
Abstract
Mycobacterium leprae, an infectious agent of chronic infection so-called Leprosy. It is a prime healthconcern in various countries including India. India is currently running one of the extensive leprosy eradication programs in the globe, named as the National Leprosy Eradication Program (NLEP). Still, the situation is getting substandard because of the emergence of resistant strains. In the present study, newer approaches –like computational subtractive proteomics and reverse vaccinology has been applied in order to find out probable drug targets and vaccine candidates. The systematic workflow of the current study consists of a computational approach, where complete proteome of the bacteria is gradually reduced to find out few unique probable drug targets and reverse vaccinology, to find out probable vaccine antigens. Reverse vaccinology approach does not require a pathogen to be grown in the laboratory, encouraging its application to microorganisms that may not be easily cultivated like M. leprae but at least have an accessible genome sequence. This approach facilitates an easier and productive process ofantigen discovery. Results from the present study could facilitate selecting M. leprae proteins for drug design as well as vaccine production pipelines in the future.
Graphic Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Leprosy is a chronic infectious peripheral neuropathy caused by Mycobacterium leprae. The different types of clinical presentations of the disease (Leprosy) are determined by the grade of the host immune response (Nascimento 2013). It is also known as Hansen’s disease, named after Hansen, who is credited with the discovery of M. leprae in 1873 (Marmor 2002). It is a serious health concern in economically poor countries of Africa, Asia and Latin America (Uddin et al. 2016). It damages peripheral nerves and also affects the skin, eyes, nose and muscles. Nerve injury in leprosy can cause chronic disabling deformities. According to the official reports received from 138 countries from all WHO regions-The worldwide registered prevalence of leprosy at the end of 2015 was 176,176 cases (0.2 cases per 10 000 people). The number of new cases reported globally in 2015 was 211,973 (2.9 new cases per 100 000 people). World Health Organization reported 216,108 cases in 2016 and an increase of 20,765 cases in 2017. The number of new cases every year indicates the degree of continual transmission of infection. India is currently running one of the largest leprosy eradication programs in the world, the National Leprosy Eradication Program (NLEP). Still, 1.2–1.3 hundred thousand new cases of leprosy reported every year, 58.8% of the total amount of new cases reported every year (Lavania et al. 2018). The mode of transmission of leprosy is not well understood, although it is probably person to person via nasal droplets (Hatta et al. 1995). According to the World Health Organization, at present leprosy is treatable with multi-drug therapy, which involves three drugs to kill the pathogen and cure the victim. M. leprae had mutational changes in some of their genes like gyrA, rpoB, and folP which developed resistance against drugs like newer quinolones, rifampicin, and dapsone. Resistant strains of M. leprae appeared due to mutations in the macrolide target, the ribosome. This finding proposes the emergence of multidrug-resistant M. leprae. Hence the mycobacterial cell wall with its specific composition and structure is referred to be a major factor in promoting the natural resistance of mycobacteria to various antibiotics (Shanmugam and Natarajan 2010). The alarming emergence of the drug resistance strains among many bacterial diseases including the M. lepraeposes a big challenge to find effective cures since the existing drugs are no more active (Matsuoka et al. 2010).
Our study describes the identification of potential drug targets and vaccine candidates within the protein-pool belong to the unique metabolic pathway of M. leprae. The extensive application of computational methods provides an effortless way to identify the epitopes of high antigenic properties (Khan et al. 2019). Since the proposed drug targets and vaccine candidates are non-homologous to the human host, therefore, reducing conflicts related to any side effects associated with the inhibition of their activities. It is expected that the drugs developed against identified targets will be specific to the pathogen and of minimal toxicity for the host (Khan et al. 2019).This study is unique as we are quite assured that the study will move forward the research in a new and effectual direction to cure the deadly disease caused by M. leprae (Leprosy).
Materials and Methods
Bacterial Strain
The highly virulent strain M. leprae TN was chosen for this in silico analysis. The genome of bacterial strain is available on the NCBI database with accession number AL450380 and also listed in the Vaxign program used in the current study.
