
Abstract
Ischaemic preconditioning (IPC) protects against myocardial ischaemia–reperfusion injury. The metabolic and ionic effects of IPC remain to be clarified in detail. We aimed to investigate the effect of IPC (2 times 5 min ischaemia) on the subcellular distribution of glycogen and Ca2+-uptake and leakiness by the sarcoplasmic reticulum (SR) in response to ischaemia–reperfusion in cardiomyocytes of isolated perfused rat hearts (Wistar rats, 335 ± 25 g). As estimated by quantitative transmission electron microscopy, the pre-ischaemic contribution [%, mean (95% CI)] of three sub-fractions of glycogen relative to total glycogen was 50 (39:61) as subsarcolemmal, 41 (31:50) as intermyofibrillar, and 9 (5:13) as intramyofibrillar glycogen. After 25 min of ischaemia, the relative contribution (%) of subsarcolemmal glycogen decreased to 39 (32:47) in control hearts (Con) and to 38 (31:45) in IPC. After 15 min reperfusion the contribution of subsarcolemmal glycogen was restored to pre-ischaemic levels in IPC hearts, but not in Con hearts. IPC increased the left ventricular developed pressure following ischaemia–reperfusion compared with Con. In saponin-skinned cardiomyocyte bundles, ischaemia reduced the SR Ca2+-uptake rate, with no effect of IPC. However, IPC reduced a SR Ca2+-leakage at pre-ischaemia, after ischaemia and during reperfusion. In conclusion, subsarcolemmal glycogen was preferentially utilised during sustained myocardial ischaemia. IPC improved left ventricular function reflecting reduced ischaemia–reperfusion injury, mediated a re-distribution of glycogen towards a preferential storage within the subsarcolemmal space during reperfusion, and lowered SR Ca2+-leakage. Under the present conditions, we found no temporal associations between alterations in glycogen localisation and SR Ca2+ kinetics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
An acute myocardial ischaemic insult switches energy provision from oxidation of bloodborne substrates as fatty acids and glucose to an anaerobic turnover of myocardial glycogen stores. With prolonged ischaemia, the glycogen stores can be exhausted leading to energy deficiency, disrupted ionic homeostasis and cardiomyocyte necrosis (Stanley et al. 2005).
Ischaemic preconditioning (IPC) is known to exert a protective effect on the heart during an episode of ischaemia–reperfusion (Hausenloy and Yellon 2016). While the underlying mechanisms are not fully understood, a hall-mark of this cardioprotection is a sequence of metabolic alterations including, but not limited to, reduced glycogen stores before prolonged ischaemia, lowered net glycogen depletion during prolonged ischaemia, and increased resynthesis rate of glycogen during reperfusion (Bradamante et al. 2000; Støttrup et al. 2010). Despite these profound alterations in glycogen metabolism by IPC, the importance of the altered glycogen metabolism in the mechanism of cardioprotection by IPC remain unknown.
A potential link between glycogen metabolism and Ca2+-handling is the Ca2+-reuptake by the sarcoplasmic reticulum (SR) Ca2+-ATPase that preferentially utilises energy from glycolysis (Boehm et al. 2000; Jeremy et al. 1992) and the regulation of the Ca2+ release process by ryanodine receptors (RyRs) via glycolytic intermediates (Zima et al. 2006). Therefore, the accumulation of cytosolic Ca2+ concentration during ischaemia–reperfusion may be temporally linked to depletion of myocardial glycogen.
Glycogen particles appear most abundantly in the perinuclear region and between the myofibrils, but also within the myofibrils. Qualitative inspection by transmission electron micrographs (TEM) has demonstrated that glycogen depletion during ischaemia predominantly occurs from the perinuclear region during ischaemia (Caulfield and Klionsky 1959), suggesting that the global cellular concentration of glycogen in the heart does not reflect the glycogen content of local subcellular environments. Since these early investigations, the technique for glycogen visualisation by TEM has improved by a refinement of the post-fixation process including reduced osmium tetroxide containing potassium ferrocyanide (De Bruijn 1973; Marchand et al. 2002). Using this improved technique, others and we have shown that the utilisation of glycogen during muscular work is different between subcellular locations of skeletal muscle fibres as characterised by a severe depletion of intra-myofibrillar glycogen and only a modest depletion of inter-myofibrillar and subsarcolemmal glycogen (Marchand et al. 2007; Nielsen et al. 2011). Therefore, a comprehensive understanding of the interplay between IPC, glycogen metabolism and myocardial function requires detailed analyses at the subcellular level.
In the present study, we aimed to investigate the effect of IPC on the subcellular distribution of glycogen and the Ca2+-handling by the SR in response to ischaemia–reperfusion in cardiomyocytes of isolated perfused rat hearts. We hypothesised that intra-myofibrillar glycogen would be preferentially depleted during IPC and ischaemia and that inter-myofibrillar glycogen would be less depleted and related to the SR Ca2+ reuptake rate.
Materials and methods
Ethical approval
Experiments conformed to national (act. no. 1306 of 23/11/2007) and institutional guidelines for animal research. A total of 57 male Wistar Hannover GALAS rats (M&B Taconic, Ry, DK) weighing on average 335 g (SD 25 g) were used. Animals were kept in a thermostatically maintained environment at 23 °C with a 12 h light/12 h dark cycle. They were fed standard chow and allowed free access to food and water until the time of the experiments.
Study design
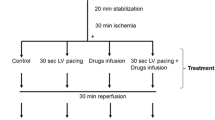
The design of the study is shown in Fig. 1. Isolated perfused rat hearts were randomly assigned to: IPC, induced by 2 cycles of 5 min global ischaemia and 5 min reperfusion, or control (Con), followed by either 0 min ischaemia (pre-ischaemia), 25 min ischaemia (Ischaemia), or 25 min ischaemia + 15 min reperfusion (ischaemia–reperfusion). All hearts underwent 20 min stabilization. Following the perfusion protocol, the hearts were quickly detached from the perfusion apparatus; the apex was removed, and the rest of the left ventricle was cut free from the heart. From the middle part of the lateral wall of the left ventricle, a small piece was obtained for TEM and another piece for the skinned fibre preparation. The remaining part of the left ventricle was immediately snap frozen in liquid N2.

