens that can induce the immunogenicity and affect the vaccine efficacy (Bertholet et al. 2009). Thus, developing a novel multi-epitope subunit vaccine by predicting T-cell epitopes (Patronov et al. 2013) and employing computational tools are efficient ways to terminate the virulence of this pathogen.
Some proteins in the proteome of Leishmania have been identified as unknown/hypothetical proteins (HPs). These proteins were potentially utilized as drug targets or vaccine candidates (Sinha et al. 2017). Since some HPs substantially affect the pathogenesis of leishmaniasis, some of these proteins are used in a recombinant vaccine against infection with Leishmania parasites (Duarte et al. 2017; Martins et al. 2016; Ribeiro et al. 2018). Besides, the membrane and secretory proteins play a crucial role in stimulating a host-specific immune response (Khatoon et al. 2019). The extracellular secretory proteins of Leishmania contain several antigenic components and induce an immune response (John et al. 2012; Khan et al. 2020). Therefore, these proteins are very helpful for novel vaccine development.
Several recent studies have investigated the secretory or membrane proteins of Leishmania using computational tools (Khan et al. 2020; Khatoon et al. 2017, 2019; Vakili et al. 2018). In these studies, a multi-epitope vaccine was designed against leishmaniasis. The aim of this study was to pinpoint the potential T-cell epitopes from three secretory HPs of the genome of L. donovani that can be further joined through proper linkers and adjuvant to design a multi-epitope subunit vaccine. Then, the designed vaccine’s physicochemical properties, antigenicity, and allergenicity were predicted by in silico approaches. The secondary and tertiary structures of the designed vaccine were also predicted and validated. Molecular docking and molecular dynamics simulation have been performed to confirm the binding interaction and stability of the docked complex. Finally, in silico cloning into the pET28a (+) vector has been performed to confirm the expression efficiency of the final vaccine construct. The proposed vaccine is also expected to be effective in stimulating humoral and cellular immunity against VL infection.
Methodology
The Collection of Hypothetical Secretory Proteins in Leishmania donovani
In this study, protein sequences of L. donovani were derived from the TriTrypDB (http://tritrypdb.org/tritrypdb/) to identify new HP candidates for vaccine design. The amino acid sequences of three HPs of L. donovani (LDBPK_120390, LDBPK_300860, and LDBPK_312610) have been chosen as secretory proteins. This procedure has been accomplished by predicting the score derived from the SignalP (Armenteros et al. 2019) server and evaluating the combined prediction of SecretomeP (Bendtsen et al. 2004) and TMHMM 2.0 (Krogh et al. 2001). Besides, CELLO2GO (Yu et al. 2014) and PSORTb v3.0 (Yu et al. 2010) have been employed to identify the subcellular localization of hypothetical L. donovani proteins. Also, the antigenic property has been measured for these HP sequences using VaxiJen 2.0 (Doytchinova et al. 2007).
The Prediction of T-cell (Cytotoxic T Lymphocyte and Helper T Lymphocyte) Epitopes
Cytotoxic T lymphocytes (CTLs) play a crucial role in host defense against foreign antigens. Thus, it is essential to predict CTL epitopes for the vaccine design. The mentioned HP sequences of L. donovani were submitted to the NetCTL 1.2 (http://www.cbs.dtu.dk/services/NetCTL/) server to predict CTLs. In this case, the threshold value was set at 0.75. This server allows prediction of CTL epitopes based on MHC-I binding peptides, proteasomal C-terminal cleavage, and Transporter Associated with Antigen Processing (TAP)-associated efficiency with antigen processing (Larsen et al. 2007).
Since the helper T cells had significant influences on adaptive immunity, the IEDB database (http://tools.iedb.org/mhcii/) has been utilized to predict helper T lymphocyte (HTL) epitopes based on their receptor binding affinity (Vita et al. 2019). In this case, epitopes with the lowest percentile rank represent a high affinity of MHC-II. Also, the 15-mer epitopes in mouse alleles were retrieved for all HP sequences in FASTA format.
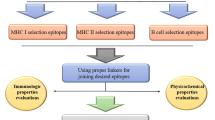
Designing a Multi-epitope Vaccine Construct
In this study, the predicted epitopes have been combined to construct a multi-epitope subunit vaccine. This procedure has been performed using suitable linkers and adjuvants. Besides, the selected CTLs and HTLs epitopes were linked via AAY and GPGPG linkers to connect various components of a multi-epitope vaccine construct. Also, the EAAAK linker has been considered to add the Mycobacterium tuberculosis 50 S ribosomal L7/L12 (Uniprot ID: P9WHE3.1) to the N-terminal sequence as an adjuvant (Lee et al. 2014). Thus, the immunogenicity of the vaccine has been enhanced.
The Prediction of Antigenicity, Allergenicity, and Toxicity
Antigenicity is defined as the ability of a chemical structure to produce an immune response. In general, it is recognized as an antigen. Besides, allergenicity is characterized as the ability of small antigens in creating immune-mediated and allergic reactions. In this case, these two properties were identified as significant factors affecting vaccine development. Also, ANTIGENpro (Magnan et al. 2010) and AllerTOP 2.0 (Dimitrov et al. 2014) servers have been measured the antigenic and allergenic properties of the designed vaccine protein. In addition, the toxicity of the designed vaccine construct has been checked by the ToxinPred (Gupta et al. 2013) server.