Subtractive Proteomic Analysis for Identification of Unique Metabolic Pathway
A database named Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto 2000) employed to gain information about different metabolic pathways of M. leprae TN. Subtractive proteomic analysis, in which the complete proteome of the bacteria is step-wise reduced to a few novel protein targets, which are mandatory for the survival of bacteria. For the identification of unique metabolic pathways, a manual comparative analysis was performed.
Prediction of Subcellular Localization
Prediction of selected proteins subcellular localization was done by using PSORTb version 3.0 (Yu et al. 2010), it predicts results for different subcellular localization like cell wall, cytoplasmic membrane, and extracellular and unknown as well and CELLO2GO (Yu et al. 2014) web-based system, which is publically available for screening of various properties of a query protein and its subcellular localization.
Functional Family and Conserved Domain Prediction
SVM-Port web-based server has been employed for the prediction of protein functional families. SVM-Prot is a server for the classification of a protein into its all major functional classes (Cai et al. 2003). Conserved domain identification was done by using NCBI CDD (A Conserved Domain Database for the functional annotation of proteins) (Marchler-Bauer et al. 2014), The results were cross-checked by InterProScan (Zdobnov and Apweiler 2001) and Pfam (Bateman et al. 2004).
Protein–Protein & Chemical-Protein Interaction Network Mapping
The prediction of protein–protein interaction was done by using the STRING database (Szklarczyk et al. 2014). The chemical-protein Interaction network was predicted via STITCH (Kuhn et al. 2011), which is a sister project of the protein–protein interactions server STRING.
Membrane Protein Pre-selection
Vaxign (He et al. 2010) was employed to shortlist proteins with potential as vaccine candidates based on their cellular localization, number of transmembrane helixes and probability of having adhesin-like characteristics. Vaxign containing 396 genomes list at present, along with fifteen Mycobacterium genomes. The probability of adhesion like characteristics is predicted by SPAAN software (Sachdeva et al. 2004) and the prediction of transmembrane helix topology is done by using Constrained Consensus Topology prediction SERVER, CMTOP (Käll et al. 2007).
Protein Antigenicity
VaxiJen v2.0 (Doytchinova and Flower 2007) was utilized to predict protein antigenicity. This software requires FASTA-submitted amino acid sequences and uses the physicochemical properties of proteins to predict their antigenicity. This feature is denoted according to an antigenic score respectively (Meunier et al. 2016).
B-Cell Epitope Prediction
BCPreds software (El-Manzalawy et al. 2012) was employed to identify B-cell epitopes in submitted amino acid sequences in FASTA format. This program provides two methods based on two different algorithms: first is (AAP) method and the BCPreds method using string kernels. These methods predict antigenic linear non-overlapping 20-mer epitopes from the whole antigen. Each selected protein was analyzed and B-cell epitopes with a score of about > 0.8 were accepted (specificity > 80%).
Trans Membrane Helix Topology and Signal Peptide Prediction
Transmembrane helices topology has been predicted via Phyre2 (Kelley and Sternberg 2009). Signal peptide prediction was predicted done by using SignalP 4.1 Server. Active binding pockets of proteins were analyzed via CASTp (Computer Altas of Surface Topography of proteins) (Dundas et al. 2006).
Homology Modeling and Structure Validation
For modeling of proteins, the sequences of proteins were retrieved from the UniprotKB protein sequence database and different templates were identified using PSI-BLAST against the RCSB (PDB). 3D-models were built using Swiss- Model Workspace (Arnold et al. 2006), accessible via the ExPASy web-based server. Validation of the model was done with the help of online tools like- ERRAT (Colovos and Yeates 1993), Verify3D (Eisenberg et al. 1997) and PROCHECK (Laskowski et al. 1993) available at the SAVES server. Ramachandran plots of preselected proteins were generated via PROCHECK. The ERRAT server provides the overall quality factor. Protparam server was employed for physio-chemical property prediction.