Study design and haemodynamic parameters during the time-course of the experimental protocol. a Isolated perfused hearts were divided into six groups (n = 9–10 in each group): IPC, conducted as 2 × 5 min ischaemia–reperfusion, or control (Con), followed by either 0 min ischaemia (Pre-ischaemia), 25 min ischaemia (Ischaemia), or 25 min ischaemia + 15 min reperfusion (ischaemia–reperfusion). Specimens of the left ventricle were collected at the end of each protocol. KH a modified Krebs–Henseleit buffer. LVDP (b) and diastolic pressure (c) during IPC (n = 27–30), 25 min ischaemia (grey area, n = 7–20) and reperfusion (n = 8–10). Dots represent means and bars represent SD. *Different from Con (P < 0.05) by ranksum with adjusted P value for multiple comparisons
Isolated perfused heart preparation
Preparation and perfusion of the isolated rat hearts using the Langendorff model have been described previously in detail (Povlsen et al. 2014). In brief, rats were anesthetized by an intraperitoneal injection of pentobarbital (60 mg/kg) and intubated and connected to a rodent ventilator (Ugo Basile 7025 rodent ventilator, Comerio, Italy). Following a laparotomy and thoracotomy, the heart was dissected free from the surrounding structures. A bolus of heparin (1000 IU/kg, Leo Pharma, Ballerup, Denmark) was administered intravenously. Subsequently, the aorta was cannulated in vivo and retrograde perfusion of the heart was commenced immediately. Hearts were quickly excised and transferred to the perfusion apparatus under continuous perfusion. Hearts were immersed in a modified Krebs Henseleit (KH) buffer at 37.0 °C during the entire perfusion protocol. The KH buffer consisted of (mmol/L): NaCl, 118.5; KCl, 4.7; NaHCO3, 25.0; glucosemonohydrate, 11.1; MgSO4·7H2O, 1.2; CaCl2, 2.0; and KH2PO4, 1.2 (Sigma, St Louis,MO, USA). The perfusion buffer was filtered (Minisart, pore size 5.00 μm, Sartorius, Goettingen, Germany) and kept at 37.0 °C. During stabilization and perfusion, the KH buffer was continuously oxygenated with 95% O2 and 5% CO2. During global no flow ischaemia, the perfusion inlet was closed, and oxygen was depleted from the immersion buffer by continuously gassing with 95% N2 and 5% CO2 to maintain a neutral pH and avoid passive oxygen diffusion to superficial layers in the heart.
Haemodynamics
The left ventricular developed pressure (LVDP) was measured throughout the perfusion protocol as described previously (Støttrup et al. 2010). A fluid-filled latex balloon (size 7) connected to a pressure transducer (Hugo Sachs Elektronik, March-Hugstetten, Germany) was inserted into the left ventricle through the left atrial appendage and mitral valve. This allowed measurement of LVDP throughout the perfusion protocol. Balloon size was adjusted to obtain an end diastolic pressure of 5–8 mm Hg during stabilization. Coronary flow was measured with an in-line flow probe (Type 2.5SB, Transonic System Inc., Ithaca, NY, USA). All data were digitally converted (DT9804; Data Translation, Marlboro, MA, USA) and stored on a laptop using Notocord Hem software (Notocord Systems, Croissy sur Seine, France).
Metabolites
Frozen heart muscle specimens were freeze-dried, dissected free of non-muscle tissue, powdered and extracted using HClO4. Heart muscle glycogen was determined by the method of Lowry and Passonneau (1972) and neutralized extracts were analysed for lactate, adenosine triphosphate (ATP) and creatine phosphate (CrP) as previously described (Harris et al. 1974).
Transmission electron microscopy
Heart muscle specimens were prepared for glycogen visualisation using TEM as previously described (Nielsen et al. 2011). One specimen (Con Pre-ischaemia) was excluded because of too low quality of the preparation. In a subset of hearts following ischaemia–reperfusion (n = 3), we found that at least 20 images (10,000× magnification) should be obtained to achieve a satisfactorily high precision of the glycogen volume estimates. Therefore, at least 20 images were obtained from both the intermyofibrillar space [median and interquartile range (IQR): 33 (28–37)] and the subsarcolemmal space [40 (36–48)].
The cellular volumetric fractions of subsarcolemmal, intermyofibrillar and intramyofibrillar glycogen were estimated by point counting and corrected for the effect of section thickness and glycogen particle diameter as proposed by Weibel (1980) (their Eq. (4.20)): VV = AA − t((1/π)BA − NA((tH)/(t + H))), where AA is glycogen area fraction, t section thickness (60 nm), BA glycogen boundary length density, NA number of particles per area, and H is the average glycogen profile diameter. It was assumed that the particles were spherical. The glycogen area fraction was estimated by point counting (using a grid with a size of 60 nm) and the average glycogen profile diameter was directly measured using iTEM™ (FEI Company, Eindhoven, The Netherlands). We could not detect glycogen particles below 5 nm. Given the exponential relationship between particle diameter and volume, this is unlikely to have significant effect on the estimates of the cellular volumetric fractions of glycogen. The glycogen boundary length density (BA) was calculated based on the direct measurement of glycogen particle diameter: BA = ((π SV)/4) + (t NV π h), where SV is the glycogen particle surface area density, and Nv is the number of particles per volume.
Subsarcolemmal glycogen volume was related to the surface area of the cardiomyocyte as estimated by measuring the length of the myocyte (multiplied by the thickness of the section). Intermyofibrillar and intramyofibrillar glycogen volume were related to the myofibrillar (inter- and intramyofibrillar) and intramyofibrillar volume, respectively. To assess the relative distribution of the different glycogen pools, subsarcolemmal glycogen is also expressed relative to the volume beneath the surface area of a cylindrically shaped myocyte (Vb) = R × 0.5 × A, where R is myocyte radius and A is the myocyte surface area. The radius was assumed to be 20 µm (Angelakos et al. 1964).
Three blinded investigators conducted all analyses. The images from the different time points and groups were distributed equally between investigators. Repeatability tests showed coefficients of variation of 7–14% evaluated as proposed by Bland and Altman (Bland and Altman 1986).
Skinned fibre preparation
A small portion of the left ventricle was placed in paraffin oil and cooled. Under a dissecting microscope, a small bundle of cardiomyocytes was isolated within 10–15 min. The bundle was tied at both ends, mounted between a force transducer (Kronex AE801, Kronex Technologies Corporation, Oakland, CA, USA) and a stationary pin. The bundle was then immersed in a standard physiological K-HDTA solution (Table 1) to ensure the fibres were fully relaxed, and then the length of the bundle was increased until force started to increase (i.e. resting length). The width of the bundle was estimated directly through the microscope with an integrated eyepiece graticule with a sensitivity of 37.5 µm. The median width of the investigated bundles was 225 µm (range: 112–337 µm). These fibre bundles could produce a force of up to 8 mN/mm2. The force responses were sampled at 1000 Hz and stored for later analysis using a custom-made software (LabView 8.0, National Instruments, Austin, TX, USA). All force transducers were calibrated prior to use.
