
Abstract
The amino terminus of RNA polymerase A (PA-N) of influenza virus is an important target for the design of new antiviral agents. In this study, molecular docking was used to screen for compounds that specifically target the deep cleft at the endonuclease active site in N-terminus of the RNA polymerase. Four potential compounds (NCI100226, NCI122653, NCI625583, and NCI403587) with high binding affinity for the active site were identified. Structural analysis of the binding conformation of each of these compound-PA-N complexes revealed that hydrophobic interaction and manganese ion chelation comprised the main interaction between the compounds and enzyme. The binding configuration stability and the number of hydrogen and ionic bonds were investigated by molecular dynamic simulations. The results indicated that NCI403587 could be a promising PA-N inhibitor, and may represent a potential new agent for the treatment of influenza.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Influenza A viruses are important human and animal respiratory pathogens responsible for both the seasonal ‘flu’ outbreaks and periodic worldwide pandemics. Antiviral drugs are particularly beneficial for combating a fast-spreading pandemic. Several drugs have been used to treat infection caused by influenza virus. These drugs are mainly directed against the M2 ion-channel protein (adamantanes) and NA protein (zanamivir and oseltamivir) (Das et al. 2010). However, drug-resistant viruses have gradually emerged, and the development of new effective anti-influenza drugs targeting different proteins of the virus is urgently needed.
The RNA polymerase of influenza virus is a heterotrimeric complex consisting of PB1, PB2 and PA subunits, and is responsible for carrying out the transcription and replication of the viral genome during infection (Kashiwagi et al. 2009). The primary roles of PB1, PB2 and PA in viral RNA synthesis have been delineated by various biochemical and genetic studies (Blaas et al. 1982; Biswas and Nayak 1994; González and Ortín 1999; Obayashi et al. 2008; Dias et al. 2009; Hara et al. 2006). PB1, the core of the RNA polymerase complex contains RNA-dependent RNA polymerase activity and it can recognize and bind to the specific conserved sequences of viral genomic RNA segments (vRNAs) and viral cRNAs (Blaas et al. 1982; González and Ortín 1999). The RNA polymerase transcribes viral RNA using short capped primers derived from host cell pre-mRNAs by a unique ‘cap snatching’ mechanism (Plotch et al. 1981). In this mechanism, the PB2 subunit binds the cap of host pre-mRNA and the PB1 subunit cleaves it at nucleotide 10-13 (Li et al. 2001; Guilligay et al. 2008). The PA subunit has endonuclease and protease activities, and has been implicated in a diverse range of functions, such as cap-binding, viral RNA binding, interaction with PB1, and replication (Maier et al. 2008; Lee et al. 2002; Hara et al. 2006; Fodor et al. 2002; Honda et al. 2002). The PA protein can be cleaved, by limited tryptic digestion, into two domains: a smaller N-terminal 25-kDa domain and a larger carboxyl terminal 55-kDa domain (Guu et al. 2008). Mutational analysis of PA suggests that N-terminal domain (PA-N) contains residues important for several functions of the polymerase complex, including protein stability, promoter binding, cap binding and endonuclease activity, which is essential for the synthesis of viral mRNAs (Hara et al. 2006; Maier et al. 2008). The crystal structures of PA-N reveals the existence of a cation-dependent endonuclease active-site core, and the amino acid residues involved in catalysis are conserved among different influenza A viruses (Yuan et al. 2009). Structural analysis of PA-N has revealed a deep cleft at the endonuclease active site, which has been considered as a promising target for the design of novel anti-influenza drugs (Dias et al. 2009). The development of new inhibitors that target PA-N can provide a new way for treating influenza virus.
Computational approaches such as molecular docking and molecular dynamics (MD) simulation have been widely applied to structure-based drug discovery and design (Morin et al. 2011; Huang and Zou 2010; Fedichev et al. 2011; Cui 2011). Molecular docking is generally used to investigate inhibitor-enzyme interactions and elucidate their binding mechanisms. In recent years, however, molecular simulation has also been widely used to investigate dynamic characteristics of inhibitor-enzyme interactions, such as the binding affinity between inhibitor and enzyme and the stability of the complex or conformational changes of the enzyme resulting from such binding, as well as any special feature associated with enzyme-inhibitor recognition. Molecular docking and MD simulation can reveal different aspects of information about drug and protein interactions. Based on this, we used both molecular docking and MD simulation in this study to analyze the interactions between enzyme and inhibitor under static and dynamic conditions.
