
Abstract
Since the postulate, 30 years ago, that phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P 2) as the precursor of inositol 1,4,5-trisphosphate (Ins(1,4,5)P 3) would be critical for skeletal muscle excitation–contraction (EC) coupling, the issue of whether phosphoinositides (PtdInsPs) may have something to do with Ca2+ signaling in muscle raised limited interest, if any. In recent years however, the PtdInsP world has expanded considerably with new functions for PtdIns(4,5)P 2 but also with functions for the other members of the PtdInsP family. In this context, the discovery that genetic deficiency in a PtdInsP phosphatase has dramatic consequences on Ca2+ homeostasis in skeletal muscle came unanticipated and opened up new perspectives in regards to how PtdInsPs modulate muscle Ca2+ signaling under normal and disease conditions. This review intends to make an update of the established, the questioned, and the unknown regarding the role of PtdInsPs in skeletal muscle Ca2+ homeostasis and EC coupling, with very specific emphasis given to Ca2+ signals in differentiated skeletal muscle fibers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phosphoinositides are either known or presumed to be involved in countless numbers of cellular functions, making it hard to actually think of a cellular mechanism that would not be modulated by one or the other of these lipid molecules. Excitation–contraction (EC) coupling in skeletal muscle—the process through which electrical activity in the transverse (t-) tubule membrane triggers the release of Ca2+ from the sarcoplasmic reticulum (SR) into the cytosol—has, until recently, been believed to be one of these few. The coupling occurs within the triad, a specialized region where two SR terminal cisternae come into close apposition with one t-tubule: t-tubule depolarization activates the voltage-sensing CAV1.1 Ca2+ channels which in turn mechanically gate the opening of the SR ryanodine receptor (RYR) Ca2+ release channel (Ríos et al. 1992; Schneider 1994). Nevertheless, EC coupling historians and aficionados know that, before the advent of the ryanodine receptor, the process was at some point postulated to rely on voltage-dependent hydrolysis of PtdIns(4,5)P 2 to produce the Ca2+ mobilizing molecule Ins(1,4,5)P 3 (Vergara et al. 1985). Current knowledge leaves little room for this possibility as the kinetics of Ins(1,4,5)P 3-induced contraction was found to be far too slow to contribute to skeletal muscle activation (Walker et al. 1987). Still, differentiated muscle fibers do possess the Ins(1,4,5)P 3 producing machinery and corresponding receptors (Hidalgo et al. 1986; Carrasco et al. 1993; Casas et al. 2010) but the role and/or the conditions of activation of this cascade remain debated. While Ins(1,4,5)P 3 lost popularity in muscle EC coupling, its precursor PtdIns(4,5)P 2 was found to have its own direct functions in other cell models where it was shown to act as a docking target or direct molecular binding partner of specific proteins, including ion channels (see Balla 2013). The possibility then exists that PtdIns(4,5)P 2 could play a role in skeletal muscle Ca2+ signaling independently from Ins(1,4,5)P 3, as recently suggested by Berthier et al. (2015). In addition, other members of the PtdInsP family were intuitively presumed to be involved when it was discovered that the 3-OH phosphoinositide phosphatase MTM1 is required for proper EC coupling function (Al Qusairi et al. 2009). While this was unexpected and the reason why EC coupling is so critically dependent upon a phosphoinositide phosphatase is still not completely understood, the current most widely accepted view is that MTM1 is necessary for the maintenance of the architecture of t-tubules and/or triads. However, this may not be all, especially along the view that certain PtdInsPs were shown to bind RYR channels and to modulate their activity (Shen et al. 2009). In the present review we have essentially confined ourselves to summarizing recent sets of data from the literature in regard to these issues, as they specifically apply to differentiated skeletal muscle fibers.
PtdIns(4,5)P 2 is the precursor of Ins(1,4,5)P 3
PtdIns(4,5)P 2 hydrolysis by phospholipase C (PLC) releases Ins(1,4,5)P 3 into the cytosol (Burgess et al. 1984); consequent activation of Ins(1,4,5)P 3 receptors in the endoplasmic reticulum membrane leads to a cytosolic Ca2+ signal that is critical for a wide array of living functions (see Berridge 2009). In cultured myotubes a large number of studies showed the presence and/or contribution of Ins(1,4,5)P 3-mediated Ca2+ release to intracellular Ca2+ signals under various conditions (Estrada et al. 2001; Jaimovich et al. 2000; Powell et al. 2001; Carrasco et al. 2003; Estrada et al. 2003; Eltit et al. 2004; Juretic et al. 2006; Eltit et al. 2006; May et al. 2006; Balghi et al. 2006a, b; Valdés et al. 2007; Liberona et al. 2008). As a specific example, G-protein coupled purinergic receptors (P2Y receptors) were shown to initiate Ca2+ release via Ins(1,4,5)P 3 signaling (Cseri et al. 2002; Cheung et al. 2003; Deli et al. 2007) and the process is believed to be involved in muscle differentiation and/or regeneration (Ryten et al. 2004; Szigeti et al. 2007).
In differentiated skeletal muscle fibers, on the other hand, since the early lines of evidence (Meissner 1986; Inui et al. 1987) that ultimately led to the definite identification of the RYR as the SR Ca2+ release channel of EC coupling, the role of Ins(1,4,5)P 3 receptors has remained poorly investigated. This issue was re-visited in recent years along three lines of possibilities: a role of Ins(1,4,5)P 3 receptors in store-operated Ca2+ entry (Launikonis et al. 2003) and a role in voltage-activated and voltage-independent cytosolic Ca2+ signals (Casas et al. 2010; Tjondrokoesoemo et al. 2013 respectively).