The Analyses of Physiochemical Properties and Solubility Prediction
ExPASy ProtParam tool (https://web.expasy.org/protparam/) has been used to calculate a set of physicochemical parameters such as theoretical isoelectric point (pI), amino acid composition, in vitro and in vivo half-life, instability, aliphatic index, molecular weight (MW), and grand average of hydropathicity (GRAVY) for the subunit vaccine protein (Gasteiger et al. 2005). Also, the Protein-Sol (https://protein-sol.manchester.ac.uk/) server has been employed to determine the solubility of the designed vaccine protein. This server predicts the vaccine solubility based on properties like isoelectric pH and amino acid compositions. In this case, protein scored ≥ 0.45 is expected to be soluble compared to the average solubility of E. coli proteins (Hebditch et al. 2017).
Furthermore, the NCBI BlastP has been considered to compare the whole multi-epitope vaccine sequence with Homo sapiens proteome for preventing autoimmunity. In this case, non-host homologous proteins were screened with default parameters (Mahram et al. 2015).
The Prediction and Validation of Multiple-Epitope Vaccine Structures
PSIPRED 4.0 (Buchan and Jones 2019) online web server has been considered to predict the secondary structure of the multi-epitope subunit vaccine construct. Besides, the homology modeling tool in the RaptorX (Kallberg et al. 2012) server has been utilized to prepare the tertiary model of the subunit vaccine protein. Also, the Galaxy Refine (Heo et al. 2013) server has been employed to refine the 3D model of the multi-epitope vaccine construct and improve the structure quality. In this study, the quality of the refined vaccine construct has been assessed by the ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php). It predicts the overall quality of the protein model in the form of a Z-score (Wiederstein et al. 2007). Also, the Ramachandran plot was generated using the PROCHECK (https://saves.mbi.ucla.edu/) server to illustrate the Ramachandran plot with allowed and disallowed regions of amino acids.
The Prediction of B-cell Epitopes and Interferon γ Inducing Epitopes
The epitopes of the humoral immune response, particularly B-cell epitopes, are antigenic determinants and considerably affect vaccine designing. In the present study, these linear B-cell epitopes (20-mer peptides) of the final vaccine construct were predicted using the BCPREDS server at default parameters. This server utilizes a support vector machine (SVM) based on kernel methods for predicting linear B-cell epitopes (El-Manzalawy et al. 2008). Also, the Ellipro server (https://tools.iedb.org/ellipro) has been employed to identify discontinuous B-cell epitopes of the refined final vaccine construct. This server implements three algorithms based on their protrusion index (PI) values and allocates a PI value to the predicted epitopes (Ponomarenko et al. 2008). Besides, interferon-gamma (IFN-γ) plays a vital role in innate and adaptive immunity. In this case, the IFNepitope (https://webs.iiitd.edu.in/raghava/ifnepitope/index.php) server has been considered for predicting the IFN-γ epitope of the final vaccine. This server predicts epitopes via the hybrid algorithms of motif and SVM (Dhanda et al. 2013).
Molecular Docking of the Designed Vaccine with the TLR-4 Receptor
The Computed Altas of Surface Topography of proteins (CASTp) server V3.0 has been employed to predict the protein binding site and hydrophobic interaction on the protein surface (Dundas et al. 2006). This situation initiates an interaction between the final vaccine construct and the TLR4 immune receptor. Then, the PatchDock web server (http://bioinfo3d.cs.tau.ac.il/PatchDock/) has been considered for the molecular docking of the final vaccine construct with TLR4 (PDB ID: 4G8A) receptor. This procedure has been carried out to demonstrate an immune response development and analyze the interaction between the ligand and receptor (Schneidman-Duhovny et al. 2005). Afterward, the Fast Interaction Refinement in Molecular Docking (FireDock) server has been utilized to refine and re-score the rigid body molecular docking complexes (Andrusier et al. 2007). This situation has been led to achieving the best structure. This server provides the ten best solutions for the procedure. Besides, the refined models are configured based on several factors such as atomic contact energy, partial electrostatics, Van Der Waals interaction (vdW), and binding energy estimations. Also, the TLR4 receptor- vaccine interaction was visualized using the UCSF Chimera (Pettersen et al. 2004) and LigPlot+ (Laskowski et al. 2011) tools.
Molecular Dynamics Simulation
In addition, the stability of the interaction between TLR-4 receptor and the vaccine construct was determined by GROMACS 5.1.1 package (Van Der Spoel et al. 2005). This process has been performed using the GROMOS96 43a1 force field parameter set to implement molecular dynamics simulations. Before simulation, residues 980 to 1200 of TLR4 receptor that were located considerably far from the vaccine-TLR4 complex (based on docking analysis) were manually removed to reduce the time of simulation and further improve MD analysis. In this case, the structure was initially solvated with transferable intermolecular potential with 3 points (TIP3P) water model. Sodium and chloride ions were also added to neutralize the system. Besides, the energy has been minimized to provide a proper molecular arrangement in space. It was conducted by eliminating high-energy configurations. Also, the simulation process has been run under canonical (NVT) and isothermal–isobaric (NPT) conditions at 300 K and 1 bar pressure with the duration of 50 ps. Then, the stabilized structure has been subjected to the MD simulation of 50 ns. Finally, the root mean square deviation (RMSD), the root mean square fluctuation (RMSF), the radius of gyration (Rg) (Lobanov et al. 2008), and hydrogen bonds (Joshi et al. 2021) were calculated for all the systems. The MD complex analysis were determined by using graphs generated by Xmgrace software.