Our goal was to screen a library of compounds for new and potent PA-N inhibitors. Therefore, molecular docking approach was applied to screen for small molecules in the library. Four best-binding compounds with higher binding affinity for PA-N were identified. MD simulation was used to investigate the dynamic binding features and the stability of the complexes resulting from inhibitor-enzyme binding. These molecules may provide the basis for designing novel antiviral drugs for treating influenza in the future.
Materials and Methods
Crystal Structure of PA-N
The crystal structure of PA-N from influenza virus strain A/Victoria/3/1975 H3N2 was obtained from Protein Data Bank (PDB ID: 2W69) (Dias et al. 2009). The structure was solved at a resolution of 2.05 Å, and consists of residues 1–209 with two manganese ions bound in the core of the endonuclease active site, which is an important feature of PA-N. This structure was used as an initial structure in molecular docking and MD simulation.
Preprocessing the Compound Library
The Maybridge Screening collection (consist of over 56,000 organic compounds) and NCI Plated Compounds (approximately 140,000 compounds) were used as the compound library in molecular docking study. The chemical toolbox software Open Babel (O’Boyle et al. 2011) was used to generate the three-dimensional atomic coordinates of the compounds in Maybridge Screening collection. In order to screen for drug-like compounds, molecules containing toxic groups, reactive groups or that are not compatible with Lipinski’s Rule of 5 (Lipinski 2000) were excluded from the library before molecular docking. These drug-like properties were estimated by XlogP3 (Cheng et al. 2007). The compounds were then processed by prepare_ligand4.py, a python script in the AutoDock Tools module, which can merge nonpolar hydrogen, add Gasteiger atomic charges, and set atom type to the AutoDock supported type. This process yielded 164,745 compounds as the compound library for virtual screening. All the compounds in the library were docked into the PA-N endonuclease active site in the molecular docking procedure.
Molecular Docking
The molecular docking procedure was performed by the molecular docking program AutoDock 4.2, run on a high performance computing platform at Liaoning University (Morris et al. 1998; Morris et al. 2009; Park et al. 2006). The settings of docking region were based on the key residues of PA-N endonuclease active site. Amino acid residues His41, Glu80, Leu106, Pro107, Asp108, and Glu119 of the PA-N were treated as the active residues of the binding cavity.
AutoGrid was used to calculate the lattice energy between the receptor and all types of atoms for each compound in the library. All ligands in the compound library used the same calculated lattice energy, thus accelerating the speed of virtual screening. During the docking procedure, the ligands were flexible and the receptor was fixed. Lamarckian genetic algorithm was used to optimize compound conformation and 10 docking runs were performed for each compound in the database. The population size was set to 150, the number of generations was set to 27,000, and the maximum number of energy evaluation was set to 2,500,000. The docking conformations was evaluated using a semi-empirical free energy force field (Huey et al. 2007).
Following the docking analysis, some promising candidates were chosen from the compound library according to the following criteria: (1) the selected compounds should show only one big cluster in the cluster analysis section of the docking result, and the binding pose with lowest binding energy should included in that cluster; (2) the binding mode of the candidate compounds and the known PA-N inhibitors were compared by AutoDock Tools, and the compounds have a binding mode that is similar to the known PA-N inhibitors were selected as promising candidates; (3) the selected compounds should contain an anionic group that can easily chelate with the manganese ion in the active site of PA-N, and contain a relatively large hydrophobic group that can bind to the hydrophobic residues in the active site; (4) the formation of hydrogen bonds between PA-N and the candidate compounds were checked by AutoDock Tools, the selected compounds should have the ability to form hydrogen bonds with key residues of the PA-N active site. The overall virtual screening process used in this study is shown as a flow chart in Fig. 1.

Flow chart of the process of in silico strategies used for virtual screening
Molecular Dynamics Simulation
Molecular dynamics (MD) simulation was conducted to investigate the dynamic properties of the inhibitor-protein complexes. All MD simulations were performed with NAMD 2.9 (Phillips et al. 2005) using the AMBER all-atom force field (Wang et al. 2004). The topology and force field parameters of the candidates were generated by antechamber package of AmberTools 1.5 using AM1-BCC charges (Jakalian et al. 2002) and GAFF force field. The Amber force field for manganese was obtained from AMBER parameter database (Bradbrook et al. 1998). The structural models were individually solvated in a TIP3P water model with a minimum solute wall distance of 10 Å. Sodium cations were added to each system to neutralize the overall charge of the system. And then, 10,000 steps energy minimization were carried out for each solvated inhibitor-protein system. After minimization, the systems were slowly heated to 310 K within 620 ps, followed by a 400 ps equilibration phase. The equilibrated systems were then subjected to production stage for 10 ns with NPT canonical ensemble at a constant temperature of 310 K and a constant pressure of 1 atm through the Langevin piston method. The integration time step was set to 2 fs, and long-range electrostatics were treated with PME method for all MD stages.