Molecular Docking
The modeled proteins under our study were docked with their respective chemical ligands for their interaction study using AutoDock4.2.5.1 (Morris et al. 2009). The structures of chemical ligands were shown in Supp. File Fig. 1. Pre-docking preparation was carried out for ligand preparation, the energy minimization through USCF Chimera (https://www.cgl.ucsf.edu/chimera/). AutoDock involves an empirical free energy force field with a Lamarckian Genetic Algorithm. Only Polar hydrogen was added to the protein and Kollman and Gastegier charges were assigned. The spacing between grid points was set to default value of 0.558 Å. The grid size selected for docking was huge enough to include all the domains of protein. A total of 50 independent runs were performed with a step sizes of 0.2 Å for translations and 50 for orientations and torsions. The maximum number of generations was set to 1000 and maximum number of top individuals that automatically survived was set to 1 with mutation rate of 0.02, crossover rate of 0.8, cluster tolerance 0.5 Å, external grid energy 1000.0.
Molecular Simulation
The simulation was executed with GROMACS 2018.1 package with the force field as Gromos43a1. The protein salvation was executed with SPC water model in a cubic box (10.8 × 10.8 × 10.8 nm3). The solvated system of protein was processed for energy minimization using the steepest algorithm up to a maximum 25,000 steps or until the maximum force (Fmax) is not greater than 1000 kJ/ mol nm which is the default threshold. The NVT and NPT ensembles for 50,000 steps (100 ps) at 300 K and 1 atm. Here, the system was firstly equilibrated using NVT ensemble followed by NPT ensemble. Then after the final Molecular Dynamic Simulation dock complex of phosphotidylmyo inositol dimannoside synthase with d-mannosamine and phosphotidylinositol dimannoside acyltransferase with 6-O-palmitoyl-alpha-d-mannopyranoside. Finally, the simulations were evaluated RMSD and RMSF were calculated for complete episode of simulations. The protein–ligand simulation of docked protein was also performed to study interaction pattern of ligands and change in protein after molecular docking with respective ligand. The all steps are kept similar except the final the molecular dynamics simulation was carried out for 50 ns long.
Results
Identification and Selection of Unique Metabolic Pathway and Non-homologous Proteins
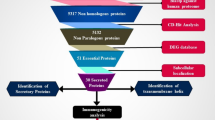
Knowledge regarding the different metabolic pathways of M. leprae and its human host is gained from the KEGG pathway database. Manual comparative analysis is performed in order to identify unique pathways of M. leprae and out of 100 metabolic pathways of M. leprae, 29 pathways were found to be unique (that is absent in H. sapiens). It was reported earlier that the immunomodulatory properties of LAM (Lipoarabinomannan) and related glycolipids contribute to the survival of M. leprae the causative agent of leprosy (Information available on KEGG Database). Because of this critical role, the LAM pathway was selected as the target pathway for drug and vaccine candidate’s prioritization in our study (Supp. File Fig. 4). Proteins involved in the LAM pathway were searched via NCBI BLASTp search tool against the human proteome. Only four proteins were found unique comparative to human proteome and so these four proteins selected for further study (Table 1).
Prediction of Sub-Cellular Localization
Localization prediction was implemented in the present study to prioritize promising drug target and vaccine candidates. All selected four non-homologous proteins involved in the LAM pathway were exposed to the prediction of subcellular localization by employing PSORTb version 3.0 and CELLO2GO (Table 1). The subcellular localization of a protein can provide information about its function (Yu et al. 2004) and also cytoplasmic or membrane localization of the target proteins helps to determine the ease of purification steps to be followed in the study (Yu et al. 2004). The membrane proteins alpha-1,2-mannosyltransferase and mannosyltransferase were selected as vaccine candidate identification; cytoplasmic proteins phosphatidyl-myo-inositol dimannoside synthase and phosphatidylinositol dimannoside acyl transferase were selected as drug target proteins for further respective characterization in the current study.
Drug Target and Vaccine Candidate’s Prioritization
We performed several systematic steps in order to characterize drug targets as well as vaccine candidates. This step involves the use of several bioinformatics tools, databases as well as target prioritization parameters. For drug target prioritization, conserved domain identification was performed using NCBI CDD (A Conserved Domain Database for the functional annotation of proteins). All selected proteins were found conserved in different strains; results were cross analyzed by InterProScan and Pfam. Protein–protein interaction was predicted done by using the STRING database, the outcome is shown in Fig. 1 and Table 2. Along with this, the chemical-protein interaction network was also predicted via STITCH (Search Tool for Interactions of Chemicals). Protein functional families were predicted through (SVMProt server) data available for all three proteins except (phosphatidylinositol dimannosideacyltransferase). Active binding pockets of proteins were analyzed via CASTp (Computer Altas of Surface Topography of proteins). The CASTp result revel that a total 67,235 and 16 number of pockets are present in phosphatidyl-myo-inositol dimannosidesynthase, phosphatidylinositol dimannosideacyltransferase, alpha-1,2-mannosyltransferase, and mannosyltransferase respectively (Supp. File Fig. 2). Out of these pockets, the largest pocket has a surface area of 817.266, 1334.289, 21.861, 275.998 and volume 1060.502, 1175.936, 5.941, 156.827 for protein phosphatidyl-myo-inositol dimannoside synthase, phosphatidylinositol dimannosideacyltransferase, alpha-1,2-mannosyltransferase, and mannosyltransferase respectively.