The first proposal was derived from measurements of t-tubule Ca2+ content following activation of EC coupling in skinned skeletal muscle fibers (Launikonis et al. 2003). They showed that the activation and subsequent depletion of SR Ca2+ is followed by rapid loss of t-tubule Ca2+ content, which was taken as evidence for store operated Ca2+ entry (SOCE). The fact that SOCE was blocked by heparin led the authors to propose that Ins(1,4,5)P 3 receptors play a role as SR Ca2+ sensors in the process, possibly through physical coupling with the plasma membrane. Results also suggested that Ins(1,4,5)P 3 needs to bind to the receptor for SOCE to be operative. Although quite provocative, this possibility received quite limited support from subsequent studies on the existence and function of SOCE in differentiated skeletal muscle (see for instance Allard et al. 2006; Dirksen 2009).
The second proposal is that voltage-activation of skeletal muscle fibers is responsible for a slow Ca2+ transient that depends on Ins(1,4,5)P 3 production and serves the excitation-transcription machinery (Casas et al. 2010). This possibility emerged from a long series of studies that demonstrated this mechanism to operate in cultured myotubes (Jaimovich et al. 2000; Powell et al. 2001; Araya et al. 2003; Carrasco et al. 2003; Eltit et al. 2004; Juretic et al. 2006). The experiments by Casas et al. (2010) showed that in differentiated mouse muscle fibers, the slow Ca2+ signal was only present in fast-type muscle fibers, that it was inhibited by blockers of Ins(1,4,5)P 3 receptors and that it was most prominent following a tetanic stimulation at 10–20 Hz that promotes and represses expression of slow- and fast-type troponin I, respectively. Sensitivity of the slow Ca2+ signal to nifedipine was consistent with the DHPR being the voltage sensor for this process, which was then confirmed by showing that Ins(1,4,5)P 3 production is controlled by the DHPR in a frequency dependent manner (Jorquera et al. 2013).
The potency of Ins(1,4,5)P 3 to generate a significant global Ca2+ signal in differentiated mammalian skeletal muscle was, nevertheless, challenged by Blaauw et al. (2012) through a series of experiments performed in parallel in C2C12 myotubes and in differentiated muscle fibers: these included activation of purinergic receptors, injection of Ins(1,4,5)P 3 and photolysis of caged Ins(1,4,5)P 3. While these different protocols generated Ca2+ signals in C2C12 myotubes they did not give rise, or to a much smaller extent, to Ca2+ transients in adult muscle fibers. Blaauw et al. took these results as evidence that Ins(1,4,5)P 3 cannot trigger global Ca2+ release in differentiated muscle fibers. However, as indicated by Jorquera et al. (2013), there may be several reasons why the detection of a global Ins(1,4,5)P 3-related Ca2+ signal may be hard in muscle fibers, including possible dependence on membrane voltage and/or intracellular Ca2+ concentration. Indeed, results by Casas et al. (2010) associated the slow, Ins(1,4,5)P 3-dependent Ca2+ signal with normally voltage-activated RYR-mediated SR Ca2+ release. It could then well be that only under such conditions, i.e. when SR calcium release via RYR is stimulated by membrane depolarization, would a significant Ca2+ release flux through sensitized Ins(1,4,5)P 3 receptors occur.
The third proposal is that Ins(1,4,5)P 3 receptors could contribute to Ca2+ sparks. Ca2+ sparks are SR Ca2+ release events confined in space and time, generated by a group of RYRs operating coherently. These events are almost non-existent in adult mammalian muscle fibers but their frequency is strongly enhanced by certain stress conditions (for review see Csernoch 2007). Among these is osmotic challenge (Wang et al. 2005) which generates Ca2+ sparks near the periphery of the muscle fibers. Tjondrokoesoemo et al. (2013) reported that osmotic-stress induced Ca2+ sparks were suppressed by the inhibition of either PLC or Ins(1,4,5)P 3 receptors and were facilitated following photolysis of caged Ins(1,4,5)P 3. The contribution of Ins(1,4,5)P 3 receptors was confirmed by short-hairpin (sh)RNA-mediated knockdown of type 1 Ins(1,4,5)P 3 receptor which eliminated the occurrence of osmotic-stress induced Ca2+ sparks. Authors proposed that initial Ca2+ release through Ins(1,4,5)P 3 receptors could activate neighboring RYR channels formerly sensitized by the osmotic stress so as to be able to generate sparks. An interesting aspect also highlighted by the authors is that hyperosmotic stress has been shown to increase PtdIns(4,5)P 2 level in HeLa cells (Yamamoto et al. 2006). If applicable to skeletal muscle, it could then also facilitate the occurrence of Ca2+ sparks in this particular condition. Overall interest of these results faces a recurrent challenge from a sparko-skeptic community when it refers to physiological relevance of these events in mammalian muscle: Ca2+ spark scarcity at rest together with the fact that EC coupling does not use Ca2+ sparks (in contrast to mammalian cardiac and amphibian skeletal muscle) should limit the interest of these events in this preparation. We, nevertheless, believe that one should not dismiss the importance of Ca2+ sparks in muscle and stay ready for potential future findings of physiological and/or pathological conditions that make them occur in large numbers in mammalian skeletal muscle. In the meantime, understanding what makes, promotes and depresses Ca2+ sparks should still be considered of primary importance for our understanding of muscle Ca2+ signaling and RYR function. In the present case, the fact that knockdown of Ins(1,4,5)P 3 type 1 receptors completely suppressed osmotic-stress induced Ca2+ sparks should instead prompt investigation of the potential contribution of Ins(1,4,5)P 3 receptors to Ca2+ sparks observed under other stress conditions.