Immune Response Simulation
The immunological responses of the designed vaccine construct have been evaluated by in silico method C-ImmSim (https://www.iac.cnr.it/~filippo/c-immsim/index.html) online dynamic immune simulation server (Rapin et al. 2010).
Codon Adaptation and In Silico Cloning of the Final Vaccine
Reverse translation and codon optimization of the final vaccine construct were conducted by the Java Codon Adaptation Tool (JCat) server (http://www.jcat.de/) to attain an improved nucleotide sequence and make it compatible with widely used prokaryotic expression system; E. coli K12 (Grote et al. 2005). Besides, three options of avoiding rho-independent transcription terminators, prokaryote ribosome binding sites, and cleavage sites restriction enzymes were selected. The JCat output includes the codon adaptation index (CAI) value and the percentage of GC content. The desired CAI value is ranged from 0.8 to 1.0, and the optimal percentage of GC content ranges from 30 to 70%. Also, SnapGene 5.1.5 software cloned the optimized codon sequence into E. coli pET-28a (+) vector. Afterward, the restriction sites of XhoI and NdeI were incorporated in the N- and C-terminal sequences, respectively.
Results
Sequence Retrieval of Leishmania donovani Protein
In this case, the amino acid sequences of three secretory proteins of L. donovani (LDBPK_120390, LDBPK_300860, and LDBPK_312610) were retrieved from the NCBI protein database to design an immunogenic subunit vaccine. The CELLO2GO (Yu et al. 2014) and PSORTb v3.0 (Yu et al. 2010) servers predicted that these secreted proteins were localized in extracellular location. These HPs were found with an antigenic probability of greater than 0.5. This value was in agreement with VaxiJen 2.0 (Doytchinova et al. 2007).
T-cell Epitopes Prediction
A total of five CTL epitopes with high-ranked binding affinity scores were predicted for the three nominated proteins (Table 1). This process has been completed by the NetCTL 1.2 server (Larsen et al. 2007). Likewise, nine HTL epitopes based on their lowest percentile rank with MHC-II were predicted from the IEDB server (Vita et al. 2019) for mouse alleles (H2-1Ad, H2-1Ed, and H2-1Ab) to design the vaccine (Table 2).
Developing a Multi-epitope Vaccine Protein
The vaccine sequence has been constructed by combining five CTL epitopes and nine HTL epitopes with the AAY and GPGPG linkers, respectively. Also, the 50 S ribosomal L7/L12 (P9WHE3) was added to the N-terminal of the construct using the EAAAK linker, which is ideal for achieving maximum immune response (Fig. 1).

Schematic diagram of the multi-epitope vaccine construct. The 372- amino acid multi-epitope vaccine containing an adjuvant (blue) at N-terminal end linked followed by CTL and HTL epitopes. EAAAK linker (pink) has joined adjuvant and CTL epitope. Whereas, AAY (Cyan) and GPGPG (yellow) linkers were used to join the CTL (red) and HTL (grey) epitopes, respectively (Color figure online)
The Prediction of Antigenicity, Allergenicity, and Toxicity of Subunit Vaccine
In the designed vaccine construct, the immunological properties such as antigenicity, allergenicity, and toxicity were evaluated by the three servers of ANTIGENpro (Magnan et al. 2010), AllerTOP 2.0 (Dimitrov et al. 2014), and ToxinPred (Gupta et al. 2013), respectively. The results demonstrated that the designed vaccine protein was antigenic, non-allergenic, and non-toxic.
Physiochemical Characterization and Solubility Prediction of Subunit Vaccine
In this case, the ProtParam (Gasteiger et al. 2005) tool has been considered to evaluate the physicochemical properties of the designed subunit vaccine sequence containing 372 amino acids. The molecular weight, theoretical isoelectric point (pI), and instability index values were 38078.35, 5.84, and 25.4, respectively. The estimated half-life was assessed 30 h in mammalian reticulocytes, > 20 h in yeast, and > 10 h in E. coli, in vivo. The aliphatic index (II) and GRAVY were 84.17 and 0.108, respectively. The results indicated that the designed vaccine was significantly acidic, stable, thermostable, and hydrophobic. Also, the scaled solubility value of the vaccine construct was 0.496. This value was higher than the average soluble E. coli protein derived from the experimental solubility dataset (Niwa et al. 2009).
Besides, the NCBI BlastP has been utilized to search for similarities between the multi-epitope vaccine and the Homo sapiens proteome. The results showed no similarities between them. This situation reveals that the designed vaccine was non-homologous with the host proteome.