Protein–protein interaction network mapping- predicted via STRING database: a Phosphatidyl-myo-inositol dimannoside synthase. b Phosphatidylinositol dimannosideacyltransferase. c Alpha-1,2-mannosyltransferase. d Mannosyltransferase
For vaccine candidate’s prioritization, length of B-cell epitope, position and score were predicted by BCPreds software through FASTA-submitted amino acid sequences. Amino acid pair (AAP) antigenicity, an algorithm was utilized. Each selected protein was analyzed and epitopes with a score of more than 0.8 and specificity more than 80% were selected. The number of transmembrane helix and antigenicity was predicted via Vaxign, a web-based pipeline. Both preselected proteins were found to be probable antigenic (Table 3). The trans-membrane helices topology has been predicted via Phyre2 (Fig. 2) and the presence of signal peptide was predicted by SignalP 4.1 Server respectively (Fig. 3).

Phyre2 topology prediction (transmembrane helices have been predicted and adopted topology is shown above for membrane protein a (Alpha-1,2-mannosyltransferase) b (Mannosyltransferase)

Prediction of signal peptide: presence of signal peptide predicted in alpha-1,2-mannosyltransferase via SignalP 4.1 Server
Homology Modeling and Validation
The homology modeling method was subjected to construct three-dimensional models of preselected proteins of M. leprae, as their three-dimensional structures are unavailable in PDB. For homology modeling, the sequences of protein were retrieved from the UniprotKB protein sequence database and different templates were identified using PSI-BLAST against the RCSB (PDB). Template IDs 3okp.1.A, 5oce.1.A, 3rce.1.A, 5ezm.1.A selected from the organisms- Corynebacterium glutamicum, Mycobacterium smegmatis, Campylobacter lari, Cupriavidus metallidurans respectively (Table 4). The 3D-models were built usingSwiss- Model Workspace and shown in Fig. 4. Assessments of 3D- models were performed on the premise of QMEAN score, E-Value and Z-Mean. The online tools ERRAT, Verify3D were utilized for further structure validation, available at Structure Analysis and Verification Server (SAVES v 5.0). PROCHECK server and RAMPAGE serverwere implemented for Ramachandran plot statistics analysis (Supp. File Fig. 3 and Table 1). Resulted, 0.0% of residues fall in the category of the disallowed regions except for protein (mannosyltransferase = 2.7%). The physio-chemical properties of all preselected proteins of M. leprae were predicted via the Protparam server shown in Table 5.

Modelled protein structures: a Phosphatidyl-myo-inositol dimannoside synthase b Phosphatidylinositol dimannosideacyltransferase c Alpha-1,2-mannosyltransferase d Mannosyltransferase
Molecular Docking
Molecular docking was performed with the respective ligand of selected protein to depict the binding pattern of inhibitors with protein under study. Phosphotidylmyo inositol dimannoside synthase was docked withd-mannosamine, phosphotidylinositoldimannosideacyltransferase was docked with 6-O-palmitoyl-alpha-d-mannopyranoside, alpha-1,2 mannosyltransferase was docked against deoxycholic acid and taurocholic acid, mannosyltransferase was docked against 5-[[3,4-bis (phenyl methoxy) phenyl] methylene]-4-oxo-2-thioxo-3-thiazolidine acetic acid and{5-[3-(2-Hydroxy-1-phenyl-ethoxy)-4-phenethyloxy-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-yl}-acetic acid. Result analyses of molecular docking were shown in Table 6 and Docking pose analysis via LigPlot (Wallace et al. 1995) shown in Fig. 5.