Aside from the two above lines of results there has been no clear evidence yet in differentiated skeletal muscle that either activation of receptors coupled to G-proteins or tyrosine phosphorylation could activate PLC so as to generate a substantial Ins(1,4,5)P 3-related intracellular Ca2+ signal. Interestingly though, PLC-δ, which is dominantly expressed in skeletal muscle (Milting et al. 1996), is essentially known to be activated by a rise in intracellular Ca2+ and the possibility then that a positive feedback from RYR-mediated Ca2+ release could generate Ins(1,4,5)P 3-mediated and a consequent Ca2+ release signal remains unexplored.
Ins(1,4,5)P 3-independent functions for PtdIns(4,5)P 2
PtdIns(4,5)P 2 is expected to be present in several subcellular membrane compartments of living cells. Expression of the pleckstrin-homology (PH) domain from PLC-δ1 fused to a fluorescent protein has though essentially reported PtdIns(4,5)P 2 to be localized at the plasma membrane (Várnai and Balla 1998; see Sun et al. 2013). This is also the case in skeletal muscle where the probe was found to nicely decorate fibers with a transverse-striated triadic-consistent pattern. This pattern could be completely disrupted by activation of a voltage sensitive PtdInsP phosphatase (VSP) in the t-tubule membrane, attesting the t-tubule localization of PtdIns(4,5)P 2 (Berthier et al. 2015).
PtdIns(4,5)P 2 in the plasma membrane has become famous for its potency to modulate the activity of several types of ion channels through either direct interactions with the channel protein or modulation of the channel interaction with other molecular partners (see Balla 2013; Hille et al. 2015). Such modulatory processes should be active in skeletal muscle fibers because the plasma- and/or t-tubule- membranes are endowed with several types of potentially PtdIns(4,5)P 2-sensitive ion channels and transporters including voltage-gated, inward rectifying, Ca2+-activated and ATP-sensitive K+ channels, Na+/Ca2+ exchanger and Ca2+ ATPase. Very limited information is yet available as to whether these, in their native muscle environment, are modulated by changes in PtdIns(4,5)P 2 and as to whether such modulations are physiologically relevant. Interestingly though, certain pathological mutations in the gene encoding the inward-rectifying K+ channel Kir2.1 were shown to affect residues involved in PtdIns(4,5)P 2 binding; this led to the suggestion that disruption of the corresponding modulation may constitute a major pathogenic mechanism (Donaldson et al. 2003).
Although PtdIns(4,5)P 2 is essentially detected in the t-tubule membrane, the first ion channel that was suspected to be modulated by PtdIns(4,5)P 2 in skeletal muscle is the RYR in the SR membrane. Indeed, in the 1990s several studies suggested that PtdIns(4,5)P 2 could directly activate SR Ca2+ release because its application was shown to increase the activity of RYR channels incorporated in artificial lipid bilayers and to either elicit or potentiate Ca2+ release from SR vesicles or tension in skinned muscle fibers (Kobayashi et al. 1989; Ogawa and Harafuji 1989; Chu and Stefani 1991; Ohizumi et al. 1999). No clear correlate regarding the physiological function of RYR channels was given at the time: on the one hand, it seems hard to consider that PtdIns(4,5)P 2 in the t-tubule could directly act on RYRs in the SR membrane, although this may be regarded with caution since there is evidence, for instance, that a PtdInsP in a given membrane can be degraded by a phosphatase in a closely apposed but distinct membrane compartment (see Kim et al. 2013). In other words, it cannot be totally excluded that PtdIns(4,5)P 2 could interact with either a cytoplasmic region of the RYR, or with a partner protein. On the other hand, it is difficult to imagine how PtdIns(4,5)P 2 in the t-tubule membrane could be released towards the SR membrane to act on RYRs. One alternative is that PtdIns(4,5)P 2 is also constitutively present in the SR membrane either in much lower amount than in the t-tubule or/and under a configuration that makes it undetected by the PH probe.
The possibility that t-tubule PtdIns(4,5)P 2 plays a role in EC coupling in intact skeletal muscle fibers was more directly tested by Berthier et al. (2015): authors investigated the consequences of t-tubule PtdIns(4,5)P 2 depletion on the properties of voltage-activated Ca2+ transients. Depletion was achieved by activation of a VSP triggered by strong membrane depolarizing pulses and, as mentioned above, could be assessed by disruption of the t-tubule pattern of a fluorescent PH probe. It was found that activation of the VSP was accompanied by a depression of voltage-activated SR Ca2+ release, demonstrating that t-tubule PtdIns(4,5)P 2 is necessary for proper EC coupling function. The mechanism for this effect remains yet unidentified: PtdIns(4,5)P 2 could be envisioned to enhance the voltage-sensing function of CAV1.1, possibly through interaction with a polybasic cluster within the cytoplasmic I-II linker region close to the β1a subunit binding domain (Flucher 2015; Kaur et al. 2015) or to promote membrane targeting of an accessory protein of EC coupling. Interestingly, the other important question behind the need for PtdIns(4,5)P 2 in EC coupling is whether physiological and/or pathological situations would exist that substantially change its level in the t-tubule membrane so as to affect EC coupling. Physiological conditions could encompass any signaling mechanism acting on the PtdIns(4,5)P 2 processing enzymes as PLC and PtdIns 3-kinase. Alternatively PtdIns(4,5)P 2 could also be envisioned to mediate a physical stretch-dependent effect between the lipid bilayer and the EC-coupling machinery during contraction (Flucher 2015). In terms of pathology, there is evidence in neurons that amyloid β peptide decreases the level of PtdIns(4,5)P 2 and that this may be relevant in Alzheimer’s disease (Berman et al. 2008). Intracellular accumulation of amyloid β in muscle fibers is also believed to be a critical pathogenic process in Inclusion Body Myositis and recent data suggest that disrupted Ca2+ handling is an early event of this disease (Shtifman et al. 2010; Lopez and Shtifman 2010). It would thus be interesting to determine whether amyloid β accumulation also results in decreased PtdIns(4,5)P 2 levels in muscle fibers.