Results and Discussion
Validation of Molecular Docking Methods
In order to estimate the feasibility and validity of the screening process which employed molecular docking, eight positive inhibitors (and their IC50 values) were selected from the literatures (Tomassini et al. 1994; Hastings et al. 1996), and each was docked into PA-N endonuclease active-site using the same docking parameters as for virtual screenig. The IC50 values for the inhibition of transcription activity and the calculated binding energy of the inhibitors are listed in Table 1. The logIC50 values of these inhibitors were well correlated (R2 = 0.83) with their corresponding binding energy (Fig. 2), suggesting that molecular docking is feasible for the screening of potential inhibitors for the PA subunit of influenza A virus.

Regression analysis of logIC50 of known inhibitors and their binding free energy
Molecular Docking
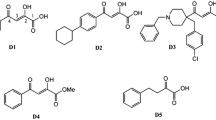
Molecular docking-based virtual screening was used to seek for compounds that specifically target the endonuclease active site of PA-N. Compounds with higher binding energy than that of the positive inhibitors were evaluated by the four criteria mentioned above. The top four compounds, NCI100226, NCI122653, NCI625583, and NCI403587, were selected as potential candidates for further analysis by docking and molecular simulation. For simplicity, NCI100226, NCI122653, NCI625583, and NCI403587 will henceforth be referred to as L26, L53, L83 and L87, respectively. The structures, names and binding energy of these four compounds are shown in Fig. 3. All four compounds had less than −20 kcal/mol binding energy, indicating that they have strong binding affinity for the active site of PA-N endonuclease. Interestingly, all candidates had similar structural features, in which each contains at least one big hydrophobic group, some polar substituents, and some anion groups, suggesting that in addition to being able to bind to the hydrophobic pocket and the prothetic manganese ions of PA-N, these compounds could also form hydrogen bond with the hydrophilic residues in the active site of the enzyme. With the exception of L87, which could form more hydrogen bonds with PA-N, all candidates shared a similar docking mode in the active pocket of PA-N, as shown in Fig. 3.

Compounds identified by molecular docking as potential PA-N inhibitors
Binding Mode of Candidate Compounds
The docking conformation and the hydrogen bonds formed between each of the four compounds and PA-N are shown in Fig. 4. The endonuclease active site is composed of a dinuclear manganese center and a hydrophobic cavity. The endonuclease activity of PAN is known to be highly dependent on the presence of metal ions in the active site, suggesting that compounds that can chelate the manganese ions may have the ability to inhibit the endonuclease activity of PA-N (Doan et al. 1999; Dias et al. 2009). The four compounds were bound to the active pocket in a similar mode, and each made contact with the manganese ions via its anion group (Fig. 4), indicating that these compounds may chelate the manganese ions. The large hydrophobic groups of these compounds were located in the deep cavity on the right side of the active pocket, and were in contact with Ala20, Tyr24 and Ile38 via van der Waals interaction. The hydrophobic interaction formed between any of these compounds and PA-N active site should result in a desolvation entropy gain, which is favorable for the stabilization of the complex. The anion group and other polar substituents could form two or more hydrogen bonds with the key residues (His41,Glu80,Leu106,Pro107,Asp108 and Glu119) in the active site of the enzyme. The hydrogen bond formed between the compounds and these PA-N key residues could result in a more stable complex. The binding energy of L53 was the lowest among the four compounds, but it appeared to have no hydrophobic contact with the active pocket. Instead, it formed more hydrogen bonds with the enzyme (Fig. 4b).

Docking modes of different compounds in PA-N. a L26. b L653. c L83. d L87. Small molecules are shown in stick format and carbon atoms are indicated in yellow, while oxygen, nitrogen and manganese atoms are indicated in red, blue, and darkorchid, respectively. Hydrogen bonds are shown in dotted red lines
Based on these results, we proposed that the above four compounds could chelate with the two manganese ions and form hydrogen bond and hydrophobic contacts with some key amino acid residues in the active site to stabilize the inhibitor-enzyme complex, consequently leading to the inhibition of endonuclease activity of PA-N.