Molecular Docking result showing protein–ligand interaction; where oxygen (O), nitrogen (N) and carbon (C) atoms are represented in red, blue and black circles: a Lig-plot of d-mannosamine with phosphotidylmyo inositol dimannosidesynthetase. b 6-O-palmitoyl-α-d-mannopyranoside with phosphatidylinositol dimannosideacyltransferase c Deoxycholic acid with protein alpha 1,2-mannosyltransferase d Taurocholic acid with protein alpha 1,2-mannosyltransferase. e 5-[[3,4-bis(phenylmethoxy)phenyl] methylene]-4-oxo-2-thioxo-3 thiazolidine acetic acid with protein mannosyltransferse. f {5-[3-(2-Hydroxy-1-phenyl-ethoxy)-4-phenethyloxy-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-yl}-acetic acidwith protein mannosyltransferse (Color figure online)

Molecular dynamic simulation study: RMSD and RMSF graph of a Phosphotidylmyo inositol dimannoside synthase was docked with d-mannosamine and b Phosphotidylinositol dimannoside acyltransferase docked with 6-O-palmitoyl-alpha-d-mannopyranoside
Molecular Dynamics Simulation
The protein was energy minimized using Gromos43a1. The root mean square fluctuation (RMSF) is the time-average of root mean squared deviation (RMSD) for all residues that were also calculated for Phosphotidylmyo inositol dimannoside synthase was docked withd-mannosamine and Phosphotidylinositoldimannosideacyltransferase docked with 6-O-palmitoyl-alpha-d-mannopyranoside, the RMSD, and RMSF graph are shown in Fig. 6a, b respectively. The energy minimization of protein Alpha-1, 2 mannosyltransferase and Mannosyltransferase was performed and results are shown in Figs. 7, 8 respectively. Protein conformation was almost stable throughout the simulation.

Molecular dynamic simulation study protein alpha 1,2-mannosyltransferase showing various parameter molecular dynamic simulation during, NPT and NVT

Molecular dynamic simulation study protein mannosyltransferase showing various parameter molecular dynamic simulation, NPT and NVT
Discussion
Leprosy has emerged as a major problem, reasons for a large number of deaths in the globe. In spite of this, there is no persistent cure and prevention for M. leprae, which is multi-drug resistant. In the last decade, bioinformatics has revolutionized vaccine development strategy; Reverse vaccinology allows vaccines to be designed even for non-cultivable pathogens. Therefore, this strategy comparably reduces the time needed to develop new vaccines (Meunier et al. 2016; Sette and Rappuoli 2010). Recently, the search for probable drug targets using computational methods and integrated “omics” data, such as genomics, proteomics, and metabolomics, has awarded much mindfulness and has been increasing rapidly (Chawley et al. 2014; Damte et al. 2013). Comparative, subtractive proteomics and genomics studies have been broadly used for the prediction and identification of probable therapeutic/drug targets and vaccine candidate proteins in various pathogenic microorganisms including bacteria and fungi (Amineni et al. 2010; Chong et al. 2006; Duffield et al. 2010; Johri et al. 2006; Volker and Brown 2002). In the current study LAM (Lipoarabinomannan) pathway was a center of attraction. For the same, these computational approaches coupled with the information available at KEGG Database provide us the information and prospect to perform the computational analysis aimed towards the identification of novel drug targets and vaccine candidate proteins belong to M. leprae.
Nowadays, where modernization overlaps traditional time consuming and laborious methods; a newer approach like reverse vaccinologycames into play. In addition reverse vaccinology based immunoinformatics approach is one of another sophisticated method. It can be an alternative approach for developing an epitope-based vaccine that can clarify humoral and cell-mediated immune response inside the host body. Reverse vaccinology approach analyses the entire protein sequences of the pathogen using modern bioinformatics tools to select target proteins for their high-throughput expression and validation (Gupta et al. 2018).