Newcomers
PtdIns can also be phosphorylated at position 3- of the inositol ring and this makes a total number of seven possible inter-convertible PtdInsP species. They are generated by an array of kinases and phosphatases that tightly regulate their respective amount and sub-cellular localization. The last 20 years have witnessed an overwhelming interest for the 3-phosphorylated PtdInsPs because of their implication in a broad array of cellular functions (e.g. Jean and Kiger 2014) and the potential therapeutic interest of targeting their metabolism in several types of diseases (Marone et al. 2008). Myotubularin (MTM1) is a PtdInsP 3-phosphatase that dephosphorylates PtdIns(3)P and PtdIns(3,5)P 2 (Blondeau et al. 2000; Tronchère et al. 2004), two PtdInsPs usually associated with membrane trafficking events in relation with the endosomal/lysosomal cell compartments (see Di Paolo and De Camilli 2006). Mutations in the MTM1 encoding gene are responsible for X-linked myotubular myopathy, a centronuclear myopathy phenotypically active from birth and characterized by deadly muscle weakness (Barth et al. 1975; De Angelis et al. 1991). A transgenic MTM1-deficient mouse model (Buj-Bello et al. 2002) was found to reproduce the main features of the human disease: following birth MTM1-deficient mice develop a severe progressive centronuclear myopathy with amyotrophy that considerably reduces their life expectancy. Examination of the ultrastructure and functional features of muscle fibers from 5 week-old MTM1-deficient mice revealed defects in t-tubule and triad organization and a severe impairment of EC coupling (Al-Qusairi et al. 2009): specifically, there was an increased ratio of longitudinal to total t-tubules and a decrease in the total number of tubules per Z line as compared to normal fibers. At the functional level, the peak amplitude of voltage-clamp depolarization-activated Ca2+ transients was reduced by a factor of more than 2 which is likely one main reason for the disease-associated loss of muscle force. The protein levels of CAV1.1 and RYR1 were reduced by a factor of 1.3 and 3 in MTM1-def0069cient fibers, respectively. This, in combination with the structural defects, likely contributes to the impairment of SR Ca2+ release and the decreased CAV1.1 and RYR1 may actually be thought to be a consequence of the structural disorganization. How MTM1 deficiency is responsible for these effects is still unclear. Tight regulation of PtdIns(3)P by MTM1 enzymatic activity was suggested to play a key role in SR remodeling, and the disruption of this mechanism to be a primary cause of the myopathy (Amoasii et al. 2013). However, triad defects are also a feature of the centronuclear myopathies linked to mutations in the genes encoding the proteins amphiphysin 2 (BIN1) and dynamin 2 (DNM2) (Toussaint et al. 2011) and this is also where a link with PtdInsPs is found. Indeed, BIN1 plays a role in membrane tubulation through a PtdInsP binding module (Lee et al. 2002). BIN1 knock-down in adult mouse muscle was achieved by Tjondrokoesoemo et al. (2011) through an shRNA strategy: muscle fibers expressing the shRNA-BIN1 plasmid suffered from disrupted t-tubule structures, from a 20 % depression of the peak amplitude of voltage-activated Ca2+ transients and from a reduced frequency of osmotic stress-induced Ca2+ sparks, highlighting the role of BIN1 in the maintenance of t-tubule structure and proper Ca2+ homeostasis. BIN1 is a binding partner of MTM1 and MTM1 promotes BIN1-mediated membrane tubulation (Royer et al. 2013) which underlies a possible common pathological mechanism in the two corresponding centronuclear myopathies. The importance of MTM1 and BIN1 in structure maintenance and EC coupling function was confirmed in zebrafish models (Dowling et al. 2009; Smith et al. 2014). As for DNM2, it is a protein partner of BIN1, and this partnership was suggested to be promoted upon PtdIns(4,5)P 2 clustering by BIN1 (Picas et al. 2014). Also quite convincing along this line is that decreased DNM2 expression rescues MTM1-related myopathy (Cowling et al. 2014). Finally, the neuronal Wiskott-Aldrich Syndrome Protein (N-WASP) was recently shown to interact with BIN1 and mutant forms of BIN1 and DNM2 (but not MTM1) associated with centronuclear myopathy were suggested to prevent accumulation of N-WASP at the triad (Falcone et al. 2014). Results also showed that triad disorganization due to down-regulation of BIN1 and DNM2 could be reverted by expression of active N-WASP. Interestingly, N-WASP is known to bind PtdIns(4,5)P 2 and to be activated by this interaction (Papayannopoulos et al. 2005). Altogether there is thus substantial evidence that t-tubule and triad maintenance in BIN1, MTM1, and DNM2 related myopathies may suffer from a common defect in relation with PtdIns(4,5)P 2 and with BIN1 making a link between MTM1 and DNM2 (Toussaint et al. 2011).
