
Abstract
Acinetobacter baumannii is a well-recognized cause of nosocomial infections. This organism is recognized to be among the most difficult antimicrobial-resistant gram-negative bacilli to control and treat. One of the main challenges we face is Carbapenem resistance in A. baumannii. Carbapenem resistance in A. baumannii associated with the loss of an outer membrane protein designated CarO (Carbapenem resistance outer membrane protein). This protein is a membrane porin of A. baumannii. Using specific antibodies against this protein exert a bacteriostatic or bactericidal effect in vitro. Attempts should be made to discover peptides that could mimic protein epitopes and possess the same immunogenicity as the complete protein. Subsequently, bioinformatics methods for epitope prediction have been developed leading to synthesis of such peptides that are important for development of vaccine. This study provides a basis for the design of pathogen specifically, B cell epitope-based vaccine that is targeted to diseases caused by A. baumannii in the global human population. A combination of available bioinformatics tools are used to understand and characterize the Baumannii Acinetobactin utilization structure of A. baumannii and appropriate selection regions as effective B cell epitopes and functional exposed amino acids. In conclusion, amino acids 19–158 were selected as vaccine candidate.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Acinetobacter baumannii is an aerobic, pleomorphic, rod-shaped and Gram-negative bacterium. The organism has low virulence but is capable of causing infection in organ transplantation and febrile neutropenia (Blue-Hnidy and Allen 2006; Weinstein et al. 2005). A. baumannii has known as an important and common pathogen creating nosocomial pneumonia and bacteremia among patients who admitted in the intensive care unit (ICU) worldwide, followed by skin, soft tissue, and urinary tract infection, and secondary meningitis over the past few decades (Kempf et al. 2012; Longo 2006; Peleg et al. 2008; Sánchez-Encinales et al. 2017; Wong et al. 2017). The resistance of A. baumannii to the commonly used antimicrobial drugs such as Aminopenicillins, Ureidopenicillins, Cephalosporins and Cephamycins has observed for the last 25 years (García-Patiño et al. 2017; Kang et al. 2017).
Since then, strains of A. baumannii have gained resistance to new antimicrobial drugs (Knezevic et al. 2016). Multidrug-resistant (MDR) A. baumannii is an emerging pathogen in healthcare and nosocomial infection rapidly. (Dijkshoorn et al. 2007; Pagdepanichkit et al. 2016). Carbapenem-resistant A. baumannii (CRAB) presently is a serious infection control challenge (Higgins et al. 2009; Livermore et al. 2016).
Carbapenem are regarded as the most powerful antibiotics because of its extremely effective antibacterial activity and low toxicity, but the emergence of Carbapenem resistance in A. baumannii has become a global concern recently. Non-enzymatic molecular mechanisms act in synergy with Carbapenem-hydrolyzing b-lactamases to provide high-level resistance, but they have been barely described (Shaker and Shaaban 2017; Wong et al. 2017). The Carbapenem-associated outer membrane protein, also called CarO, is the most characterized porin of A. baumannii (Mussi et al. 2005; Poirel and Nordmann 2006). CarO allows the selective uptake of the ornithine, other basic amino acids, and uptake of Carbapenem based on structural homologies (Gordon and Wareham 2010). MDR strains showed disruptions in the CarO gene by the various insertion elements (Catel-Ferreira et al. 2011).
This paper briefly discusses and explores bioinformatics tools in vaccine design. The best antigenic region of CarO has been determined as a novel construct for vaccine or diagnostics. That could be used as peptide vaccines against A. Baumannii.
Methods
Sequence Availability, Homology Alignment and Template Search
The CarO protein sequence was obtained from NCBI at http://www.ncbi.nlm.nih.gov/protein and saved in FASTA format for further analyses; then Protein BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) against reference sequence on the database was performed using Blosum80 matrix to collect homologous sequences among different bacteria. For Template Search, the protein sequence of CarO was used as an input data for the PSI-BLAST against protein data bank (PDB) at http://blast.ncbi.nlm.nih.gov/Blast.cgito identify its homologous structures. In order to choose the best template, lower E value, higher query coverage and maximum identity was considered. The best result of PSI-BLAST was selected as template (Altschul et al. 1990).
T-COFFEE at http://tcoffee.crg.cat/apps/tcoffee/do:regular and PRALINE (Notredame et al. 2000) at http://www.ibi.vu.nl/programs/pralinewww/ (Pirovano et al. 2008) were used for generating of alignments. Alignments can demonstrate the conservancy of the protein residues among various strains. In this regard, suitable vaccine candidates are those effective against all strains of a given pathogen. Conservancy of the amino acids among bacteria other than Acinetobacter implies probable cross-reactivity levels.