In the present study, the initial information of metabolic pathways obtained from the KEGG database described the details about the common pathway as well as the number of pathways among the M. leprae and its human host. LAM, possible virulence factor involved in the persistence of Mycobacterium within macrophages (Chan et al. 1991). Detailed studies related to structure and function over the last decade has led to present recognition of the mycobacterial lipoarabinomannan like a phosphatidylinositol anchored lipoglycan with diverse biological activities (Chatterjee and Khoo 1998). It has been shown that the immunomodulatory properties of LAM and related glycolipids contribute to the survival of M.leprae the causative agent of leprosy (Information available on KEGG Database). As a result, the LAM pathway wasselected as the target pathway for drug and vaccine candidate's prioritization.As a result, the LAM pathway wasselected as the target pathway for drug and vaccine candidate's prioritization.
Conclusion
We applied a computational subtractive and reverse vaccinology approach in order to propose novel and potential drug targets as well as unique vaccine candidates against M. leprae. Target proteins were selected based on non-homology and essentiality. Alpha 1,2-mannosyltransferase and mannosyltransferase proteins (related to LAM pathway) were identified as essential proteins that could serve as probable vaccine candidates. Results from the present study could ease selecting these M. leprae proteins for vaccine production pipelines in future. To conclude, reverse vaccinology justified as a powerful tool for identifying new vaccine candidates. We are quite assured that the study will move forward the research in a new and effectual direction to cure leprosy.
References
Amineni U, Pradhan D, Marisetty H (2010) In silico identification of common putative drug targets in Leptospira interrogans. J Chem Biol 3(4):165–173
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22(2):195–201
Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Studholme DJ, Sonnhammer EL (2004) The Pfam protein families database. Nucleic Acids Res, 32(suppl_1): D138–D141.
Cai C, Han L, Ji ZL, Chen X, Chen YZ (2003) SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res 31(13):3692–3697
Chan J, Fan X, Hunter S, Brennan P, Bloom B (1991) Lipoarabinomannan, a possible virulence factor involved in persistence of Mycobacterium tuberculosis within macrophages. Infect Immun 59(5):1755–1761
Chatterjee D, Khoo K-H (1998) Mycobacterial lipoarabinomannan: an extraordinary lipoheteroglycan with profound physiological effects. Glycobiology 8(2):113–120
Chawley P, Samal HB, Prava J, Suar M, Mahapatra RK (2014) Comparative genomics study for identification of drug and vaccine targets in Vibrio cholerae: MurA ligase as a case study. Genomics 103(1):83–93
Chong C-E, Lim B-S, Nathan S, Mohamed R (2006) In silico analysis of Burkholderia pseudomallei genome sequence for potential drug targets. In Silico Biol 6(4):341–346
Colovos C, Yeates T (1993) ERRAT: an empirical atom-based method for validating protein structures. Protein Sci 2:1511–1519
Damte D, Suh J-W, Lee S-J, Yohannes SB, Hossain MA, Park S-C (2013) Putative drug and vaccine target protein identification using comparative genomic analysis of KEGG annotated metabolic pathways of Mycoplasma hyopneumoniae. Genomics 102(1):47–56
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform 8(1):4
Duffield M, Cooper I, McAlister E, Bayliss M, Ford D, Oyston P (2010) Predicting conserved essential genes in bacteria: in silico identification of putative drug targets. Mol BioSyst 6(12):2482–2489
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res, 34(suppl_2): W116–W118.
Eisenberg D, Lüthy R, Bowie JU (1997) [20] VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol Elsevier 277:396–404
El-Manzalawy Y, Dobbs D, Honovar V (2012) BCPREDS: B-cell epitope prediction server. Artificial Intelligence Research Laboratory, Department of Computer Science, Iowa State University of Science and Technology. https://webs.iiitd.edu.in/raghava/bcepred/bcepred_submission.html
Gupta N, Khan F, Kumar A (2018) Exploring highly antigenic protein of Campylobacter jejuni for designing epitope based vaccine: immunoinformatics approach. Int J Pept Res Ther 25(3):1159–1172
Hatta M, van Beers SM, Madjid B, Djumadi A, de Wit MY, Klatser PR (1995) Distribution and persistence of Mycobacterium leprae nasal carriage among a population in which leprosy is endemic in Indonesia. Trans R Soc Trop Med Hyg 89(4):381–385
He Y, Xiang Z, Mobley HL (2010) Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. Biomed Res Int. https://doi.org/10.1155/2010/297505
Johri AK, Paoletti LC, Glaser P, Dua M, Sharma PK, Grandi G, Rappuoli R (2006) Group B Streptococcus: global incidence and vaccine development. Nat Rev Microbiol 4(12):932
Käll L, Krogh A, Sonnhammer EL (2007) Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res, 35(suppl_2): W429-W432.