The Prediction of Secondary and Tertiary Structures of Subunit Vaccine Construct
Figure 2 depicts the secondary structure prediction of the subunit vaccine construct using the PSIPRED (Buchan and Jones 2019) server. It contains alpha-helix (38%), beta strands (17%), and coils (45%). Besides, the RaptorX (Kallberg et al. 2012) server has been utilized to construct a 3D structure of the subunit vaccine. The best template for modeling the designed vaccine was the crystal structure of Thermotoga maritime (PDB ID: 1dd3A) with the p-value of 6.19e−05. Then, the GalaxyRefine (Heo et al. 2013) server refined the vaccine structure and generated five models. Among these generated models, model 1 (Fig. 3a) has been chosen due to its properties such as GDT-HA (0.9348), RMSD (0.458), MolProbity (1.734), Clash score (7.2), Poor rotamers (0.4), and Rama favored (95.1).

Schematic illustration of secondary structure of the subunit vaccine construct including alpha-helix, beta strands, and coils

The tertiary structure of the final vaccine construct and its validation. a The 3D model of the final vaccine construct was obtained by RaptorX. b ProSA-web z-score plot indicating a Z-score = − 5.67. c Ramachandran plot showing 98.3% of the residues are present in the favored and allowed regions, and 1.7% of residues are in disallowed region
In this case, the final vaccine construct has been validated by the ProSA-web and Ramachandran plots after the model refinement. As shown in Fig. 3b, the ProSA-web server provided a Z-score of − 5.67 for the input vaccine protein. Figure 3c illustrates the Ramachandran plot of the vaccine construct, and only 1.7% of the residues are in the disallowed regions. These issues confirm the quality of the designed vaccine.
Analysis of B-cell Epitopes and Interferon γ Inducing Epitopes Prediction
Two servers, BCPred (El-Manzalawy et al. 2008) and ElliPro (Ponomarenko et al. 2008), have been employed to predict the linear and conformational B-cell binding epitopes among the primary input sequence of the final vaccine construct. Figure 4 depicts information about the eight epitopes. These epitopes had a BCPreds cut-off score of more than 0.9. A total of 66 residues were predicted as the conformational B-cell epitopes with a probability score greater than 0.8 and residue numbers ranging from 303 to 370. These scores start with Tyrosine residue (score 0.85) and end with Tryptophan residue (score 0.98). The findings showed that the designed vaccine could enhance humoral immunity (Khatoon et al. 2017; Shi et al. 2015).

Schematic representation of predicted B-cell epitopes and IFN-γ inducing epitopes. a Red color ribbons show the liner B-cell epitopes, b Purple mesh surface color sequences show the discontinuous B-cell epitopes, and c Black color ribbons illustrate IFN-γ inducing epitopes in the constructed vaccine (Color figure online)
The IFNepitope (Dhanda et al. 2013) server has been utilized to predict 364 epitopes (15-mer) based on SVM. This case included both negative and positive predictions. In this regard, two IFN-γ inducing epitopes were selected. These epitopes had a prediction score of one (Fig. 4).
Molecular Docking of the Final Vaccine Construct with the Immune Receptor (TLR-4)
The CASTp server analyzed the active binding sites of the protein. It could serve as a possible site for interacting with the TLR4 receptor. Besides, this server predicted the potential binding site with 2073.8 Å2 surface area and 2980.2 Å3 molecular surface volume. Also, the PatchDock (Schneidman-DuhovnyInbar et al. 2005) server has been considered to perform the molecular docking of the vaccine construct against TLR4 at the predicted binding pocket. In this case, the top ten models were refined via their binding energy using the FireDock (Andrusier et al. 2007) server. Figure 5 displays the selected final model (model No. 2) for molecular docking based on the binding energy.

Molecular docking of the final vaccine construct with human TLR4 receptor. a Graphical representation Patch-docking of the vaccine construct (red color) and TLR-4 receptor (blue color). b The interaction map of the TLR4-vaccine complex showing residues involved in hydrophobic (red dotted lines) and hydrogen bonding (green dotted lines) (Color figure online)
Molecular Dynamics Simulation of Vaccine- TLR4 Complex
In order to validate structural stability of the subunit vaccine-TLR4 complex, molecular dynamics simulation was performed using a 50 ns MD simulation study. Obtained potential energy for the complex was − 7.72982e + 6 KJ/mol. The complex attained equilibrium during the simulation trajectory after about 20 ns of simulation and acquired a value between 1.9 and 2.23 nm. The RMSD plot shows the complex stabilized in the last 30 ns of the simulation (Fig. 6a). The RMSF plot of the TLR4- vaccine complex indicated that most of the peaks are belong to residues, which are located in the loop regions and the terminal residues of strand (Fig. 6b). Because the loop structure is much more flexible, the loop region consisting of Val288-Ala372 residues showed larger RMSF values compared to α-helix structures. The overall predicted RMSF graph suggested, the ligand have a little fluctuated (less than 2 nm) throughout the simulation time. The compactness of vaccine and TLR4 complex evaluated and the average Rg was found to be 3.34 Å. As shown in Fig. 6c, we could assume that this complex did not induce noticeable fluctuation after 50 ns of simulation and remain compact after 25 ns that is in agreement with the RMSD values. The number of hydrogen bonds formed between the vaccine and the TLR4 through 50 ns simulation period is shown in Fig. 6d. The observed bonding graph, demonstrated that hydrogen bond interaction patterns increased slightly and then remained constant during the last 30 ns of simulation time. All evidences indicate that receptor tends to bind to vaccine and remain stable as previous RMSD and Rg represented.