Protein Topology and Signal Peptide Prediction
Topology prediction servers predict the transmembrane and outer membrane region of proteins. Transmembrane helix and signal peptides are not suitable regions as B cell epitope. PRED-TMBB2 at http://www.compgen.org/tools/PRED-TMBB2 (Bagos et al. 2004) was employed to predict the transmembrane beta-strands of the gram-negative bacteria outer membrane proteins, and discriminating such proteins from water-soluble ones when screening large datasets. The transmembrane regions was also determined by SPOCTOPUS at http://octopus.cbr.su.se/ (Viklund et al. 2008).
SignalP 4.1 server at http://www.cbs.dtu.dk (Petersen et al. 2011) used to determine the presence and location of signal peptide cleavage sites in amino acid sequences. This property was also predicted by SOSUI at http://harrier.nagahama-i-bio.ac.jp/sosui/ (Hirokawa et al. 1998).
Functional Annotation of Protein Domain
Recognition of protein domains helps us recognize functionally important domains within a protein. Antibodies appointed against such domains could impair CarO functions and result in virulence reduction.
Dompred at http://bioinf.cs.ucl.ac.uk/psipred/dompred (Bryson et al. 2007) was used to predict Protein domains and detect homolog domains. InterProscan at http://www.ebi.ac.uk/interpro/about.html (Zdobnov and Apweiler 2001) was used to detect functional analysis of protein sequences by classifying them into families and predict the presence of domains and important sites.
Orientation of the Protein 3D Structure in Membrane
Rotational and translational positions of transmembrane and peripheral proteins in membranes using their 3D structure (PDB coordinate file) as input were calculated by the PPM server at http://opm.phar.umich.edu/server.php. It can be applied to newly determine experimental protein structures or theoretical models. Many membrane-associated proteins from the PDB have already been pre-calculated and can be found in the OPM database.
Predicting Functionally and Structurally Important Residues
Functional conserved amino acids allow more realism and robustness in the description of epitope prediction and protein binding surfaces. There are different ways to predict functionally and structurally important residues. In this study, conserved functional and structural amino acids were determined by Consurf at http://consurf.tau.ac.il (Berezin et al. 2004). Consurf predicts functional residues according to the neural-network algorithm. InterProSurf at http://curie.utmb.edu/pattest9.html (Negi et al. 2007) used to predict functional sites on protein surface using patch analysis.
Physicochemical Properties of Protein
Parameters such as hydrophilicity, flexibility, accessibility, turns, exposed surface, polarity and antigenic propensity of polypeptides chains have been correlated with the location of B cell epitopes. This has led to a search for empirical rules that would allow the position of B cell epitopes to be predicted from certain features of the protein sequence. Bcepred at http://www.imtech.res.in/raghava/bcepred/ (Saha and Raghava 2004) was used to predict linear B-cell epitopes in a protein sequence. Average score of physico-chemical properties (hydrophobicity, flexibility/mobility, accessibility, polarity, exposed surface and turns) of proteins were predicted with IEDB at http://tools.iedb.org/bcell/ (Vita et al. 2014).
Prediction of Linear B Cell Epitope
The identification and characterization of B-cell epitopes play an important role in vaccine design, immunodiagnostic tests, and antibody production. There are many tools act as B cell epitope predictor. Since each software uses special algorithm and individual methods in epitope prediction. Therefore, usage of several tools to predict linear B-cell epitopes in protein sequences are highly desirable.
BepiPred 1.0 server at http://www.cbs.dtu (Jespersen et al. 2017) was used to predict linear B-cell epitopes based on hidden Markov model. Bcpred server at http://ailab.ist.psu.edu (EL‐Manzalawy et al. 2008) with 90% specificity was also employed with report of overlapping epitopes. Bcpred explores two machine learning approaches for predicting flexible length linear B-cell epitopes. antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity scores (SVMTriP) in order to achieve the higher accuracy and specificity were predicted by Svmtrip at http://sysbio.unl.edu/SVMTriP/prediction.php (Yao et al. 2012).
Prediction of Conformational B Cell Epitope
Ellipro at http://tools.immuneepitope.org (Ponomarenko et al. 2008) predicted linear and discontinuous antibody epitopes based on a protein antigen’s 3D structure. Discotope at http://.cbs.dtu.dk/services/DiscoTope/(Kringelum et al. 2012) determined discontinuous B cell epitopes from protein 3D structures. The method utilizes calculation of surface accessibility and a novel epitope propensity amino acid score. The final scores are calculated by combining the propensity scores of residues in spatial proximity and the contact numbers.