The role of MTM1 enzymatic activity in this framework is thus still uncertain because, on one hand, PtdIns(4,5)P 2 seems to be the preferential PdtInsP binding partner of BIN1 in the t-tubule membrane (although see also the model proposed by Fugier et al. 2011) and, on the other hand, there is no clear evidence that MTM1 substrates would be present and play a role in the t-tubule membrane of muscle fibers. This may be taken as one indication that, besides the contribution of t-tubule and triad defects to EC coupling failure in MTM1-deficient muscle fibers, the enzymatic activity of MTM1 could play another role in the disease. Along this line, substantial triad alterations are also observed in other muscular deficiency conditions (Nishi et al. 1999; Wu et al. 2012) and a decreased level of RYR1 is a hallmark of several human diseases due to RYR1 mutations (Monnier et al. 2003; Zhou et al. 2006, 2007, 2010) but these conditions are not ineludibly associated with a dramatic loss of force as observed in the absence of MTM1.
Also interesting in this context are studies from MTMR14 (or JUMPY)-deficient models. MTMR14 is a PtdInsP phosphatase that uses the same substrates as MTM1 (Tosch et al. 2006). Mice deficient in MTMR14 suffer from muscle weakness and fatigue (Shen et al. 2009) but, seemingly, with rather limited structural defects (Hnia et al. 2012). Furthermore, knockdown of MTMR14 in zebrafish impairs EC coupling with no associated structural defects (Dowling et al. 2010). This suggests that defective PtdInsP dephosphorylation may affect EC coupling through a mechanism independent of maintaining the structure. Finally, it is worth stressing that in MTM1-deficient muscle fibers resting [Ca2+], rate of cytosolic Ca2+ removal, and SR Ca2+ content are unchanged (Al-Qusairi et al. 2009), so the disease cannot be envisioned as a complete collapse of Ca2+ homeostasis: it is very specifically affecting RYR1-mediated Ca2+ release.
One possibility is that either the accumulation of MTM1 substrates or the deprivation of MTM1 products would have a more direct effect on EC coupling. For instance, Shen et al. (2009) reported that all PtdInsPs bind—to some extent—to RYR1 and that PtdIns(3,4)P 2, and PtdIns(3,5)P 2 directly activate its Ca2+ channel activity. They suggested that the increased level of these two PtdInsPs in MTMR14-deficient muscle is responsible for spontaneous SR Ca2+ leak. Conversely, Rodriguez et al. (2014) found that injection of PtdIns(3,5)P 2 or PtdIns(3)P into intact muscle fibers depresses voltage-clamp depolarization-activated SR Ca2+ release. Also, application of PtdIns(3,5)P 2 or PtdIns(3)P but also of PtdIns(5)P was found to lower the frequency of spontaneous Ca2+ sparks detected in permeabilized muscle fibers, with PtdIns(3,5)P2 completely annihilating their presence. Altogether, although these sets of functional data are not completely coherent and do not provide a simple picture, they highlight the potency of altered PtdInsP metabolism to directly affect EC coupling function, independently from structural effects. Further investigations will be needed to establish whether this contributes, and to what extent, to the altered EC coupling function in MTM1-deficient muscle.
Concluding remarks
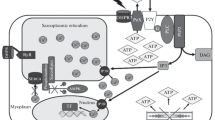
There is increasing evidence that PtdInsPs play a role in muscle Ca2+ signaling in differentiated muscle with SR Ca2+ release being a major target mechanism: this appears to occur through acute regulation of RYR-mediated SR Ca2+ release by t-tubule PtdIns(4,5)P 2 but also through existence of a correlated Ins(1,4,5)P 3-mediated Ca2+ release process with potential role in excitation-transcription coupling and in excitation-independent local SR Ca2+ release events. PtdIns(3)P and PtdIns(3,5)P 2 have now also entered the picture: their proper metabolism is critical for healthful maintenance of the membrane organization necessary for voltage-activated SR Ca2+ release. T-tubule PtdIns(4,5)P 2 is also a likely critical component of this maintenance process through a role in plasma membrane tubulation. How exactly the different PtdInsPs operate and cooperate in this remains to be clarified. In addition, there is also evidence that several PtdInsPs, including PtdIns(3)P and PtdIns(3,5)P 2 may directly affect RYR channel activity. Figure 1 summarizes these different mechanisms. Altogether, it is thus clear that EC coupling function requires well-regulated control of several PtdInsPs. Besides the structure maintenance issue and its role in centronuclear myopathies, one key question is whether physiological conditions that trigger changes in PtdInsPs levels—so as to transiently or chronically affect EC coupling—exist or not.