Molecular dynamics (MD) simulation analysis of the vaccine- TLR4 complex. a Root mean square deviation (RMSD) of the vaccine–receptor complex backbone, b root mean square fluctuations (RMSF) of the docked complex side chains, c radius of gyration (Rg) of the docked complex, and d Number of hydrogen bonds formed during the 50 ns MD simulations
Immune Response Simulation
The refined vaccine protein was subjected to in silico immune simulations to check the proficiency of the vaccine construct to the adaptive immune system. The simulation results showed that responses (secondary and tertiary) produced high levels of IgG and IgM antibodies (IgM + IgG and IgM), while the antigen level decreased (Fig. 7a). Besides, Fig. 7b exhibits an effective antiviral response by the vaccine in the elevated levels of IFN-γ and interleukin‐2. Also, Fig. 7c displays the estimation of the B-cell population in the elevated levels of B memory cells and B-cell isotype IgM. In addition, a high response was observed in the T cells (both Th and Tc) populations (Fig. 7d and e). The obtained results suggest that the proposed vaccine construct can effectively trigger the immune response and provide immunity against VL infections.

In silico immune simulation of the final vaccine construct. a Immunoglobulins levels upon antigen exposure, b The level of cytokines and interleukins, c Population of B -cells, d and e Population of T lymphocytes (T-helper and T-cytotoxic)
Virtual Cloning of the Final Vaccine Construct
The Jcat (Grote et al. 2005) server has been utilized to adapt the codon for multi-epitope vaccine as per E. coli K12 strain. In this case, the Codon Adaptation Index (CAI) of the improved sequence was 0.968 with GC contents of 56.45%. Also, the length of the optimized codon sequence was 1,116 nucleotides. The results showed the possibility of good expression of the vaccine candidate in the E. coli host. Afterward, the SnapGene software generated the sequence of the recombinant plasmid to insert the optimized sequence into the pET28a (+) vector (Fig. 8). This procedure has been performed for in silico cloning.

The in silico cloning of the optimized vaccine (red lines) into pET28a (+) expression vector (black lines) (Color figure online)
Discussion
The VL infection is a serious public health problem, which annually affects millions of people across the globe. Leishmaniasis treatment is confined to several anti-leishmaniasis drugs, but these drugs showed increased drug resistance and toxicity. Therefore, there is no effective controlling approach for VL (Nagill et al. 2011). Despite the efforts of scientists to develop a vaccine against leishmaniasis, there is still no effective vaccine against this disease. Several antigens (e.g., cysteine protease C and kinetoplastid membrane protein-11) have been evaluated as targets for vaccines against Leishmania (Basu et al. 2005; Das et al. 2014). Also, recombinant vaccines (e.g., Leish111f and Leishmune) have been developed as vaccine candidates against leishmaniasis (Jain et al. 2015). Nevertheless, no vaccine has been approved against leishmaniasis for use in humans (Kedzierski et al. 2006).
Advances in bioinformatics tools have been led to the development of subunit vaccines containing specific immunogenic components of pathogens that stimulated an immune response (Adhikari et al. 2018; Pandey et al. 2018; Rahman et al. 2020; Shey et al. 2019). Since T-cells play a crucial role in controlling VL infection, most researchers have designed the vaccine via identifying L. donovani antigenic epitopes and their interaction with host MHC alleles and immune cells (Stager et al. 2012). In this regard, most recent studies have focused on the L. donovani genome sequences to identify specific immunogenic components for designing novel vaccines using immunoinformatics methods (Atapour et al. 2021; Khan et al. 2020; Singh et al. 2020; Vakili et al. 2018). Study on the pathogenic secretory proteins revealed that these proteins were antigenic in nature and had diverse functions during infection (Khatoon et al. 2017). Identification of these proteins and understanding of their antigenic epitopes with host MHC alleles can be used to produce an effective vaccine against leishmaniasis (Kaye et al. 2011). For example, the literature survey showed that amastigote-associated potential secretory proteins were an effective vaccine design against VL (Khan et al. 2020). Also, the KMP-11 protein sequence (as a target antigen) and B-type flagellin (as adjuvant) have been used for designing vaccine construct (Atapour et al. 2021).
This study emphasized the computational approaches for predicting the binding of peptides to MHC molecules. This procedure is performed to select the three hypothetical secretory proteins in L. donovani. These are used as effective multi-epitope vaccine candidates. In the present study, three L. donovani hypothetical secretory proteins have been scanned to predict the presence of a signal peptide, subcellular localization, and antigenicity. The potential epitopes of these proteins were selected against T-cell (Cytotoxic and Helper T-lymphocyte) epitopes using the available online tools. In this case, five CTL and nine HTL epitopes were chosen as part of the developed multi-epitope vaccine construct. This procedure was conducted by joining suitable linkers and an adjuvant together. None of these predicted epitopes have been used earlier that shows the novelty of our study.