Epitopia at http://epitopia.tau.ac.il/ (Rubinstein et al. 2009) used to detect immunogenic regions in a given protein structure or sequence. The input is analyzed with regards to its physico-chemical and structural-geometrical properties.
Pocket and Binding Site Detection
GHECOM (Grid-based HECOMi finder) at http://strcomp.protein.osaka-u.ac.jp/ghecom/ (Zhang et al. 2011) was used to find multi-scale pockets on protein surfaces. In addition, Depth (http://mspc.bii.a-star.edu.sg/tankp/help.html) was employed to calculate/predict depth, cavity sizes, ligand binding sites and PKA. Depth measures the closest distance of a residue/atom to bulk solvent (Tan et al. 2011).
Immunogenic Regions Selection
A region with the largest gatherings of linear and conformational epitopes could be selected as vaccine candidates. This region should be qualified as single-scale amino acid properties assay. Some specifications such as probability of antigenicity, physiochemical properties average, PI and etc. should also be considered in region selection. Hence, one region was selected as appropriate antigenic candidates. Further analyses were performed on the selected regions to validate the selection.
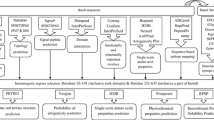
The flow chart of experimental procedure which summarizes an overview of the methodology used in this study is shown in Fig. 1.

Flow chart showing an overview of the methodology pipeline
Results
Protein Sequencing and Alignments
CarO protein sequence with accession No. AKL79738.1 is available as a query for protein BLAST against Acinetobacter producing a set of sequences containing various species of Acinetobacter. Out of 100 alignments, 50 sequences with the highest similarity (identity ≥ 90%, query coverage: 100% and E value: 0) were selected as PRALINE and T-COFFEE alignment input. BLAST search of Acinetobacter revealed numerous sequences of CarO. These hits showed that the CarO protein is specific for Acinetobacter species. The results of PSI-BLAST revealed that there are limited proteins similar to CarO with resolved structures. The structure with highest score under the PDB code of 4FUV_A (73.68% identity, 91% Query Cover) was selected as the template for homology modelling.
The image of T-COFFEE alignment results with conservation color scheme is shown in Fig. 2. PRALINE confirms T-COFEE results.

CarO sequence alignment with 45 sequences obtained from protein BLAST against Acinetobacter. The superposition was made with the T-coffee program and adjusted manually. Residues conservancy is depicted by blue to pink colors (Color figure online)
Protein Topology and Signal Peptide Prediction
Cleavage site of signal peptide was predicted between positions 21 and 22 of protein sequence by Signalp and SOSUI server. PRED-TMBB2 server and SPOCTOPUS identify eight transmembrane segments. 2D topology prediction of PRED-TMBB2server is shown in Fig. 3a, b.

Topology prediction and detection of beta-barrel outer membrane proteins by PRED-TMBB 2 server. The diagram shows (a) the estimated preference of a particular residue to be located either on the transmembrane (red) or on the periplasmic (blue) or extracellular (pink). Schematic picture (b) shows residues from 39–46, 53–59, 61–69, 96–104, 116–124, 171–181, 185–195 and 237–249 are transmembrane regions (Color figure online)
Functional Annotation of Protein Domain
Domain annotation shows that CarO protein is composed of one domain that it contains 2 stranded b-barrel at positions 47 to 135 and 163 to 202 of protein.
Orientation of the Protein 3D Structure in Membrane
The OPM database currently includes all unique structures of transmembrane protein complexes and selected monotopic, peripheral proteins and membrane-bound peptides from PDB with their calculated membrane boundaries. OPM explores orientations of quaternary complexes formed by a number of interacting proteins, rather than orientations of individual subunits or domains. The precision of calculated hydrophobic thicknesses and tilt angles are ~ 1 Å and 2°, respectively, as judged from their deviations in different crystal forms of the same proteins. The fluctuations of these parameters calculated within 1 kcal/mol around the global minimum of transfer energy are usually smaller than 2 Å and 4°, respectively (± values in all Tables of the database). The calculated tilt angles in homologous proteins differ by 2–16° depending on the size of the protein, its oligomeric state and percentage of sequence identity (Fig. 4).