Role of phosphoinositides in Ca2+ signaling and excitation–contraction coupling (ECC). This schematic representation illustrates how PtdInsPs may act to regulate triad membrane structure and SR Ca2+ release in skeletal muscle. ETC, excitation-transcription coupling. InsP 3R, Ins(1,4,5)P 3 receptor
References
Allard B, Couchoux H, Pouvreau S, Jacquemond V (2006) Sarcoplasmic reticulum Ca2+ release and depletion fail to affect sarcolemmal ion channel activity in mouse skeletal muscle. J Physiol 575:69–81
Al-Qusairi L, Weiss N, Toussaint A, Berbey C, Messaddeq N, Kretz C, Sanoudou D, Beggs AH, Allard B, Mandel JL, Laporte J, Jacquemond V, Buj-Bello A (2009) T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci USA 106:18763–18768
Amoasii L, Hnia K, Chicanne G, Brech A, Cowling BS, Müller MM, Schwab Y, Koebel P, Ferry A, Payrastre B, Laporte J (2013) Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo. J Cell Sci 126:1806–1819
Araya R, Liberona JL, Cárdenas JC, Riveros N, Estrada M, Powell JA, Carrasco MA, Jaimovich E (2003) Dihydropyridine receptors as voltage sensors for a depolarization-evoked, IP3R-mediated, slow calcium signal in skeletal muscle cells. J Gen Physiol 121:3–16
Balghi H, Sebille S, Constantin B, Patri S, Thoreau V, Mondin L, Mok E, Kitzis A, Raymond G, Cognard C (2006a) Mini-dystrophin expression down-regulates overactivation of G protein-mediated IP3 signaling pathway in dystrophin-deficient muscle cells. J Gen Physiol 127:171–182
Balghi H, Sebille S, Mondin L, Cantereau A, Constantin B, Raymond G, Cognard C (2006b) Mini-dystrophin expression down-regulates IP3-mediated calcium release events in resting dystrophin-deficient muscle cells. J Gen Physiol 128:219–230
Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93:1019–10137
Barth PG, Van Wijngaarden GK, Bethlem J (1975) X-linked myotubular myopathy with fatal neonatal asphyxia. Neurology 25:531–536
Berman DE, Dall’Armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G (2008) Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci 11:547–554
Berridge MJ (2009) Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta 1793:933–940
Berthier C, Kutchukian C, Bouvard C, Okamura Y, Jacquemond V (2015) Depression of voltage-activated Ca2+ release in skeletal muscle by activation of a voltage-sensing phosphatase. J Gen Physiol 145:315–330
Blaauw B, Del Piccolo P, Rodriguez L, Hernandez Gonzalez VH, Agatea L, Solagna F, Mammano F, Pozzan T, Schiaffino S (2012) No evidence for inositol 1,4,5-trisphosphate-dependent Ca2+ release in isolated fibers of adult mouse skeletal muscle. J Gen Physiol 140:235–241
Blondeau F, Laporte J, Bodin S, Superti-Furga G, Payrastre B, Mandel JL (2000) Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet 9:2223–2229
Buj-Bello A, Laugel V, Messaddeq N, Zahreddine H, Laporte J, Pellissier JF, Mandel JL (2002) The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci USA 99:15060–15065
Burgess GM, Godfrey PP, McKinney JS, Berridge MJ, Irvine RF, Putney JW Jr (1984) The second messenger linking receptor activation to internal Ca release in liver. Nature 309:63–66
Carrasco MA, Sierralta J, Hidalgo C (1993) Phospholipase C activity in membranes and a soluble fraction isolated from frog skeletal muscle. Biochim Biophys Acta 1152:44–48
Carrasco MA, Riveros N, Ríos J, Müller M, Torres F, Pineda J, Lantadilla S, Jaimovich E (2003) Depolarization-induced slow calcium transients activate early genes in skeletal muscle cells. Am J Physiol 284:C1438–C1447
Casas M, Figueroa R, Jorquera G, Escobar M, Molgó J, Jaimovich E (2010) IP(3)-dependent, post-tetanic calcium transients induced by electrostimulation of adult skeletal muscle fibers. J Gen Physiol 136:455–467
Cheung KK, Ryten M, Burnstock G (2003) Abundant and dynamic expression of G protein-coupled P2Y receptors in mammalian development. Dev Dyn 228:254–266
Chu A, Stefani E (1991) Phosphatidylinositol 4,5-bisphosphate-induced Ca2+ release from skeletal muscle sarcoplasmic reticulum terminal cisternal membranes. Ca2+ flux and single channel studies. J Biol Chem 266:7699–7705
Cowling BS, Chevremont T, Prokic I, Kretz C, Ferry A, Coirault C, Koutsopoulos O, Laugel V, Romero NB, Laporte J (2014) Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest 124:1350–1363
Cseri J, Szappanos H, Szigeti GP, Csernátony Z, Kovács L, Csernoch L (2002) A purinergic signal transduction pathway in mammalian skeletal muscle cells in culture. Pflugers Arch 443:731–738
Csernoch L (2007) Sparks and embers of skeletal muscle: the exciting events of contractile activation. Pflugers Arch 454:869–878
De Angelis MS, Palmucci L, Leone M, Doriguzzi C (1991) Centronuclear myopathy: clinical, morphological and genetic characters. A review of 288 cases. J Neurol Sci 103:2–9
Deli T, Szappanos H, Szigeti GP, Cseri J, Kovács L, Csernoch L (2007) Contribution from P2X and P2Y purinoreceptors to ATP-evoked changes in intracellular calcium concentration on cultured myotubes. Pflugers Arch 453:519–529
Di Paolo Gl, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657