The designed vaccine protein has been subjected to BLASTP search against human proteins to prevent the undesired pathogen cross-reactivity with the host. The analysis results demonstrated that the designed vaccine did not have any homology with the host proteome. Besides, the secondary structure of the vaccine construct including alpha-helices, beta-sheets, and coils was predicted. Also, the RaptorX and GalaxyRefine servers have been utilized to generate the 3D structure and model refinement, respectively. The Ramachandran plot showed that the refined tertiary structure of the vaccine improved to the desired level. Meanwhile, the final vaccine construct had physicochemical and antigenic properties with no probable allergenicity or toxicity.
Besides, molecular docking and MD simulation were performed to understand the binding affinity and stability of the TLR4- vaccine complex. The vaccine- TLR4 complex showed stability and compactness during MD simulation. Also, an immune simulation was carried out for the designed vaccine. This procedure has been accomplished to characterize its ability to produce immunological responses against the VL. The simulation results revealed that the final multi-epitope vaccine construct elicited unique immune responses. In addition, the in silico optimization of codon has been implemented for the final vaccine construct. The values of CAI and GC contents demonstrated that the vaccine construct could be expressed in the E. coli host. The in silico cloning of the optimized vaccine showed its better integration for the expression in a bacterial system. This study focuses on the design of a multi-epitope subunit vaccine using immunoinformatics tools, which suggests that the final vaccine construct may have a good protective effect against VL infection.
Conclusions
One of the major causes of mortality in tropical and subtropical regions across the world is VL. In this study, we utilized immunoinformatics tools to design a novel multi-epitope vaccine against VL. Based on the results, our multi-epitope subunit vaccine is immunogenic and non-allergic, which has the potential to induce cellular and humoral immune responses. Although in silico results showed the effectiveness of this vaccine, immunogenic potency of the proposed vaccine needs to be experimentally validated.
References
Adhikari UK, Tayebi M, Rahman MM (2018) Immunoinformatics approach for epitope-based peptide vaccine design and active site prediction against polyprotein of emerging oropouche virus. J Immunol Res 2018:6718083. https://doi.org/10.1155/2018/6718083
Afrin F, Ali N (1997) Adjuvanticity and protective immunity elicited by Leishmania donovani antigens encapsulated in positively charged liposomes. Infect Immun 65(6):2371–2377. https://doi.org/10.1128/iai.65.6.2371-2377.1997
Alvar J, Yactayo S, Bern C (2006) Leishmaniasis and poverty. Trends Parasitol 22(12):552–557. https://doi.org/10.1016/j.pt.2006.09.004
Andrusier N, Nussinov R, Wolfson HJ (2007) FireDock: fast interaction refinement in molecular docking. Proteins 69(1):139–159. https://doi.org/10.1002/prot.21495
Armenteros JJA, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37(4):420–423. https://doi.org/10.1038/s41587-019-0036-z
Atapour A, Ghalamfarsa F, Naderi S, Hatam G (2021) Designing of a novel fusion protein vaccine candidate against human visceral leishmaniasis (vl) using immunoinformatics and structural approaches. Int J Pept Res Ther. https://doi.org/10.1007/s10989-021-10218-8
Basu R, Bhaumik S, Basu JM, Naskar K, De T, Roy S (2005) Kinetoplastid membrane protein-11 DNA vaccination induces complete protection against both pentavalent antimonial-sensitive and -resistant strains of Leishmania donovani that correlates with inducible nitric oxide synthase activity and IL-4 generation: evidence for mixed Th1- and Th2-like responses in visceral leishmaniasis. J Immunol 174(11):7160–7171. https://doi.org/10.4049/jimmunol.174.11.7160
Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S (2004) Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel 17(4):349–356. https://doi.org/10.1093/protein/gzh037
Bertholet S, Goto Y, Carter L, Bhatia A, Howard RF, Carter D, Coler RN, Vedvick TS, Reed SG (2009) Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine 27(50):7036–7045. https://doi.org/10.1016/j.vaccine.2009.09.066
Buchan DW, Jones DT (2019) The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res 47(W1):W402–W407. https://doi.org/10.1093/nar/gkz297
Das A, Ali N (2014) Combining cationic liposomal delivery with MPL-TDM for cysteine protease cocktail vaccination against Leishmania donovani: evidence for antigen synergy and protection. PLoS Negl Trop Dis 8(8):e3091. https://doi.org/10.1371/journal.pntd.0003091
Dhanda SK, Vir P, Raghava GP (2013) Designing of interferon-gamma inducing MHC class-II binders. Biol Direct 8:30. https://doi.org/10.1186/1745-6150-8-30
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014) AllerTOP v. 2—a server for in silico prediction of allergens. J Mol Model 20(6):1–6. https://doi.org/10.1007/s00894-014-2278-5
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8(1):1–7. https://doi.org/10.1186/1471-2105-8-4