Orientation of protein in cell membrane. The origin of coordinates corresponds to the center of lipid bilayer. Z axis coincides with membrane normal; atoms with the positive sign of Z coordinate are arranged in the “outer” leaflet as defined by the user-specified topology. Positions of DUMMY atoms correspond to locations of lipid carbonyl groups
Functionally and Structurally Important Residues
ConSurf and interproSurf annotated functional residues on the 3D structure of CarO. Results are shown in Figs. 5 and 6, respectively. In ConSurf server Protein structure shows in ribbon. Functional residues which were predicted by ConSurf at the protein structure differentiated with Purple filling space model. InterproSurf revealed residues number 95, 152, 154, 156, 166, 19, 161, 223, 224, 225, 82, 83, 84, 91, 92, 93 by auto patch analysis.

ConSurf, identification of functionally and structurally important residues results. Functional residues which were predicted by ConSurf at the protein structure differentiated with Purple filling space model (Color figure online)

Functional residues at the protein structure surface predicted by Interprosurf. Protein structure shows in ribbon. Functional residues which were predicted by InterProSurf at the protein structure differentiated with red and green filling space model (Color figure online)
Physicochemical Properties of Protein
IEDB and Bcepred servers predict properties such as hydrophilicity, accessibility, antigenicity, flexibility and beta turn secondary structure in the protein sequence. Although single-scale amino acid properties were acquirable in all sequence length, most salient regions of higher probability were located in regions 45–60, 100–120 and 145–160. Peaks in the plot indicate putative susceptible epitope boundaries (Fig. 7).

Graphical display of linear epitopes predicted by various software. The length and position of linear epitopes are graphically illustrated on a gray rod. The epitopes with the highest score are shown in red and the remainders are in blue. The consensus predictions of more or all servers shown in violet (Color figure online)
Linear and Conformational B Cell Epitope Prediction
Linear B cell epitopes predicted by Bepipred server are more concentrated in the regions of 45–50, 74–82, 148–159 and 212–229. A region at region 19–38 shows the presence of a high density of linear epitopes. The highest score is related to ‘‘MKVLRVLVTTTALLAAGAAMADEAVVHDSYAFDKNQLIP’’ sequence at region 1–39 (Fig. 7). ABCpred result shows 20 hits of 16 meric peptide sequences as B-cell epitopes ranking based on scores (Fig. 7).
BCpred predicted 6 linear B-cell epitopes in CarO sequence ranking based on scores (Fig. 7). The most reliable linear epitopes predicted by BCpred (score = 0.994 and 0.987) are ‘‘SIDGKNYQQAVPGQEGGVRG’’ and ‘‘LNAEIRPWGASTNPWAQGLY’’ started at position 142 and 98 respectively. Svmtrip predicted 3 linear B cell epitopes ranking based on score (Fig. 7). The best epitope recommended by this server is ‘‘AMADEAVVHDSYAFDKNQLI’’ at region 19–38. ‘‘GAAYLDNDYDLAKRIGNGDT’’ at region 121-140 is the second best epitope predicted by Svmtrip. Six linear along with 2 discontinuous B cell epitopes were predicted by ElliPro software (Tables 1, 2). Two linear and 2 discontinuous epitopes with the highest PI (protrusion index) are shown in Fig. 8a, b.

Epitope mapping on 3D models. Discovery Studio Visualizer 2.5.5 software was used. From left to right, 2 linear (a) and 2 discontinuous epitopes (b) with the highest PI score predicted by Ellipro server are shown
Discontinuous B cell epitopes predicted from the 3D structure of protein by Discotope are shown in Fig. 9. All these servers highlighted outer membrane loops as conformational B cell epitope. Epitopia predicted 5 immunogenic regions. These regions are located at positions between 417 and 436, 287 and 307, 470 and 482, 554 and 570 and 609 and 619 respectively ranking based on scores.

Discontinuous B cell epitopes predicted from protein 3D structures by Discotope. Protein 3D structure is showing in ribbon in gray. Discontinuous B cell epitopes at the protein structure differentiated with yellow (Color figure online)
Protein Pocket Detection
GHECOM server found 11 pockets on protein surfaces using mathematical morphology. A residue in a deeper and larger pocket has a greater chance to be a true pocket. The pockets of small-molecule binding sites and active sites were higher than the average value; specifically, the values for the active sites were much higher. This suggests that pockets contribute to the formation of binding sites and active sites of protein. GHECOM results are shown in Fig. 10a, b.