Solutions for skinned fibre experiments
Experimental solutions were prepared as described previously (Nielsen et al. 2007; Spencer et al. 2006). In order to empty the SR of Ca2+, a “release solution” was prepared by adding 30 mM caffeine and 0.25 mM free EGTA to the K-HDTA solution opening the RyRs (Stephenson and Wendt 1986) and the higher EGTA concentration used enables chelating of the released Ca2+ while being able to obtain a force recording (Stephenson and Wendt 1986). A reloading of SR with Ca2+ was performed by immersing the bundle into a “load solution”, where the pCa was set at 6.3 and total EGTA at 1 mM by adding 0.25 mM free EGTA and 0.75 mM Ca-EGTA to the K-HDTA solution. Spontaneous force oscillations were investigated by exposing the bundles to a weakly buffered K-HDTA solution (“spontaneous solution) with a total EGTA of 0.25 mM and pCa set at either 7.0 (0.167 mM free EGTA and 0.083 mM Ca-EGTA) or 6.3 (0.063 mM free EGTA and 0.187 mM Ca-EGTA). The weakly buffering of Ca2+ was chosen to achieve recordings of spontaneous oscillations in force (Stephenson and Wendt 1986). The submaximal and maximal Ca2+ activated force was determined in solutions similar to the K-HDTA but with 50 mM EGTA and pCa set at either 5.7 (4.5 mM free EGTA and 45.5 mM Ca-EGTA) or 4.6 (50 mM Ca-EGTA), respectively. Chemicals were acquired from Sigma-Aldrich (St Louis, MO, USA), unless otherwise stated.
Sarcoplasmic reticulum Ca2+ handling of skinned fibre bundles
Chemical skinning the bundles was performed by 25 min immersion in a K-HDTA solution (Table 1) containing saponin (50 µg/ml) to preferentially permeabilize the surface membrane, leaving the SR membrane functionally intact. After skinning, the bundles were washed in a new K-HDTA solution without saponin for 5 min. The endogenous Ca2+ content of SR was released by moving the bundle to a release solution containing 30 mM caffeine (Table 1) followed by washout of caffeine for 1 min in a K-HDTA solution. To stabilize subsequent force responses, the bundle was initially loaded with Ca2+ for 12 min in the load solution before a full release (high force output).
Rate of SR Ca2+ loading was assessed by exposing the bundles to repeated cycles of load-release solutions (Lamboley et al. 2016) (Fig. 5a):
-
(1)
Loading the SR with calcium for 18, 4 and 2 min in the load solution (pCa 6.3).
-
(2)
Pre-equilibrating the Ca2+ buffering capacity for 30 s in K-HDTA solution with 0.25 mM EGTA.
-
(3)
Full release of SR Ca2+ content for > 2 min in the release solution.
-
(4)
Washout of caffeine in K-HDTA solution with 0.25 mM EGTA.
The rate of SR Ca2+ loading was calculated as the ratio between the force-time integral at full release after the 4 and 2 min of loading (Fig. 5a) and expressed as a percentage of maximal loading (force-time integral after 18 min of loading). This ratio between 4 and 2 min loading takes into account any differences in diffusion properties of Ca2+ within fibre bundles of different thickness, since the 2 min loading time is adequate for Ca2+ to accomplish equilibrium. The 18 min of loading corresponds to near maximal loading capacity as verified in a pilot study showing a force-time integral of 97% (2% SD) after 12 min of loading compared with 16 min of loading (n = 4).
To investigate SR Ca2+ leakiness, spontaneous force oscillations were assessed by exposing the bundles to weakly Ca2+ buffered K-HDTA solutions (“spontaneous solutions”, Table 1) after loading the SR for 4 min in the loading solution and then exposing the bundles to a series of solutions according to the following scheme:
-
(1)
Loading the SR with calcium for 4 min in the loading solution
-
(2)
Resting in weakly buffered low Ca2+ (pCa 7.0) for 2 min in the spontaneous solution (Fig. 6Aa)
-
(3)
Resting in weakly buffered high Ca2+ (pCa 6.3) for 2 min in the spontaneous solution (Fig. 6Aa)
-
(4)
Pre-equilibrating the Ca2+ buffering capacity for 30 s in K-HDTA with 0.25 mM EGTA
-
(5)
Full release of SR Ca2+ content for > 2 min in the release solution
-
(6)
Washout of caffeine in K-HDTA solution with 0.25 mM EGTA
Spontaneous force oscillation analyses were conducted during the last 90 s of the exposure time. A spontaneous force development was defined as a sudden increase in force by at least 0.7 nN without a plateau in force before a drop in force also of at least 0.7 nN. The frequency was measured as the number of force developments per min and the mean half-force duration as the time (ms) from the half maximal force at the ascending phase to the half maximal force at the descending phase of each force development. Mean amplitude was measured as the force development from the initiation to the peak of each spontaneous force. The amplitude was normalized to the maximal Ca2+ activated force production by the bundle. The force-time product was evaluated as the ratio of the force-time integral between cytosolic high and low [Ca2+] to take into account the noise of the force signal. Pre-ischaemia force was defined as a moving median of the 5 lowest values within 10 s intervals. Submaximal and maximal Ca2+ activated force was measured as the force production in pCa 5.7 and 4.6, respectively.
From the 57 investigated hearts, a skinned fibre bundle was successfully obtained from 52 hearts. In those hearts, maximal Ca2+ activated force was measured in 51 hearts and myofibrillar Ca2+ sensitivity was measured in 45 hearts (the force transducer broke during the experiment of one heart and in six hearts no stable submaximal force could be obtained). SR Ca2+ uptake properties were measured in 41 hearts. In this measurement, four bundles were excluded due to unstable force response [Con Ischaemia (n = 1), Con Ischaemia-Reperfusion (n = 1), IPC Pre-ischaemia (n = 2)] and 7 bundles due to negative uptake rate [Con Pre-ischaemia (n = 2), Con Ischaemia (n = 1), Con Ischaemia-Reperfusion (n = 1), IPC Pre-ischaemia (n = 1), IPC Ischaemia (n = 2)]. Analyses of spontaneous force oscillations were measured in 44 hearts. One bundle (Con Pre-ischaemia) was excluded due to unstable force response and 7 bundles were excluded due to absence of any detectable force oscillations [Con Pre-ischaemia (n = 1), Con Ischaemia (n = 1), Con Ischaemia-Reperfusion (n = 2), IPC Pre-ischaemia (n = 2), IPC Ischaemia-Reperfusion (n = 1)].
Statistics
Statistical analyses were performed using Stata, version 15 (StataCorp LP, College Station, TX, USA). All interactions or main effects were tested using a linear mixed-effects model, with time and group as fixed effects. Assumptions on heteroscedasticity and normal distribution were evaluated by inspecting the distribution of residuals and a standardized normal probability plot, respectively. Variables with skewed distributions were log-transformed prior to analysis or, if the assumptions are not met, a non-parametric ranksum test (Mann–Whitney two-sample statistic) was applied. Associations between variables were evaluated using Pearson’s correlation coefficient. Concordance was evaluated using Lin’s concordance test (Lin 1989) Values are presented as geometric means and 95% confidence intervals (CI), unless stated otherwise. The significance level was set at α = 0.05.