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30
Kelley LA, Sternberg MJ (2009) Protein structure prediction on the web: a case study using the Phyre server. Nat Protoc 4(3):363
Khan F, Srivastava V, Kumar A (2019) Computational identification and characterization of potential T-cell epitope for the utility of vaccine design against enterotoxigenic Escherichia coli. Int J Pept Res Ther 25(1):289–302
Kuhn M, Szklarczyk D, Franceschini A, Von Mering C, Jensen LJ, Bork P (2011) STITCH 3: zooming in on protein–chemical interactions. Nucleic Acids Res 40(D1):D876–D880
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291
Lavania M, Singh I, Turankar RP, Ahuja M, Pathak V, Sengupta U, Das L, Kumar A, Darlong J, Maseey A, Nathan R (2018) Molecular detection of multidrug-resistant Mycobacterium leprae from Indian leprosy patients. J Glob Antimicrob Resist 12:214–219
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Lanczycki CJ, Hurwitz DI (2014) CDD: NCBI's conserved domain database. Nucleic Acids Res 43(D1):D222–D226
Marmor MF (2002) The ophthalmic trials of GHA Hansen. Surv Ophthalmol 47(3):275–287
Matsuoka M, Suzuki Y, Garcia IE, Fafotis-Morris M, Vargas-Gonzalez A, Carreno-Martinez C, Fukushima Y, Nakajima C (2010) Possible mode of emergence for drug-resistant leprosy is revealed by an analysis of samples from Mexico. Jpn J Infect Dis 63(6):412–416
Meunier M, Guyard-Nicodème M, Hirchaud E, Parra A, Chemaly M, Dory D (2016) Identification of novel vaccine candidates against Campylobacter through reverse vaccinology. J Immunol Res. https://doi.org/10.1155/2016/5715790
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
Nascimento OJ (2013) Leprosy neuropathy: clinical presentations. Arq Neuro-psiquiatria 71(9B):661–666
Sachdeva G, Kumar K, Jain P, Ramachandran S (2004) SPAAN: a software program for prediction of adhesins and adhesin-like proteins using neural networks. Bioinformatics 21(4):483–491
Sette A, Rappuoli R (2010) Reverse vaccinology: developing vaccines in the era of genomics. Immunity 33(4):530–541
Shanmugam A, Natarajan J (2010) Computational genome analyses of metabolic enzymes in Mycobacterium leprae for drug target identification. Bioinformation 4(9):392
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Kuhn M, Tsafou KP (2014) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(D1):D447–D452
Uddin R, Azam SS, Wadood A, Khan W, Farooq U, Khan A (2016) Computational identification of potential drug targets against Mycobacterium leprae. Med Chem Res 25(3):473–481
Volker C, Brown JR (2002) Bioinformatics and the discovery of novel anti-microbial targets. Curr Drug Targets Infect Disord 2(4):279–290
Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel 8(2):127–134
Yu CS, Lin CJ, Hwang JK (2004) Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci 13(5):1402–1406
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Brinkman FS, Foster LJ (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics, 26(13): 1608–1615.
Yu C-S, Cheng C-W, Su W-C, Chang K-C, Huang S-W, Hwang J-K, Lu C-H (2014) CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 9(6):e99368
Zdobnov EM, Apweiler R (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17(9):847–848
Acknowledgements
The authors would like to acknowledge the Institutional Research Scientific Committee (IRSC) of Dr. B. Lal Institute of biotechnology for financial and infrastructural support.
Funding
This research did not receive any specific grant from funding agencies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare there is no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gupta, E., Gupta, S.R.R. & Niraj, R.R.K. Identification of Drug and Vaccine Target in Mycobacterium leprae: A Reverse Vaccinology Approach. Int J Pept Res Ther 26, 1313–1326 (2020). https://doi.org/10.1007/s10989-019-09936-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-019-09936-x