Pocket detection of CarO protein by GHECOM server. a Graph residue-based pocketness. The height of the bar shows the value of pocketness [%] for each residue. The color of pocketness bar indicates cluster number of pocket (red: cluster 1, blue: cluster 2, green: cluster 3, yellow: cluster 4, cyan: cluster 5). b Jmol view of pocket structure based on pocketness color (Color figure online)
The potential binding sites (PBS) of proteins are part of residue or pocket which bind to ligands directly on protein surface, they are near to the ligand binding sites. Binding cavity is a protein sub-structure of conserved geometrical and chemical properties complimentary to its bound ligand. The algorithm estimates the probability value of form-ing part of a binding cavity for every residue of the protein. The plot shows both mean and standard deviation of depth values. Probability of residue forming a binding site and residue depth plot and a 3D rendition of the cavity prediction is shown in Fig. 11a, b.

Prediction of Probability of resi-due forming a binding site and residue depth plot and a 3D rendition of the cavity by depth server. a Probability of residue forming a binding site and residue depth plot. b A 3D rendition of the cavity prediction is shown using Jmol. Residues of the predicted bind-ing cavity are colored red and the rest of the protein is colored blue (Color figure online)
Immunogenic Region Selection
One region covering residues 19–158 was selected as vaccine candidate by various software (Fig. 12) and several properties was compared to her parent protein (CarO).Vaxijen antigenicity score, PI, instability index, solubility, hydrophilicity, accessibility, flexibility and secondary structure properties calculated for vaccine candidate. All results for candidate and CarO protein were summarized in Table 3. Parameters such as hydrophilicity, flexibility, accessibility, turns, exposed surface, polarity and antigenic propensity of polypeptides chains have been correlated with the location of continuous epitopes. This has led to a search for empirical rules that would allow the position of continuous epitopes to be predicted from certain features of the protein sequence. All prediction calculations are based on propensity scales for each of the 20 amino acids. Each scale consists of 20 values assigned to each of the amino acid residues on the basis of their relative propensity to possess the property described by the scale.

Graphical display of immunogenic regions predicted by various software. The region with the highest score are shown in red and the remainders are in blue. The consensus predictions of more or all servers shown in violet (Color figure online)
Discussion
Nosocomial infections caused by strains A. baumannii are a serious clinical problem. CarO from A. baumannii is a small membrane porin with a monomeric eight-stranded barrel lacking an open channel and a large extracellular domain (Longo 2006). CarO functions as an uptake channel for small molecules such as Carbapenem (Zahn et al. 2015).
A proteomic study was performed on outer membrane vesicles (OMVs) of A. baumannii ATCC 19606. Immunization with OMVs induces protective immunity against challenge with A. baumannii. Among OMV proteins, six proteins were nominated as highly immunogenic, one of which was CarO (McConnell et al. 2011).Using several criteria to assess immunogenicity and B epitope densities CarO was selected as an immunogen in A. baumannii which contributes to sidrophore mediated iron uptake (Bazmara et al. 2019).
This protein participates in the selective uptake of l-ornithine, Carbapenem, and other basic amino acids in A. baumannii (Mussi et al. 2007). Thus, employment of bioinformatics tools to select appropriate region as a vaccine candidate seems logical. CarO exists in all pathogenic and clinical strains of A. baumannii (Siroy et al. 2005). Vaccine developed based on this protein could be effective against all pathogenic strains of A. baumannii. Alignment of sequences from various strains suggests that such vaccines could trigger antibodies with complete specificity for A. baumannii. CarO is an acidic outer membrane protein with pI about 4.66. Acidic nature of CarO as well as its localization are stimulating for B-cell responses (Rahbar et al. 2012a). Tertiary structure of CarO is like other outer membrane transport proteins with determined structure. Topology predictions illustrated that 21 amino acids at the N-terminus of the protein were predicted as signal peptide. This region could not be exposed and suitable to participate in B cell epitopes. InterProSurf result shows that most of the functional conserved amino acids are located in selected region (Ashkenazy et al. 2010). Therefore, it could be inferred that this region is the most important functional site of the protein. Thus, antibodies raised against these domains could block Acinetobactin uptake by CarO.
New genome analysis tools based on bioinformatics and immunoinformatics approaches help us select suitable antigens or epitopes directly from the genomes of pathogens in order to design a vaccine. These tools could be employed for epitope selection and vaccine design. Moreover, prediction of protein structures is one of their wide applications (Jahangiri et al. 2017; Rahbar et al. 2012b; Sefid et al. 2013, 2015). Some of these designed vaccine candidates validate with experimental analyses. In vivo analyses confirmed in silico predictions (Esmaeilkhani et al. 2019; Fattahian et al. 2011; Noori et al. 2014; SANGROODI et al. 2015).