Results
Haemodynamics
We found no difference in LVDP following the initial 20 min stabilization (P = 0.70) or during the prolonged 25 min ischaemia between the IPC and the Con group (P = 0.44) (Fig. 1b). During the early phase of the prolonged ischaemia, the diastolic pressures increased earlier (after 5 min) in the IPC hearts than in the Con hearts (P = 0.01; Fig. 1c). Following 15 min reperfusion, the LVDP was threefold higher in the IPC hearts than in the Con hearts (P = 0.008; Fig. 1b), implying a cardioprotective effect of IPC on myocardial contractile function. The difference was caused by a lower diastolic pressure, i.e. less myocardial ischaemic contracture, (Fig. 1c) during reperfusion in the IPC hearts (P < 0.02).
Metabolites
The cellular concentrations of glycogen, lactate, CrP and ATP are shown in Table 2.
The glycogen concentration was 30% lower in IPC hearts than in Con hearts at pre-ischaemia (P < 0.001), decreased to similar very low absolute levels after the prolonged ischaemia in IPC and Con hearts (ischaemia vs pre-ischaemia: P < 0.001), and increased by fourfold across groups during reperfusion (vs ischaemia, P < 0.001). The glycogen concentrations following ischaemia–reperfusion were reduced to 57% of the pre-ischaemic level in Con hearts (P < 0.001) but re-established to the pre-ischaemic level in IPC hearts (P = 0.20).
IPC mediated an attenuation of the lactate accumulation after ischaemia (P < 0.001), and lower levels after ischaemia–reperfusion (P = 0.034). The CrP concentration was higher in IPC hearts than in Con hearts at pre-ischaemia (P < 0.001) and decreased to similar absolute levels after ischaemia (P < 0.001). After ischaemia–reperfusion the concentration of CrP was higher in IPC hearts than in Con hearts (P < 0.001). The ATP concentration was lower in IPC hearts than in Con hearts at pre-ischaemia (P < 0.001) and decreased to similar very low absolute levels after ischaemia in both groups (P < 0.001). After ischaemia–reperfusion the ATP concentration was higher in IPC hearts than in Con hearts (P = 0.01).
Subcellular glycogen distribution
Representative TEM images are shown in Fig. 2. In Con hearts at pre-ischaemia, the TEM based analyses of the subcellular distribution of glycogen revealed a heterogeneous distribution, with approximately 50% located in the subsarcolemmal space, 40% located in the intermyofibrillar space and only 10% in the intramyofibrillar space (Fig. 2d). The diameter of individual glycogen particles was normally distributed with an average size of 20-22 nm and with no difference between localisations (P = 0.93, Fig. 2e).

Glycogen visualisation by transmission electron microscopy. a, b Images were obtained both in the subsarcolemmal space (a) and in the myofibrillar space (b). Glycogen particles are seen as black dots both in the subsarcolemmal space (short white arrow in a), between the myofibrils (intermyofibrillar, short white arrow in b), and within the myofibrils (intramyofibrillar, long white arrow in b). Arrow, sarcolemma. Mi, mitochondria. Z, Z-disc. M, M-band. Scale bar = 500 nm. c The TEM estimated glycogen content is plotted against the biochemically determined glycogen content with added lines of regression (dotted) and agreement (solid). R2, indicate coefficient of determination for the regression line. R 2c , indicate squared concordance correlation coefficient. d The average relative subcellular distribution of glycogen of Con hearts at pre-ischaemia shows two major depositions. e The kernal density plots of the glycogen particle diameter of three subcellular locations. Color coding as in (d)
While the volumetric content of subsarcolemmal glycogen was 55% lower in IPC hearts than in Con hearts at pre-ischaemia (P = 0.01), we found no difference between the two groups after ischaemia (P = 0.65) or after ischaemia–reperfusion (P = 0.54; Group × time interaction: P = 0.046. Figure 3a). Because the pre-ischaemia volumetric content of subsarcolemmal glycogen in the IPC hearts was reduced, ischaemia mediated a smaller decrease in subsarcolemmal glycogen in IPC hearts than in Con hearts (52% vs 81%, respectively. P = 0.04, Figure 3a). After ischaemia–reperfusion the increase in subsarcolemmal glycogen was not different between groups (P = 0.91, Fig. 3a). Across groups the volumetric content increased by 69% (P = 0.03).

Quantitative estimation of three subcellular localisations of glycogen. Three subfractions of glycogen, subsarcolemmal (a, b), intermyfibrillar (c, d), and intramyofibrillar (e, f), were estimated in control (black circles) and IPC (white circles) hearts before ischaemia (Pre-ischaemia), after 25 min ischaemia (Ischaemia) and after 15 min reperfusion (ischaemia–Reperfusion). In the absolute values of glycogen (a, c, e) horizontal and vertical lines represent geometric means and 95% CI. In the relative values of glycogen (b, d, f) horizontal and vertical lines represent means and 95% CI. n = 8, 10, 9, 10, 9, and 10 (from left to right). *Different from Con. †Different from pre-ischaemia. ‡Different from ischaemia. a Different from pre-ischaemia (main effect). b different from ischaemia (main effect). Comparisons were made by mixed effects model. The absolute values of glycogen (a, c, e) were log-transformed before statistics were performed. See text for exact P values
The volumetric content of intermyofibrillar glycogen was not different between IPC and Con hearts at any time point (Group × time interaction: P = 0.12. Main effect of IPC: P = = 0.13). Across groups the volumetric content decreased by 55% following ischaemia (P < 0.0001) and did not change after ischaemia–reperfusion (P = 0.56).
While the volumetric content of intramyofibrillar glycogen was 72% lower in IPC hearts than in Con hearts at pre-ischaemia (P = 0.01), we found no difference between the two groups after ischaemia (P = 0.28) or after ischaemia–reperfusion (P = 0.92; Group × time interaction: P = 0.03, Fig. 3e). Ischaemia mediated no further reduction of intramyofibrillar glycogen in IPC hearts (P = 0.32) but mediated a 90% reduction in Con hearts (P < 0.001). After ischaemia–reperfusion the increase in intramyofibrillar glycogen was not different between groups (P = 0.40, Fig. 3e). Across groups the volumetric content increased by 207% (P = 0.001).
The differential effects of IPC and ischaemia–reperfusion on the spatially distinct glycogen stores altered the relative distribution of glycogen by a decrease (P = 0.02) in the relative contribution of the volumetric content of subsarcolemmal glycogen and an increase (P = 0.002) in the relative contribution of the volumetric content of intermyofibrillar glycogen following ischaemia in both groups. After ischaemia–reperfusion the relative contribution of the volumetric content of intramyofibrillar glycogen increased in both groups (P = 0.001). In IPC hearts only, the relative contribution of the volumetric content of subsarcolemmal glycogen increased (P = 0.007) and the relative contribution of the volumetric content of intermyofibrillar glycogen decreased after ischaemia–reperfusion (P < 0.001).