Molecular Dynamics Simulation
The dynamic properties of the complexes between PA-N and each of the compounds were investigated by molecular dynamics simulation. A 10 ns MD simulation was performed for each of the four compounds (L26, L53, L83 & L87) and one positive inhibitor (2,4-dioxo-4-phenylbutanoic acid, L05). The root mean square deviations (RMSDs) of the complex as well as of the ligands obtained from MD are summarized in Fig. 5. The RMSDs of the complexes formed between PA-N and one of the compounds or the positive inhibitor (L05) stabilized at 5 ns during the 10 ns of MD simulation, suggesting that all the complexes had reached energy equilibrium. RMSDs of the complexes formed between PA-N and L26, L87, L05, L53 and L83 started to fluctuate around 2.57, 2.76, 3.36, 3.24 and 3.49 Å, respectively. According to the RMSD of the complex for each system, L26 and L87 appeared to have a significantly lower value than L53, L83 and L05, indicating that L26 and L87 could bind to PA-N to yield a more stable complex than the complex formed between PA-N and its positive inhibitor. The RMSDs of L05, L26 and L87 were also stabilized at 5 ns, but then fluctuated at around 2.58, 2.66 and 1.99 Å, respectively (Fig. 5b). These results indicated that L87 can form a more stable complex with PA-N than the other candidates or the positive inhibitor.

Complex RMSD (a) and ligand RMSD (b) profile of putative and positive inhibitors of PA-N
The formation of hydrogen bond between PA-N and L26, L87 or L05 was also investigated. The hydrogen bond profile of each complex is summarized in Fig. 6. Four hydrogen bonds were observed between L05 (positive inhibitor) and PA-N (Fig. 6c). The distance between the oxygen atom of L05 and Oε1 atom of Glu80 fluctuated from 2.60 to 4.37 Å, indicating that it is an unstable hydrogen bond. In contrast, four hydrogen bonds were detected between L26 and Glu80, Ile120, Asp108 and His41 of PA-N (Fig. 6a). The length of the four hydrogen bonds remained at 2.36–4.04 Å throughout the simulation, indicating a strong interaction between L26 and PA-N active site. L87 formed five hydrogen bonds with PA-N, and two of these were formed with Glu80 whereas the other three were with His41, Asp108 and Glu119 (Fig. 6b). All the hydrogen bonds were stable during the simulation. The stronger hydrogen-bond interaction between L87 and PA-N suggested a tighter binding between the compound and enzyme. The observed profiles of hydrogen-bond distance suggested that L87 could be a promising inhibitor for inhibiting PA-N endonuclease activity.

Time-dependent-hydrogen bond distance (Å) for a L26, b L87 and c L05 in the 10 ns MD simulation
Conclusion
The spread of influenza virus has posed a serious threat to the health of the general public worldwide, so the development of effective anti-influenza drugs is considered to be a matter of urgency. Viral polymerases play an important role in viral replication cycle, and PA is a part of the polymerase. For this reason, PA-N has become an ideal target for identifying new inhibitors. Here, we employed molecular docking to screen 200,000 small molecules for compounds which can directly bind to the subunit of PA-N. We identified four potential PA-N endonuclease active site inhibitors from Maybridge Screening collection and NCI Plated Compounds library. All inhibitors appeared to adopt a similar binding mode and interacted with key residues in the binding pocket of the enzyme active site via hydrogen bonds. The putative PA-N inhibitors identified from the screening process appeared to possess lower binding energy than all the known inhibitors. The mode of binding between compound and enzyme revealed that these compounds may inhibit the endonuclease activity of PA-N by chelating with the manganese ions in the active center. Further analysis revealed that the hydrophobic interaction and the formation of hydrogen bond between each compound and key residues of PA-N could stabilize the inhibitor-enzyme complex. The 10 ns MD simulation identified L87 as the most promising candidate based on the behavior of their RMSD and hydrogen bond profile. All four putative PA-N inhibitors could be further developed into drugs for combating infection caused by influenza A virus.