Propensity scale methods assign a propensity value to each amino acid which measures the tendency of an amino acid to be part of a B-cell epitope (as compared to the background). To reduce fluctuations, the score for each target amino acid residue in a query sequence is computed as the average of the propensity values of the amino acids in a sliding window centered at the target residue. Hydrophilicity, accessibility, antigenicity, flexibility and secondary structure properties have fundamental role in B cell epitope prediction (Rubinstein et al. 2008). Relying on just one of these properties, reliable results could not be achieved. Amino acid pair (AAP) antigenicity scale is based on the finding that B-cell epitopes favor particular AAPs. Using SVM classifier, the AAP antigenicity scale approach has much better performance than the scales based on the single amino acid propensity (Sefid et al. 2015). Thus, we combined all the data obtained from various servers and software to predict the B-cell epitopes. Region suggested by single-scale amino acid properties assay (50–240) includes our selected region. This region could efficiently induce B cell responses. B-cell epitopes are regions on the surface of antigens recognized by B-cell receptors or specific antibodies. These epitopes can be categorized into two types: linear and conformational epitopes. Linear epitope is recognized by its linear sequence of amino acids, In contrast conformational epitope recognized by specific three-dimensional shape and its protein structure (Ansari and Raghava 2010). Linear and conformational epitopes in CarO protein predict by several software and various algorithms to achieve consensus epitopes. Consensus epitopes obtained from various algorithms are more reliable for selection. The highest scored linear B cell epitopes are located in region 19–38. The second best linear B cell epitope introduced by Svmtrip is located in region 121–140. Fifteen of 20 hits (*75%) predicted as B cell epitopes were within amino acids 19–160 (selected region) by ABCpred (Saha and Raghava 2007). Linear and conformational B cell epitopes predicted by Ellipro were located within selected region (Dehghani and Sefid 2016). Depth server result shows that higher possibility of residue forming a binding site are located in region 80–85 and 130–154. All servers predicting conformational B cell epitopes based on 3D structure such as Discotope demonstrated that residue (70, 108, 138, 140, 178, 176, 201, 202, and 205) have the most important role to form discontinues epitopes (Reimer 2009).
The humoral immune system response is based on the interactions between antibodies and antigens for the clearance of pathogens and foreign molecules (Parkin and Cohen 2001). An epitope, also known as antigenic determinant, is the part of an antigen that is recognized by the immune system. The epitope is the specific piece of the antigen to which an antibody binds (Peters et al. 2005). Epitopia server can predict immunogenic regions on protein structure. Fifty percent of immunogenic regions predicted by the server are located in region 107–225 of CarO. All these analyses reveal that majorities as well as the best B cell epitopes are located in 20–160 region. So, this region was selected as vaccine candidate. Average of each single scale amino acid propensity (hydrophilicity, accessibility, flexibility, beta turn and B cell epitope averages) was increased in the candidate vaccines than their parent protein, CarO. The present study was designed to in silico resolving the major obstacles in the control or in prevention of the Infection caused by A. baumannii. We exploited bioinformatics tools for better understanding and characterizing the CarO structure of A. baumannii and selection of appropriate region as effective B cell epitopes.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Ansari HR, Raghava GP (2010) Identification of conformational B-cell epitopes in an antigen from its primary sequence. Immunome Res 6:6
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533
Bagos PG, Liakopoulos TD, Spyropoulos IC, Hamodrakas SJ (2004) PRED-TMBB: a web server for predicting the topology of β-barrel outer membrane proteins. Nucleic Acids Res 32:W400–W404
Bazmara H, Rasooli I, Jahangiri A, Sefid F, Astaneh SDA, Payandeh Z (2019) Antigenic properties of iron regulated proteins in Acinetobacter baumannii: an in silico approach. Int J Pept Res Ther 25:205–213
Berezin C et al (2004) ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics 20:1322–1324
Blue-Hnidy DE, Allen SD (2006) Microbiology for the surgical pathologist. In: Essentials of anatomic pathology. Springer, New York, pp 155–229