The relative contribution of the subsarcolemmal to the total volumetric content of glycogen was associated with the LVDP after ischaemia–reperfusion in Con hearts, but not in IPC hearts (Fig. 4a). This association was not statistically significant with the diastolic pressure (Fig. 4b). No other correlations were found between the volumetric content of glycogen or the relative contribution of glycogen in the subfractions and the haemodynamic variables (LVDP, systolic and diastolic pressure).

Association of LVDP and diastolic pressure with subcellular glycogen distribution after reperfusion. LVDP (a) and diastolic pressure (b) plotted against the relative contribution of subsarcolemmal glycogen to total glycogen following ischaemia–reperfusion. Closed and open symbols represent control and IPC hearts, respectively. Line represents best fit. The P value is computed by linear regression analyses. The corresponding R2 and P values for LVDP in IPC hearts were R2 = 0.003, P = 0.88, for diastolic pressure in control hearts R2 = 0.24, P = 0.19, and for diastolic pressure in IPC hearts R2 = 0.02, P = 0.77
Force and SR Ca2+ handling of skinned fibre bundles
In the IPC as well as the Con groups, the specific maximal Ca2+ activated force was lower following ischaemia–reperfusion than at pre-ischaemia (P = 0.05) and ischaemia (P = 0.005). IPC had no effect on the specific maximal Ca2+ activated force at a single time point of the ischaemia–reperfusion protocol (P = 0.36) or across all time points (P = 0.41). We found no significant effects of IPC (P = 0.26), the ischaemia–reperfusion protocol (P = 0.99), or the combination of IPC and the ischaemia–reperfusion protocol (P = 0.73) on the submaximal force production. The specific maximal Ca2+ activated force and submaximal force is shown in Table 3.
The SR Ca2+ uptake rate (Fig. 5b) decreased by 51% from pre-ischaemia to ischaemia (P = 0.03) and increased to pre-ischaemic levels after 15 min reperfusion (P = 0.02). IPC had no effect on the SR Ca2+ uptake rate at a single time point of the ischaemia–reperfusion protocol (P = 0.58) or across all time points (P = 0.49).

SR Ca2+ uptake. a Representative force responses from a skinned fibre bundle when emptying the SR of Ca2+ after maximal loading (18 min exposure) and submaximal loading (4 and 2 min exposure). b Rate of SR Ca2+ loading. n = 6–8 per group. Horizontal lines represent geometric means and vertical lines represent 95% CI. Con control, IPC ischaemic preconditioning. †Different from pre-ischaemia. ‡Different from ischaemia. a, different from pre-ischaemia (main effect). b Different from ischaemia (main effect). Comparisons were made by mixed effects model using log-transformed data
The frequency of spontaneous force developments increased by 44% from pre-ischaemia to after ischaemia (P = 0.03) and decreased to 44% lower levels after ischaemia–reperfusion than at pre-ischaemia (P = 0.04, Fig. 6B). The mean half-force duration time did not change throughout the ischaemia–reperfusion protocol (P = 0.99) but was 20% lower in IPC hearts than in Con hearts (P = 0.01, Fig. 6C). The mean amplitude (Fig. 6D) did not change throughout the ischaemia–reperfusion protocol (P = 0.89) and was not affected by IPC (P = 0.40) or the combination of both (P = 0.67). The force-time integral did not change throughout the ischaemia–reperfusion protocol (P = 0.46 by ranksum) but was 23% lower in IPC hearts than in Con hearts (P = 0.04 by ranksum, Fig. 6E).

Spontaneous force oscillations. A Representative force responses from a skinned fibre bundle when exposed to low (A) or high (B) cytosolic [Ca2+]. The morphology of the spontaneous force oscillations was evaluated as frequency (B), half duration (C), and amplitude (D) at high cytosolic [Ca2+]. The force-time integral (E) was evaluated as the ratio of force-time integral between cytosolic high and low [Ca2+]. Con control, IPC ischaemic preconditioning. In B, horizontal lines represent means and vertical lines represent 95% CI. In C–E, horizontal lines represent geometric means and vertical lines represent 95% CI. n = 7, 7, 8, 9, 6 and 7 from left to right. *Different from Con. †Different from pre-ischaemia. ‡Different from ischaemia. A Different from pre-ischaemia (main effect). B Different from ischaemia (main effect). Comparisons were made by mixed effects model. In C–E, values were log-transformed before statistics were performed
We detected no significant correlations between these SR Ca2+ handling properties, the subcellular glycogen distribution and content, the LVDP, systolic pressure and diastolic pressure.
Discussion
Using quantitative TEM, the results of the present study demonstrate that the cardioprotective effect of IPC is linked to alterations in the subcellular distribution of glycogen particles. Specifically, we demonstrated that (1) IPC mediates a preferential resynthesis of subsarcolemmal glycogen during reperfusion after prolonged ischaemia; (2) the relative contribution of subsarcolemmal glycogen correlates with the recovery of the LVDP during reperfusion; and (3) subsarcolemmal glycogen is the main glycogen source for energy provision during prolonged ischaemia. Next, we investigated whether these alterations in subcellular glycogen distribution during ischaemia–reperfusion were associated with changes in SR Ca2+ uptake and leakiness. While IPC reduced the leakage of Ca2+ out of the SR and ischaemia decreased the Ca2+ uptake rate by the SR, these changes were not associated with the alterations in the subcellular distribution of glycogen particles.
Subcellular glycogen distribution
The measured metabolites (Table 2) of the present study confirm the marked effects of IPC on intracellular heart metabolism (Bradamante et al. 2000; Støttrup et al. 2010). The results are in line with a cardioprotective effect of 2 × 5 min preconditioning (Barbosa et al. 1996; Povlsen et al. 2014) and with a high resynthesis rate of glycogen during early reperfusion previously demonstrated in similar ischaemia-reperfusion protocols (Depre and Hue 1997; Doenst et al. 1998; Bradamante et al. 2000). If the ischaemic period is sustained for more than the 25 min used in the present study, several studies have demonstrated that resynthesis of glycogen is negligible (King and Opie 1996; Fraser et al. 1998; Støttrup et al. 2010), presumably due to a higher degree of irreversible ischaemic damage (Depre et al. 1999). At the subcellular level, our quantitative approach also confirms that subsarcolemmal glycogen is the major deposition of glycogen in cardiomyocytes (Caulfield and Klionsky 1959; Ferrans et al. 1964, 1969; Todd et al. 1979) and preferentially depleted during ischaemia (Caulfield and Klionsky 1959). This contrasts with skeletal muscle fibres, where subsarcolemmal glycogen is less depleted during both prolonged moderate intensity and supramaximal exercise compared with inter- and intra-myofibrillar glycogen (Gejl et al. 2017; Marchand et al. 2007; Nielsen et al. 2011). In the absence of data on work-matched glycogen consumption in cardiac and skeletal muscles, this difference suggests that the utilisation of local subcellular glycogen stores is muscle type-specific. One obvious explanation is the difference in spatial energy requirements. The heart has a high energy-turnover in the subsarcolemmal space to cover the extrusion of Na+ and Ca2+, while the energy-turnover in skeletal muscles is preferentially related to the myofibrillar apparatus and SR Ca2+ handling (Rolfe and Brown 1997). Therefore, the storage of glycogen in the subsarcolemmal space may be more important in the heart than in skeletal muscles.