Duarte MC, Lage DP, Martins VT, Costa LE, Carvalho A, Ludolf F, Santos TTO, Vale DL, Roatt BM, Menezes-Souza D, Fernandes AP, Tavares CAP, Coelho EAF (2017) A vaccine composed of a hypothetical protein and the eukaryotic initiation factor 5a from Leishmania braziliensis cross-protection against Leishmania amazonensis infection. Immunobiology 222(2):251–260. https://doi.org/10.1016/j.imbio.2016.09.015
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 34:W116-118. https://doi.org/10.1093/nar/gkl282
El-Manzalawy Y, Dobbs D, Honavar V (2008) Predicting linear B-cell epitopes using string kernels. J Mol Recognit 21(4):243–255. https://doi.org/10.1002/jmr.893
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Springer, Berlin, pp 571–607
Ghorbani M, Farhoudi R (2018) Leishmaniasis in humans: drug or vaccine therapy? Drug Des Dev Ther 12:25–40. https://doi.org/10.2147/DDDT.S146521
Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:W526-531. https://doi.org/10.1093/nar/gki376
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Open Source D, Discovery C, Raghava GP (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8(9):e73957. https://doi.org/10.1371/journal.pone.0073957
Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J (2017) Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics 33(19):3098–3100. https://doi.org/10.1093/bioinformatics/btx345
Heo L, Park H, Seok C (2013) GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res 41:W384-388. https://doi.org/10.1093/nar/gkt458
Jain K, Jain N (2015) Vaccines for visceral leishmaniasis: a review. J Immunol Methods 422:1–12. https://doi.org/10.1016/j.jim.2015.03.017
John L, John GJ, Kholia T (2012) A reverse vaccinology approach for the identification of potential vaccine candidates from Leishmania spp. Appl Biochem Biotechnol 167(5):1340–1350. https://doi.org/10.1007/s12010-012-9649-0
Joshi T, Joshi T, Sharma P, Chandra S, Pande V (2021) Molecular docking and molecular dynamics simulation approach to screen natural compounds for inhibition of Xanthomonas oryzae pv. Oryzae by targeting peptide deformylase. J Biomol Struct Dyn 39(3):823–840. https://doi.org/10.1080/07391102.2020.1719200
Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J (2012) Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7(8):1511–1522. https://doi.org/10.1038/nprot.2012.085
Kaye PM, Aebischer T (2011) Visceral leishmaniasis: immunology and prospects for a vaccine. Clin Microbiol Infect 17(10):1462–1470. https://doi.org/10.1111/j.1469-0691.2011.03610.x
Kedzierski L, Zhu Y, Handman E (2006) Leishmania vaccines: progress and problems. Parasitology 133(S2):S87. https://doi.org/10.1017/s0031182006001831
Khan MAA, Ami JQ, Faisal K, Chowdhury R, Ghosh P, Hossain F, Abd El Wahed A, Mondal D (2020) An immunoinformatic approach driven by experimental proteomics: in silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasites Vectors 13:1–21. https://doi.org/10.1186/s13071-020-04064-8
Khatoon N, Pandey RK, Ojha R, Aathmanathan VS, Krishnan M, Prajapati VK (2019) Exploratory algorithm to devise multi-epitope subunit vaccine by investigating Leishmania donovani membrane proteins. J Biomol Struct Dyn 37(9):2381–2393. https://doi.org/10.1080/07391102.2018.1484815
Khatoon N, Pandey RK, Prajapati VK (2017) Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep 7(1):1–12. https://doi.org/10.1038/s41598-017-08842-w
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580. https://doi.org/10.1006/jmbi.2000.4315
Kumar R, Engwerda C (2014) Vaccines to prevent leishmaniasis. Clin Transl Immunol 3(3):e13. https://doi.org/10.1038/cti.2014.4
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 8(1):1–12. https://doi.org/10.1186/1471-2105-8-424
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model 51(10):2778–2786. https://doi.org/10.1021/ci200227u
Lee SJ, Shin SJ, Lee MH, Lee MG, Kang TH, Park WS, Soh BY, Park JH, Shin YK, Kim HW, Yun CH, Jung ID, Park YM (2014) A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS ONE 9(8):e104351. https://doi.org/10.1371/journal.pone.0104351
Lemesre JL, Holzmuller P, Cavaleyra M, Goncalves RB, Hottin G, Papierok G (2005) Protection against experimental visceral leishmaniasis infection in dogs immunized with purified excreted secreted antigens of Leishmania infantum promastigotes. Vaccine 23(22):2825–2840. https://doi.org/10.1016/j.vaccine.2004.11.061
Lobanov MY, Bogatyreva NS, Galzitskaya OV (2008) Radius of gyration as an indicator of protein structure compactness. Mol Biol 42:623–628. https://doi.org/10.1134/S0026893308040195
Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, Baldi P (2010) High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 26(23):2936–2943. https://doi.org/10.1093/bioinformatics/btq551
Mahram A, Herbordt M (2015) Fast and accurate NCBI BLASTP: acceleration with multiphase FPGA-based prefiltering. NCBI BLASTP High-Perform Reconfigurable Comput Syst 7(4):1–20. https://doi.org/10.1145/2629691
Martins VT, Lage DP, Duarte MC, Costa LE, Garde E, Rodrigues MR, Chávez-Fumagalli MA, Menezes-Souza D, Roatt BM, Tavares CAP (2016) A new Leishmania-specific hypothetical protein, LiHyT, used as a vaccine antigen against visceral leishmaniasis. Acta Trop 154:73–81. https://doi.org/10.1016/j.actatropica.2015.11.006