Bryson K, Cozzetto D, Jones DT (2007) Computer-assisted protein domain boundary prediction using the Dom-Pred server. Curr Protein Pept Sci 8:181–188
Catel-Ferreira M et al (2011) Structure–function relationships of CarO, the carbapenem resistance-associated outer membrane protein of Acinetobacter baumannii. J Antimicrob Chemother 66:2053–2056
Dehghani E, Sefid F (2016) Evaluation of lamb linear and conformational epitopes on the vibrio Harveyi. Int J Adv Biotechnol Res 7(3):667–671
Dijkshoorn L, Nemec A, Seifert H (2007) An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951
El-Manzalawy Y, Dobbs D, Honavar V (2008) Predicting linear B-cell epitopes using string kernels. J Mol Recognit 21:243–255
Esmaeilkhani H, Rasooli I, Hashemi M, Nazarian S, Sefid F (2019) Immunogenicity of cork and loop domains of recombinant Baumannii acinetobactin utilization protein in murine model. Avicenna J Med Biotechnol 11:180
Fattahian Y, Rasooli I, Gargari SLM, Rahbar MR, Astaneh SDA, Amani J (2011) Protection against Acinetobacter baumannii infection via its functional deprivation of biofilm associated protein (Bap). Microb Pathog 51:402–406
García-Patiño MG, García-Contreras R, Licona-Limón P (2017) The immune response against Acinetobacter baumannii, an emerging pathogen in nosocomial infections. Front Immunol 8:441
Gordon NC, Wareham DW (2010) Multidrug-resistant Acinetobacter baumannii: mechanisms of virulence and resistance. Int J Antimicrob Agents 35:219–226
Higgins PG, Dammhayn C, Hackel M, Seifert H (2009) Global spread of carbapenem-resistant Acinetobacter baumannii. J Antimicrob Chemother 65:233–238
Hirokawa T, Boon-Chieng S, Mitaku S (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379
Jahangiri A, Rasooli I, Owlia P, Fooladi AAI, Salimian J (2017) In silico design of an immunogen against Acinetobacter baumannii based on a novel model for native structure of outer membrane protein A. Microb Pathog 105:201–210
Jespersen MC, Peters B, Nielsen M, Marcatili P (2017) BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res 45(W1):W24–W29
Kang AD, Smith KP, Eliopoulos GM, Berg AH, McCoy C, Kirby JE (2017) Invitro apramycin activity against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Diagn Microbiol Infect Dis 88:188–191
Kempf M et al (2012) Cell surface properties of two differently virulent strains of Acinetobacter baumannii isolated from a patient. Can J Microbiol 58:311–317
Knezevic P, Aleksic V, Simin N, Svircev E, Petrovic A, Mimica-Dukic N (2016) Antimicrobial activity of Eucalyptus camaldulensis essential oils and their interactions with conventional antimicrobial agents against multi-drug resistant Acinetobacter baumannii. J Ethnopharmacol 178:125–136
Kringelum JV, Lundegaard C, Lund O, Nielsen M (2012) Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol 8:e1002829
Livermore DM, Mushtaq S, Warner M, Woodford N (2016) In-vitro activity of eravacycline against carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii. Antimicrob Agents Chemother 60(6):3840–3844
Longo DL (2006) Idiotype vaccination in follicular lymphoma: knocking on the doorway to cure. Oxford University Press, Oxford
McConnell MJ, Domínguez-Herrera J, Smani Y, López-Rojas R, Docobo-Pérez F, Pachón JJI (2011) Vaccination with outer membrane complexes elicits rapid protective immunity to multidrug-resistant Acinetobacter baumannii. Infect Immunity 79:518–526
Mussi MA, Limansky AS, Viale AM (2005) Acquisition of resistance to carbapenems in multidrug-resistant clinical strains of Acinetobacter baumannii: natural insertional inactivation of a gene encoding a member of a novel family of β-barrel outer membrane proteins. Antimicrob Agents Chemother 49:1432–1440
Mussi MA, Relling VM, Limansky AS, Viale AM (2007) CarO, an Acinetobacter baumannii outer membrane protein involved in carbapenem resistance, is essential for l-ornithine uptake. FEBS Lett 581:5573–5578
Negi SS, Schein CH, Oezguen N, Power TD, Braun W (2007) InterProSurf: a web server for predicting interacting sites on protein surfaces. Bioinformatics 23:3397–3399
Noori E, Rasooli I, Owlia P, Gargari SLM, Ebrahimizadeh W (2014) A conserved region from biofilm associated protein as a biomarker for detection of Acinetobacter baumannii. Microb Pathog 77:84–88
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217
Pagdepanichkit S, Tribuddharat C, Chuanchuen RJ (2016) Distribution and expression of the Ade multidrug efflux systems in Acinetobacter baumannii clinical isolates. Can J Microbiol 62:794–801
Parkin J, Cohen B (2001) An overview of the immune system. The Lancet 357:1777–1789
Peleg AY, Seifert H, Paterson DL (2008) Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582
Peters B et al (2005) The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol 3:e91