Interestingly, during reperfusion the resynthesis of subsarcolemmal and intramyofibrillar glycogen was most pronounced compared with intermyofibrillar glycogen (Fig. 3). In IPC hearts, no resynthesis of intermyofibrillar glycogen could be detected (Fig. 3c) resulting in an increase in the share of subsarcolemmal glycogen from 40 to 50% (Fig. 3b). This shift in glycogen distribution was not observed in Con hearts, but correlation analyses revealed that the hearts with the highest share of subsarcolemmal glycogen also had a more pronounced recovery of the LVDP during reperfusion (Fig. 4a), indicating a relationship between the cardioprotective effect of IPC and the ability to store glycogen in the subsarcolemmal space. It could be speculated that a higher net resynthesis of subsarcolemmal glycogen during reperfusion is the result of a lower local energetic demand in the subsarcolemmal space with IPC. Indeed, IPC reduces ischaemia–induced acidosis (Bradamante et al. 2000), which may attenuate both the activation of Na+/H+ exchanger and the reverse mode activation of sarcolemmal Na+/Ca2+ exchanger (Tani and Neely 1989). This results in less intracellular Na+ and Ca2+ overload and thereby lower activation of the energy demanding sarcolemmal Na+, K+-ATPase and Ca2+ ATPase with IPC. On the other hand, the higher share of subsarcolemmal glycogen results also from no net resynthesis of intermyofibrillar glycogen. Whether this is due to an increased glycogenolytic activity or decreased resynthesis rate in this location remains to be investigated. Indeed, during the early phase of reperfusion (15 min) cardioprotection has in addition to enhanced glucose uptake also been ascribed to increased anaerobic glycolysis (Heywood et al. 2017) suggesting that a higher turnover of glycogen could be important for cardiomyocyte survival during this critical phase of reperfusion.
SR Ca2+ uptake and leak
An excessive increase in cytosolic calcium level after ischaemia–reperfusion is a major trigger of cardiomyocyte injury (Garcia-Dorado et al. 2012). The balance between cytosolic Ca2+ buffering and Ca2+ influx and efflux across the plasma membrane and the SR determines the cytosolic calcium level. Disturbances in these fluxes, causing cytosolic Ca2+ accumulation during ischaemia–reperfusion are predominantly caused by leakage from the SR (Valverde et al. 2010). In addition to the influx of Ca2+ across the plasma membrane initiating excitation-contraction coupling, the SR is the main intracellular Ca2+ store in cardiac myocytes, regulating excitation-contraction coupling via Ca2+ uptake by the SR Ca2+ ATPases and Ca2+ release via RyRs (Orchard and Brette 2008). In the present study, we take advantage of a direct measure of SR Ca2+ handling obtained in isolated cardiomyocyte bundles chemically skinned by the detergent saponin (Stephenson and Wendt 1986). The advantage of this technique is the ability to investigate SR Ca2+ uptake and release events during controlled and clamped conditions using known concentrations of metabolites (ATP and CrP) and ions (Ca2+, Mg2+, H+).
We demonstrate in such saponin-skinned cardiomyocyte bundles that the SR Ca2+ uptake rate decreases during prolonged ischaemia and restores to pre-ischaemic levels during reperfusion (Fig. 6). This is in line with most studies using different techniques (Hohl et al. 1992; Kaplan et al. 1992; Krause and Hess 1984; Zucchi et al. 1996), but not all (Luciani et al. 1993). In the latter study, SR Ca2+ uptake rate was investigated at low cytosolic calcium levels (pCa 7.0), at which the effect of ischaemia may be difficult to detect (Hohl et al. 1992). Therefore, our results confirm that the SR Ca2+ uptake process is impaired with ischaemia also by a mechanism independent of global metabolic alterations.
Changes in SR Ca2+ uptake rate manifest in changes in cytosolic Ca2+ concentration, particularly during diastole and hence elevating diastolic pressure (Sankaranarayanan et al. 2017). Therefore, a decreased SR Ca2+ uptake rate (Fig. 5b) may explain the significant increase in diastolic pressure during ischaemia (Fig. 1c). The early increase in diastolic pressure during the ischaemia by IPC suggests that the SR Ca2+ uptake rate is decreased early during ischaemia in IPC hearts. Since glycolysis preferentially fuels the SR Ca2+ pumping (Boehm et al. 2000; Jeremy et al. 1992), the observed lower glycogen levels with IPC (Fig. 3) may explain the early development of diastolic pressure in IPC. Although we did not measure the cytosolic ADP level, given the decreased ATP levels with IPC (Table 2), IPC might increase the ADP levels. Such a disarray might increase SR Ca2+ leakiness via the SR Ca2+ ATPase (Inesi and de Meis 1989) and contribute to the observed early increase of diastolic pressure during ischemia in IPC hearts. During reperfusion, the reduction of diastolic pressure following IPC does not seem to be explained by higher SR Ca2+ re-uptake (Fig. 5). In accordance with modulation of RyR activity as demonstrated by a reduction in the half duration time of the spontaneous force developments, a more valid explanation is a lower leakage of Ca2+ out of the SR (Fig. 6) with a concomitant decrease in diastolic cytosolic Ca2+ level (Steenbergen et al. 1990).
We investigated SR Ca2+ leakage by a morphological characteristic of spontaneous force development (i.e. Ca2+ sparks) including the frequency, amplitude, duration and force-time integral. In saponin-skinned cardiomyocyte bundles these spontaneous force oscillations can be investigated by exposing the bundles to a weakly buffered EGTA solution (Stephenson and Wendt 1986). It has been shown that a treatment with Triton X-100, which disrupts all membrane structures, abolishes these spontaneous force developments clearly suggesting that they are the result of cyclical release and uptake of Ca2+ by the SR (Stephenson and Wendt 1986) and therefore can be interpreted as the consequence of Ca2+ waves (Cheng et al. 1996; Jeremy et al. 1992). We find that the frequency of spontaneous force developments increases with ischaemia and decreases during reperfusion but is unaffected by IPC (Fig. 6b). Since we controlled the luminal and cytosolic levels of Ca2+, the fluctuation in frequency reflects a modulation of the open probability of RyR2s with ischaemia (Cheng et al. 1993; Satoh et al. 1997).