Dirksen RT (2009) Checking your SOCCs and feet: the molecular mechanisms of Ca2+ entry in skeletal muscle. J Physiol 587:3139–3147
Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, Szepetowski P, Pereira S, Mozaffar T, Escolar DM, Fu YH, Ptácek LJ (2003) PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 60:1811–1816
Dowling JJ, Vreede AP, Low SE, Gibbs EM, Kuwada JY, Bonnemann CG, Feldman EL (2009) Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet 5:e1000372
Dowling JJ, Low SE, Busta AS, Feldman EL (2010) Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum Mol Genet 19:2668–2681
Eltit JM, Hidalgo J, Liberona JL, Jaimovich E (2004) Slow calcium signals after tetanic electrical stimulation in skeletal myotubes. Biophys J 86:3042–3051
Eltit JM, García AA, Hidalgo J, Liberona JL, Chiong M, Lavandero S, Maldonado E, Jaimovich E (2006) Membrane electrical activity elicits inositol 1,4,5-trisphosphate-dependent slow Ca2+ signals through a Gbetagamma/phosphatidylinositol 3-kinase gamma pathway in skeletal myotubes. J Biol Chem 281:12143–12154
Estrada M, Cárdenas C, Liberona JL, Carrasco MA, Mignery GA, Allen PD, Jaimovich E (2001) Calcium transients in 1B5 myotubes lacking ryanodine receptors are related to inositol trisphosphate receptors. J Biol Chem 276:22868–22874
Estrada M, Espinosa A, Müller M, Jaimovich E (2003) Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology 144:3586–3597
Falcone S, Roman W, Hnia K, Gache V, Didier N, Lainé J, Auradé F, Marty I, Nishino I, Charlet-Berguerand N, Romero NB, Marazzi G, Sassoon D, Laporte J, Gomes ER (2014) N-WASP is required for Amphiphysin-2/BIN1-dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Mol Med 6:1455–1475
Flucher BE (2015) How is SR calcium release in muscle modulated by PIP(4,5)2? J Gen Physiol 145:361–364
Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, Lopez de Munain A, Sergeant N, Laquerrière A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 17:720–725
Hidalgo C, Carrasco MA, Magendzo K, Jaimovich E (1986) Phosphorylation of phosphatidylinositol by transverse tubule vesicles and its possible role in excitation-contraction coupling. FEBS Lett 202:69–73
Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC (2015) Phosphoinositides regulate ion channels. Biochim Biophys Acta 1851:844–856
Hnia K, Kretz C, Amoasii L, Böhm J, Liu X, Messaddeq N, Qu CK, Laporte J (2012) Primary T-tubule and autophagy defects in the phosphoinositide phosphatase Jumpy/MTMR14 knockout mice muscle. Adv Biol Regul 52:98–107
Inui M, Saito A, Fleischer S (1987) Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J Biol Chem 262:1740–1747
Jaimovich E, Reyes R, Liberona JL, Powell JA (2000) IP(3) receptors, IP(3) transients, and nucleus-associated Ca2+ signals in cultured skeletal muscle. Am J Physiol 278:C998–C1010
Jean S, Kiger AA (2014) Classes of phosphoinositide 3-kinases at a glance. J Cell Sci 127:923–928
Jorquera G, Altamirano F, Contreras-Ferrat A, Almarza G, Buvinic S, Jacquemond V, Jaimovich E, Casas M (2013) Cav1.1 controls frequency-dependent events regulating adult skeletal muscle plasticity. J Cell Sci 126:1189–1198
Juretić N, García-Huidobro P, Iturrieta JA, Jaimovich E, Riveros N (2006) Depolarization-induced slow Ca2+ transients stimulate transcription of IL-6 gene in skeletal muscle cells. Am J Physiol 290:C1428–C1436
Kaur G, Pinggera A, Ortner NJ, Lieb A, Sinnegger-Brauns MJ, Yarov-Yarovoy V, Obermair GJ, Flucher BE, Striessnig J (2015) A polybasic plasma membrane binding motif in the I-II linker stabilizes voltage-gated cav1.2 calcium channel function. J Biol Chem 290:21086–21100
Kim YJ, Hernandez ML, Balla T (2013) Inositol lipid regulation of lipid transfer in specialized membrane domains. Trends Cell Biol 23:270–278
Kobayashi M, Muroyama A, Ohizumi Y (1989) Phosphatidylinositol 4,5-bisphosphate enhances calcium release from sarcoplasmic reticulum of skeletal muscle. Biochem Biophys Res Commun 163:1487–1491
Launikonis BS, Barnes M, Stephenson DG (2003) Identification of the coupling between skeletal muscle store-operated Ca2+ entry and the inositol trisphosphate receptor. Proc Natl Acad Sci USA 100:2941–2944
Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, De Camilli P (2002) Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297:1193–1196
Liberona JL, Cárdenas JC, Reyes R, Hidalgo J, Molgó J, Jaimovich E (2008) Sodium-dependent action potentials induced by brevetoxin-3 trigger both IP3 increase and intracellular Ca2+ release in rat skeletal myotubes. Cell Calcium 44:289–297
Lopez JR, Shtifman A (2010) Intracellular β-amyloid accumulation leads to age-dependent progression of Ca2+ dysregulation in skeletal muscle. Muscle Nerve 42:731–738
Marone R, Cmiljanovic V, Giese B, Wymann MP (2008) Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 1784:159–185
May C, Weigl L, Karel A, Hohenegger M (2006) Extracellular ATP activates ERK1/ERK2 via a metabotropic P2Y1 receptor in a Ca2+ independent manner in differentiated human skeletal muscle cells. Biochem Pharmacol 71:1497–1509