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Pirovano W, Feenstra KA, Heringa J (2008) PRALINE™: a strategy for improved multiple alignment of transmembrane proteins. Bioinformatics 24:492–497
Poirel L, Nordmann P (2006) Carbapenem resistance in Acinetobacter baumannii: mechanisms and epidemiology. Clin Microbiol Infect 12:826–836
Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform 9:514
Rahbar MR et al (2012) A potential in silico antibody–antigen based diagnostic test for precise identification of Acinetobacter baumannii. J Theoret Biol 294:29–39
Reimer U (2009) Prediction of linear B-cell epitopes epitope mapping protocols. Methods Mol Biol 524:335–344
Rubinstein ND, Mayrose I, Halperin D, Yekutieli D, Gershoni JM, Pupko T (2008) Computational characterization of B-cell epitopes. Mol Immunol 45:3477–3489
Rubinstein ND, Mayrose I, Martz E, Pupko T (2009) Epitopia: a web-server for predicting B-cell epitopes. BMC Bioinform 10:287
Saha S, Raghava G (2004) BcePred: prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. In: Nicosia G, Cutello V, Bentley PJ, Timmis J (eds) Artificial immune systems. ICARIS 2004. Lecture notes in computer science, vol 3239. Springer, Berlin, Heidelberg, pp 197–204
Saha S, Raghava GP (2007) Prediction methods for B-cell epitopes. Methods Mol Biol 409:387–394
Sánchez-Encinales V et al (2017) Overproduction of outer membrane protein A by Acinetobacter baumannii as a risk factor for nosocomial pneumonia, bacteremia, and mortality rate increase. J Infect Dis 215:966–974
Sangroodi YH, Rasooli I, Nazarian S, Ebrahimizadeh W, Sefid F (2015) Immunogenicity of conserved cork and ß-barrel domains of baumannii acinetobactin utilization protein in an animal model. Turkish J Med Sci 45:1396–1402
Sefid F, Rasooli I, Jahangiri A (2013) In silico determination and validation of baumannii acinetobactin utilization a structure and ligand binding site. BioMed Res Int 2013:172784
Sefid F, Rasooli I, Jahangiri A, Bazmara H (2015) Functional exposed amino acids of BauA as potential immunogen against Acinetobacter baumannii. Acta Biotheor 63:129–149
Shaker MA, Shaaban MI (2017) Formulation of carbapenems loaded gold nanoparticles to combat multi-antibiotic bacterial resistance: in vitro antibacterial study. Int J Pharm 525:71–84
Siroy A et al (2005) Channel formation by CarO, the carbapenem resistance-associated outer membrane protein of Acinetobacter baumannii. Antimicrob Agents Chemother 49:4876–4883
Tan KP, Varadarajan R, Madhusudhan MS (2011) DEPTH: a web server to compute depth and predict small-molecule binding cavities in proteins. Nucleic Acids Res 39:W242–W248
Viklund H, Bernsel A, Skwark M, Elofsson A (2008) SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics 24:2928–2929
Vita R et al (2014) The immune epitope database (IEDB) 3.0. Nucleic Acids Res 43:D405–D412
Weinstein RA, Gaynes R, Edwards JR (2005) Overview of nosocomial infections caused by gram-negative bacilli. Clin Infect Dis 41:848–854
Wong D, Nielsen TB, Bonomo RA, Pantapalangkoor P, Luna B, Spellberg B (2017) Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin Microbiol Rev 30:409–447
Yao B, Zhang L, Liang S, Zhang C (2012) SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS ONE 7:e45152
Zahn M, D’Agostino T, Eren E, Baslé A, Ceccarelli M, van den Berg BJ (2015) Small-molecule transport by CarO, an abundant eight-stranded β-barrel outer membrane protein from Acinetobacter baumannii. J Mol Biol 427:2329–2339
Zdobnov EM, Apweiler R (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848
Zhang Z, Li Y, Lin B, Schroeder M, Huang BJB (2011) Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 27:2083–2088
Acknowledgements
We thank Yazd University for granting us a permission to use its web server.
Funding
This study was supported by an unrestricted free access to Yazd University web site for data collection. The authors have received no financial support for the elaboration of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Yazd University did not play any decision-making role in the study analysis or writing of the manuscript. All authors declare no Potential Conflicts of Interest.
Ethical Approval
As the present study involved no experimental with animals or human, hence there was no need for approval by the Ethics Committee of Yazd University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tohidinia, M., Moshtaghioun, S.M., Sefid, F. et al. Functional Exposed Amino Acids of CarO Analysis as a Potential Vaccine Candidate in Acinetobacter Baumannii. Int J Pept Res Ther 26, 1185–1197 (2020). https://doi.org/10.1007/s10989-019-09923-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-019-09923-2