Since the amplitude of the spontaneous force developments largely depends on the luminal content of calcium within the SR (Cannell et al. 1995), it would be expected that reduced SR Ca2+ uptake rate would decrease the luminal SR Ca2+ content and then the amplitude. However, we find no change in the amplitude suggesting that the luminal SR Ca2+ was not affected by ischaemia. Of note, we measured the amplitudes after a 4-min loading, which should be sufficient to obtain high luminal Ca2+ levels also after ischaemia. We find that the half duration time is reduced by IPC, suggesting that IPC reduces the time that the RyRs are open and/or lowers the number of additional open RyRs at the tail of the force development (Stern et al. 2013). This effect of IPC on half duration time explains the lower force-time integral observed with IPC. Despite fluctuations in the frequency with ischaemia–reperfusion, IPC manifests by reduced SR Ca2+ leak after ischaemia–reperfusion. This is in accordance with the findings that IPC reduces cytosolic Ca2+ overload during ischaemia–reperfusion (Dekker et al. 1996; Smith et al. 1996). The possible role of ischaemia–reperfusion and the protective effect of IPC may be explained by a modulation of the RyR channel.
Conclusion
The cardioprotective effect of IPC is associated with a preferential resynthesis of subsarcolemmal glycogen, but no resynthesis of intermyofibrillar glycogen during reperfusion, and a decreased SR Ca2+ leak at pre-ischaemia, post-ischaemia and after reperfusion. During reperfusion, the recovery of the LVDP correlates with this preferable storage of glycogen in the subsarcolemmal space suggesting a compartmentalised protective effect of IPC. Even though IPC reduced the leakage of Ca2+ from SR during reperfusion, we identified no temporal association with alterations in the subcellular distribution of glycogen particles.
Authors translational perspective
Our findings extend previous findings of metabolic effects during reperfusion by IPC and reflects a favourable modulation of myocardial carbohydrate metabolism. Future studies should clarify whether a variety of pharmacological and mechanical interventions that yield cardioprotection induces this subcellular modification consistently such that it represents a uniform intracellular response to cardioprotective interventions. If so, interventions that increase subsarcolemmal glycogen content may represent a concept to achieve cardioprotection that may be translated to the clinical setting.
References
Angelakos ET, Bernardini P, Barrett WC Jr (1964) Myocardial fibre size and capillary-fibre ratio in the rigth and left ventricles of the rat. Anat Rec 149:671–676
Barbosa V, Sievers RE, Zaugg CE, Wolfe CL (1996) Preconditioning ischemia time determines the degree of glycogen depletion and infarct size reduction in rat hearts. Am Heart J 131:224–230
Bland JM, Altman DG (1986) Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1:307–310
Boehm E, Ventura-Clapier R, Mateo P, Lechène P, Veksler V (2000) Glycolysis supports calcium uptake by the sarcoplasmic reticulum in skinned ventricular fibres of mice deficient in mitochondrial and cytosolic creatine kinase. J Mol Cell Cardiol 32:891–902
Bradamante S, Marchesani A, Barenghi L, Paracchini L, de Jonge R, de Jong JW (2000) Glycogen turnover and anaplerosis in preconditioned rat hearts. Biochem Biophys Acta 1502:363–379
Cannell MB, Cheng H, Lederer WJ (1995) The control of calcium release in heart muscle. Science 268:1045–1049
Caulfield J, Klionsky B (1959) Myocardial ischaemia and early infarction: an electron microscopic study. Amer J Path 35:489–523
Cheng H, Lederer WJ, Cannell MB (1993) Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262:740–744
Cheng H, Lederer MR, Lederer WJ, Cannell MB (1996) Calcium sparks and Ca2+I waves in cardiac myocytes. Am J Physiol Cell 270:C148–C159
De Bruijn WC (1973) Glycogen, its chemistry and morphologic appearance in the electron microscope. I. A modified OsO 4 fixative which selectively contrasts glycogen. J Ultrastruct Res 42:29–50
Dekker LR, Fiolet JW, Van Bavel E, Coronel R, Opthof T, Spaan JA, Janse MJ (1996) Intracellular Ca2+, intracellular electrical coupling, and mechanical activity in ischaemic rabbit papillary muscle. Effects of preconditioning and metabolic blockade. Circ Res 79:237–246
Depre C, Hue L (1997) Inhibition of glycogenolysis by glucose analogue in the working rat heart. J Mol Cell Cardiol 29:2253–2259
Depre C, Vanoverschelde J-LJ, Taegtmeyer H (1999) Glucose for the heart. Circulation 99:578–588
Doenst T, Guthrie PH, Taegtmeyer H (1998) Ischemic preconditioning in rat heart: no correlation between glycogen content and return of function. Mol Cell Biochem 180:153–161
Ferrans VJ, Hibbs RG, Black WC, Weilbaecher DG (1964) Isoproterenol-induced myocardial necrosis. A histochemical and elctron microscopic study. Am Heart J 68:71–90
Ferrans VJ, Hibbs RG, Walsh JJ, Burch GE (1969) Histochemical and electron microscopical studies on the cardiac necroses produced by sympathomimetic agents. Ann NY Acad Sci 156:309–332
Fraser H, Lopaschuk GD, Clanachan AS (1998) Assessment of glycogen turnover in aerobic, ischemic, and reperfused working rat hearts. Am J Physiol Heart Circ Physiol 44:H1533–H1541
Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM (2012) Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res 94:168–180
Gejl KD, Ørtenblad N, Andersson E, Plomgaard P, Holmberg H-C, Nielsen J (2017) Local depletion of glycogen with supramaximal exercise in human skeletal muscle fibres. J Physiol 595:2809–2821
Harris RC, Hultman E, Nordesjö LO (1974) Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scand J Clin Lab Invest 33:109–120
Hausenloy DJ, Yellon DM (2016) Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol 13:193–209
Heywood SE, Richart AL, Henstridge DC, Alt K, Kiriazis H, Zammit C, Carey AL, Kammoun HL, Delbridge LM, Reddy M, Chen Y-C, Du X-J, Hagemeyer CE, Febbrario MA, Siebel AL, Kingwell BA (2017) High-density lipoprotein delivered after myocardial infarction increases cardiac glucose uptake and function in mice. Sci Transl Med 9:100. https://doi.org/10.1126/scitranslmed.aam6084
Hohl CM, Garleb AA, Altschuld RA (1992) Effects of simulated ischaemia and reperfusion on the sarcoplasmic reticulum of digitonin-lysed cardiomyocytes. Circ Res 70:716–723
Inesi G, de Meis L (1989) Regulation of steady state filling in sarcoplasmic reticulum. J Biol Chem 264:5929–5936
Nielsen JS, Sahlin K, Ørtenblad N (2007a) Reduced sarcoplasmic reticulum content of releasable Ca2+ in rat soleus muscle fibres after eccentric contractions. Acta Physiol 191:217–228