References
Biswas SK, Nayak DP (1994) Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J Virol 68:1819–1826
Blaas D, Patzelt E, Kuechler E (1982) Cap-recognizing protein of influenza virus. Virology 116:339–348
Bradbrook GM, Gleichmann T, Harrop SJ, Habash J, Raftery J, Kalb J, Yariv J, Hillier IH, Helliwell JR (1998) X-Ray and molecular dynamics studies of concanavalin-A glucoside and mannoside complexes relating structure to thermodynamics of binding. J Chem Soc Faraday Trans 94:1603–1611
Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L (2007) Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model 47:2140–2148
Cui Y (2011) Using molecular simulations to probe pharmaceutical materials. J Pharm Sci 100:2000–2019
Das K, Aramini JM, Ma L-C, Krug RM, Arnold E (2010) Structures of influenza A proteins and insights into antiviral drug targets. Nat Struct Mol Biol 17:530–538
Dias A, Bouvier D, Crépin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW (2009) The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918
Doan L, Handa B, Roberts NA, Klumpp K (1999) Metal ion catalysis of RNA cleavage by the influenza virus endonuclease. Biochemistry 38:5612–5619
Fedichev P, Timakhov R, Pyrkov T, Getmantsev E, Vinnik A (2011) Structure-based drug design of a new chemical class of small molecules active against influenza A nucleoprotein in vitro and in vivo. PLoS curr 3:RRN1253
Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG (2002) A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol 76:8989–9001
González S, Ortín J (1999) Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J 18:3767–3775
Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, Lewis J, Ruigrok RW, Ortin J, Hart DJ (2008) The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol 15:500–506
Guu TS, Dong L, Wittung-Stafshede P, Tao YJ (2008) Mapping the domain structure of the influenza A virus polymerase acidic protein (PA) and its interaction with the basic protein 1 (PB1) subunit. Virology 379:135–142
Hara K, Schmidt FI, Crow M, Brownlee GG (2006) Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J Virol 80:7789–7798
Hastings JC, Selnick H, Wolanski B, Tomassini JE (1996) Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob Agents Chemother 40:1304–1307
Honda A, Mizumoto K, Ishihama A (2002) Minimum molecular architectures for transcription and replication of the influenza virus. Proc Natl Acad Sci U S A 99:13166–13171
Huang S-Y, Zou X (2010) Advances and challenges in protein-ligand docking. Int J Mol Sci 11:3016–3034
Huey R, Morris GM, Olson AJ, Goodsell DS (2007) A semiempirical free energy force field with charge-based desolvation. J Comput Chem 28:1145–1152
Jakalian A, Jack DB, Bayly CI (2002) Fast, efficient generation of high-quality atomic charges. AM1-BCC model IIParameterization and validation. J Comput Chem 23:1623–1641
Kashiwagi T, Leung BW, Deng T, Chen H, Brownlee GG (2009) The N-terminal region of the PA subunit of the RNA polymerase of influenza A/HongKong/156/97 (H5N1) influences promoter binding. PLoS ONE 4:e5473
Lee MM, Bishop K, Medcalf L, Elton D, Digard P, Tiley L (2002) Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res 30:429–438
Li M-L, Rao P, Krug RM (2001) The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J 20:2078–2086
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249
Maier HJ, Kashiwagi T, Hara K, Brownlee GG (2008) Differential role of the influenza A virus polymerase PA subunit for vRNA and cRNA promoter binding. Virology 370:194–204
Morin A, Meiler J, Mizoue LS (2011) Computational design of protein–ligand interfaces: potential in therapeutic development. Trends Biotechnol 29:159–166
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998) Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19:1639–1662
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutodockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open babel: an open chemical toolbox. J Cheminform 3:1–14
Obayashi E, Yoshida H, Kawai F, Shibayama N, Kawaguchi A, Nagata K, Tame JR, Park S-Y (2008) The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 454:1127–1131
Park H, Lee J, Lee S (2006) Critical assessment of the automated autodock as a new docking tool for virtual screening. Proteins 65:549–554
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Plotch SJ, Bouloy M, Ulmanen I, Krug RM (1981) A unique cap (m7G pppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847–858
Tomassini J, Selnick H, Davies ME, Armstrong ME, Baldwin J, Bourgeois M, Hastings J, Hazuda D, Lewis J, McClements W (1994) Inhibition of cap (m7G pppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob Agents Chemother 38:2827–2837
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174
Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T (2009) Crystal structure of an avian influenza polymerase PAN reveals an endonuclease active site. Nature 458:909–913
Acknowledgments
This work was supported by the Education Department of Liaoning Province (General Research Project Foundation No. L2014001) and Research Center for Computer Simulating and Information Processing of Bio-macromolecules of Liaoning.
Conflict of interest
Haixin Ai, Fangliang Zheng, Fangbo Deng, Chunyu Zhu, Ying Gu, Li Zhang, Xuejiao Li, Alan K Chang, Jian Zhao, Junfeng Zhu and Hongsheng Liu declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Haixin Ai and Fangliang Zheng contributed equally to this work.
Rights and permissions
About this article

Cite this article
Ai, H., Zheng, F., Deng, F. et al. Structure-Based Virtual Screening for Potential Inhibitors of Influenza A Virus RNA Polymerase PA Subunit. Int J Pept Res Ther 21, 149–156 (2015). https://doi.org/10.1007/s10989-014-9442-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-014-9442-8