Moore EM, Lockwood DN (2010) Treatment of visceral leishmaniasis. J Glob Infect Dis 2(2):151–158. https://doi.org/10.4103/0974-777X.62883
Nagill R, Kaur S (2011) Vaccine candidates for leishmaniasis: a review. Int Immunopharmacol 11(10):1464–1488. https://doi.org/10.1016/j.intimp.2011.05.008
Niwa T, Ying BW, Saito K, Jin W, Takada S, Ueda T, Taguchi H (2009) Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc Natl Acad Sci USA 106(11):4201–4206. https://doi.org/10.1073/pnas.0811922106
Pandey RK, Bhatt TK, Prajapati VK (2018) Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci Rep 8(1):1125. https://doi.org/10.1038/s41598-018-19456-1
Patronov A, Doytchinova I (2013) T-cell epitope vaccine design by immunoinformatics. Open Biol 3(1):120139. https://doi.org/10.1098/rsob.120139
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, Peters B (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9:514. https://doi.org/10.1186/1471-2105-9-514
Rahman N, Ali F, Basharat Z, Shehroz M, Khan MK, Jeandet P, Nepovimova E, Kuca K, Khan H (2020) Vaccine design from the ensemble of surface glycoprotein epitopes of SARS-COV-2: an immunoinformatics approach. Vaccines (Basel). https://doi.org/10.3390/vaccines8030423
Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 5(4):e9862. https://doi.org/10.1371/journal.pone.0009862
Rappuoli R (2000) Reverse vaccinology. Curr Opin Microbiol 3(5):445–450. https://doi.org/10.1016/s1369-5274(00)00119-3
Ribeiro PA, Dias DS, Lage DP, Costa LE, Martins VT, Tavares GS, Mendonça DV, Lima MP, Oliveira JS, Steiner BT (2018) Evaluation of a Leishmania hypothetical protein administered as DNA vaccine or recombinant protein against Leishmania infantum infection and its immunogenicity in humans. Cell Immunol 331:67–77. https://doi.org/10.1016/j.cellimm.2018.05.009
Santos WR, Aguiar IA, Paraguai de Souza E, de Lima VM, Palatnik M, Palatnik-de-Sousa CB (2003) Immunotherapy against murine experimental visceral leishmaniasis with the FML-vaccine. Vaccine 21(32):4668–4676. https://doi.org/10.1016/s0264-410x(03)00527-9
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33:363. https://doi.org/10.1093/nar/gki481
Sharma SK, Dube A, Nadeem A, Khan S, Saleem I, Garg R, Mohammad O (2006) Non PC liposome entrapped promastigote antigens elicit parasite specific CD8+ and CD4+ T-cell immune response and protect hamsters against visceral leishmaniasis. Vaccine 24(11):1800–1810. https://doi.org/10.1016/j.vaccine.2005.10.025
Shey RA, Ghogomu SM, Esoh KK, Nebangwa ND, Shintouo CM, Nongley NF, Asa BF, Ngale FN, Vanhamme L, Souopgui J (2019) In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep 9(1):4409. https://doi.org/10.1038/s41598-019-40833-x
Shi J, Zhang J, Li S, Sun J, Teng Y, Wu M, Li J, Li Y, Hu N, Wang H, Hu Y (2015) Epitope-based vaccine target screening against highly pathogenic mers-cov: an in silico approach applied to emerging infectious diseases. PLoS ONE 10(12):e0144475. https://doi.org/10.1371/journal.pone.0144475
Singh G, Pritam M, Banerjee M, Singh AK, Singh SP (2020) Designing of precise vaccine construct against visceral leishmaniasis through predicted epitope ensemble: a contemporary approach. Comput Biol Chem 86:107259. https://doi.org/10.1016/j.compbiolchem.2020.107259
Sinha AK, Singh P, Prakash A, Pal D, Dube A, Kumar A (2017) Putative drug and vaccine target identification in Leishmania donovani membrane proteins using naive bayes probabilistic classifier. IEEE/ACM Trans Comput Biol Bioinform 14(1):204–211. https://doi.org/10.1109/TCBB.2016.2570217
Stager S, Rafati S (2012) CD8(+) T cells in leishmania infections: friends or foes? Front Immunol 3:5. https://doi.org/10.3389/fimmu.2012.00005
Vakili B, Eslami M, Hatam GR, Zare B, Erfani N, Nezafat N, Ghasemi Y (2018) Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int J Biol Macromol 120:1127–1139. https://doi.org/10.1016/j.ijbiomac.2018.08.125
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718. https://doi.org/10.1002/jcc.20291
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, Peters B (2019) The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 47(D1):D339–D343. https://doi.org/10.1093/nar/gky1006
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(suppl2):W407–W410. https://doi.org/10.1093/nar/gkm290
Yu CS, Cheng CW, Su WC, Chang KC, Huang SW, Hwang JK, Lu CH (2014) CELLO2GO: a web server for protein subcellular localization prediction with functional gene ontology annotation. PLoS ONE 9(6):e99368. https://doi.org/10.1371/journal.pone.0099368
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26(13):1608–1615. https://doi.org/10.1093/bioinformatics/btq249
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author(s) declare no competing interests in this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lari, A., Lari, N. & Biabangard, A. Immunoinformatics Approach to Design a Novel Subunit Vaccine Against Visceral Leishmaniasis. Int J Pept Res Ther 28, 34 (2022). https://doi.org/10.1007/s10989-021-10344-3
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-021-10344-3