Jeremy RW, Koretsune Y, Marban E, Becker LC (1992) Relation between glycolysis and calcium homeostasis in postischaemic myocardium. Circ Res 70:1180–1190
Kaplan P, Hendrikx M, Mattheussen M, Mubagwa K, Flameng W (1992) Effetc of ischaemia and reperfusion on sarcoplasmic reticulum calcium uptake. Circ Res 71:1123–1130
King LM, Opie LH (1996) Does preconditioning act by glycogen depletion in the isolated rat heart? J Mol Cell Cardiol 28:2305–2321
Krause S, Hess ML (1984) Characterization of cardiac sarcoplasmic reticulum dysfunction during short-term, normothermic, global ischaemia. Circ Res 55:176–184
Lamboley CR, Wyckelsma VL, McKenna MJ, Murphy RM, Lamb GD (2016) Ca2+ leakage out of the sarcoplasmic reticulum is increased in type I skeletal muscle fibres in aged humans. J Physiol 594:469–481
Lin LI (1989) A concordance correlation coefficient to evaluate reproducibility. Biometrics 45:255–268
Lowry OH, Passonneau JV (1972) A flexible system on enzymatic analysis. Academic Press, New York
Luciani GB, D’Agnolo A, Mazzucco A, Gallucci V, Salviati G (1993) Effects of ischaemia on sarcoplasmic reticulum and contractile myofilament activity in human myocardium. Am J Physiol 265:H1334–H1341
Marchand I, Chorneyko K, Tarnopolsky M, Hamilton S, Shearer J, Potvin J, Graham TE (2002) Quantification of subcellular glycogen in resting human muscle: granule size, number, and location. J Appl Physiol 93:1598–1607
Marchand I, Tarnopolsky M, Adamo KB, Bourgeois JM, Chorneyko K, Graham TE (2007) Quantitative assessment of human muscle glycogen granules size and number in subcellular locations during recovery from prolonged exercise. J Physiol 580:617–628
Nielsen JS, Sahlin K, Ørtenblad N (2007b) Reduced sarcoplasmic reticulum content of releasable Ca2+ in rat soleus muscle fibres after eccentric contractions. Acta Physiol 191:217–228
Nielsen J, Holmberg H-C, Schrøder HD, Saltin B, Ørtenblad N (2011) Human skeletal muscle glycogen utilisation in exhaustive exercise: role of subcellular localisation and fibre type. J Physiol 589:2871–2885
Orchard C, Brette F (2008) T-tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. Cardiovasc Res 77:237–244
Povlsen JA, Løfgren B, Dalgas C, Jespersen NR, Johnsen J, Bøtker HE (2014) Frequent biomarker analysis in the isolated perfused heart reveals two distinct phases of reperfusion injury. Int J Cardiol 171:9–14
Rolfe DFS, Brown GC (1997) Cellular energy utilisation and molecular origin of standard metabolic rate in mammals. Physiol Rev 77:731–758
Sankaranarayanan R, Kistamas K, Greensmith DJ, Venetucci LA, Eisner DA (2017) Systolic [Ca2+]i regulates diastolic levels in rat ventricular myocytes. J Physiol 595:5545–5555
Satoh H, Blatter LA, Bers DM (1997) Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol Heart Circ Physiol 272:H657–668
Smith GB, Stefenelli T, Wu ST, Wikman-Coffelt J, Parmley WW, Zaugg CE (1996) Rapid adaption of myocardial calcium homeostasis to short episodes of ischaemia in isolated rat hearts. Am Heart J 131:1106–1112
Spencer TN, Botting KJ, Morrison JL, Posterino GS (2006) Contractile and Ca2+-handling properties of the right ventricular papillary muscle in the late-gestation sheep fetus. J Appl Pysiol 101:728–733
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Steenbergen C, Murphy E, Watts JA, London RE (1990) Correlation between cytosolic free calcium, contracture, ATP, and irreversible ischaemic injury inperfused rat heart. Circ Res 66:135–146
Stephenson DG, Wendt IR (1986) Effects of procaine on calcium accumulation by the sarcoplasmic reticulum of mechanically disrupted rat cardiac muscle. J Physiol 373:195–207
Stern MD, Rios E, Maltsev VA (2013) Life and death of a cardiac calcium spark. J Gen Physiol 142:257–274
Støttrup NB, Løfgren B, Birkler RD, Nielsen JM, Wang L, Caldarone CA, Kristiansen SB, Contractor H, Johannsen M, Bøtker HE, Nielsen TT (2010) Inhibition of the malate-aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovas Res 88:257–266
Tani M, Neely JR (1989) Role of intracellular Na+ in Ca2+ overload and depressed recovery of ventricular function of reperfused ischaemic rat hearts. Possible involvement of H+-Na+ and Na+ and Ca2+ exchange. Circ Res 65:1045–1056
Todd GL, Pieper GM, Clayton FC, Eliot RS (1979) Heterogeneity in distribution of cardiac glycogen following isoproterenol infusion in the dog. Histochem J 11:425–434
Valverde CA, Kornyeyev D, Ferreire M, Petrosky AD, Mattiazzi A, Escobar AL (2010) Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc Res 85:671–680
Weibel ER (1980) Stereological methods, vol 2: theoretical foundations. Academic Press, London
Zima AV, Kockskämper J, Blatter LA (2006) Cytosolic energy reserves determine the effect of glycolytic sugar phosphates on sarcoplasmic reticulum Ca2+ release inn cat ventricular myocytes. J Physiol 577:281–293
Zucchi R, Ronca-Testoni S, Di Napoli P, Yu G, Gallina S, Bosco G, Ronca G, Calafiore AM, Mariani M, Barsotti A (1996) Sarcoplasmic reticulum calcium uptake in human myocardium subjected to ischaemia and reperfusion during cardiac surgery. J Mol Cell Cardiol 28:1693–1701
Acknowledgements
The authors thank Karin Trampedach, Susan Bøgebjerg, and Sandra Holm Riggelsen for technical assistance.
Funding
This work was supported by the Danish Council for Independent Research [DFF – 1333-00,144 to J.N.], the Danish Heart Association [72981 to J.N], the Lundbeck Foundation [R108-A10616 to J.N.], and The Danish Council for Strategic Research (11-1115818 to H.E.B.].
Author information
Authors and Affiliations
Contributions
The experiments were performed in the laboratory of the Department of Cardiology, Aarhus University Hospital. The TEM was performed at the Department of Pathology, Odense University Hospital, Denmark. The TEM image analyses were performed at the Department of Sports Science and Clinical Biomechanics, University of Southern Denmark. JN, JJ, NØ and HEB were involved in the design of this study. All authors were involved in acquisition, analysis or interpretation of data for the work. JN drafted the work and all authors revised it critically for important intellectual content. All authors approved the final, submitted version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nielsen, J., Johnsen, J., Pryds, K. et al. Myocardial subcellular glycogen distribution and sarcoplasmic reticulum Ca2+ handling: effects of ischaemia, reperfusion and ischaemic preconditioning. J Muscle Res Cell Motil 42, 17–31 (2021). https://doi.org/10.1007/s10974-019-09557-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-019-09557-3