Meissner G (1986) Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J Biol Chem 261:6300–6306
Milting H, Heilmeyer LM Jr, Thieleczek R (1996) Cloning of a phospholipase C-delta 1 of rabbit skeletal muscle. J Muscle Res Cell Motil 17:79–84
Monnier N, Ferreiro A, Marty I, Labarre-Vila A, Mezin P, Lunardi J (2003) A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Hum Mol Genet 12:1171–1178
Nishi M, Komazaki S, Kurebayashi N, Ogawa Y, Noda T, Iino M, Takeshima H (1999) Abnormal features in skeletal muscle from mice lacking mitsugumin29. J Cell Biol 147:1473–1480
Ogawa Y, Harafuji H (1989) Ca-release by phosphoinositides from sarcoplasmic reticulum of frog skeletal muscle. J Biochem 106:864–867
Ohizumi Y, Hirata Y, Suzuki A, Kobayashi M (1999) Two novel types of calcium release from skeletal sarcoplasmic reticulum by phosphatidylinositol 4,5-biphosphate. Can J Physiol Pharmacol 77:276–285
Papayannopoulos V, Co C, Prehoda KE, Snapper S, Taunton J, Lim WA (2005) A polybasic motif allows N-WASP to act as a sensor of PIP2 density. Mol Cell 17:181–191
Picas L, Viaud J, Schauer K, Vanni S, Hnia K, Fraisier V, Roux A, Bassereau P, Gaits-Iacovoni F, Payrastre B, Laporte J, Manneville JB, Goud B (2014) BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat Commun 5:5647
Powell JA, Carrasco MA, Adams DS, Drouet B, Rios J, Müller M, Estrada M, Jaimovich E (2001) IP(3) receptor function and localization in myotubes: an unexplored Ca2+ signaling pathway in skeletal muscle. J Cell Sci 114:3673–3683
Ríos E, Pizarro G, Stefani E (1992) Charge movement and the nature of signal transduction in skeletal muscle excitation-contraction coupling. Annu Rev Physiol 54:109–133
Rodríguez EG, Lefebvre R, Bodnár D, Legrand C, Szentesi P, Vincze J, Poulard K, Bertrand-Michel J, Csernoch L, Buj-Bello A, Jacquemond V (2014) Phosphoinositide substrates of myotubularin affect voltage-activated Ca2+ release in skeletal muscle. Pflugers Arch 466:973–985
Royer B, Hnia K, Gavriilidis C, Tronchère H, Tosch V, Laporte J (2013) The myotubularin-amphiphysin 2 complex in membrane tubulation and centronuclear myopathies. EMBO Rep 14:907–915
Ryten M, Yang SY, Dunn PM, Goldspink G, Burnstock G (2004) Purinoceptor expression in regenerating skeletal muscle in the mdx mouse model of muscular dystrophy and in satellite cell cultures. FASEB J 18:1404–1416
Schneider MF (1994) Control of calcium release in functioning skeletal muscle fibers. Annu Rev Physiol 56:463–484
Shen J, Yu WM, Brotto M, Scherman JA, Guo C, Stoddard C, Nosek TM, Valdivia HH, Qu CK (2009) Deficiency of MIP/MTMR14 phosphatase induces a muscle disorder by disrupting Ca2+ homeostasis. Nat Cell Biol 11:769–776
Shtifman A, Ward CW, Laver DR, Bannister ML, Lopez JR, Kitazawa M, LaFerla FM, Ikemoto N, Querfurth HW (2010) Amyloid-β protein impairs Ca2+ release and contractility in skeletal muscle. Neurobiol Aging 31:2080–2090
Smith LL, Gupta VA, Beggs AH (2014) Bridging integrator 1 (Bin1) deficiency in zebrafish results in centronuclear myopathy. Hum Mol Genet 23:3566–3578
Sun Y, Thapa N, Hedman AC, Anderson RA (2013) Phosphatidylinositol 4,5-bisphosphate: targeted production and signaling. BioEssays 35:513–522
Szigeti GP, Szappanos H, Deli T, Cseri J, Kovács L, Csernoch L (2007) Differentiation-dependent alterations in the extracellular ATP-evoked calcium fluxes of cultured skeletal muscle cells from mice. Pflugers Arch 453:509–518
Tjondrokoesoemo A, Park KH, Ferrante C, Komazaki S, Lesniak S, Brotto M, Ko JK, Zhou J, Weisleder N, Ma J (2011) Disrupted membrane structure and intracellular Ca2+ signaling in adult skeletal muscle with acute knockdown of Bin1. PLoS One 6:e25740
Tjondrokoesoemo A, Li N, Lin PH, Pan Z, Ferrante CJ, Shirokova N, Brotto M, Weisleder N, Ma J (2013) Type 1 inositol (1,4,5)-trisphosphate receptor activates ryanodine receptor 1 to mediate calcium spark signaling in adult mammalian skeletal muscle. J Biol Chem 288:2103–2109
Tosch V, Rohde HM, Tronchère H, Zanoteli E, Monroy N, Kretz C, Dondaine N, Payrastre B, Mandel JL, Laporte J (2006) A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum Mol Genet 15:3098–3106
Toussaint A, Cowling BS, Hnia K, Mohr M, Oldfors A, Schwab Y, Yis U, Maisonobe T, Stojkovic T, Wallgren-Pettersson C, Laugel V, Echaniz-Laguna A, Mandel JL, Nishino I, Laporte J (2011) Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol 121:253–266
Tronchère H, Laporte J, Pendaries C, Chaussade C, Liaubet L, Pirola L, Mandel JL, Payrastre B (2004) Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin in mammalian cells. J Biol Chem 279:7304–7312
Valdés JA, Hidalgo J, Galaz JL, Puentes N, Silva M, Jaimovich E, Carrasco MA (2007) NF-kappaB activation by depolarization of skeletal muscle cells depends on ryanodine and IP3 receptor-mediated calcium signals. Am J Physiol 292:C1960–C1970
Várnai P, Balla T (1998) Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol 143:501–510
Vergara J, Tsien RY, Delay M (1985) Inositol 1,4,5-trisphosphate: a possible chemical link in excitation-contraction coupling in muscle. Proc Natl Acad Sci USA 82:6352–6356
Walker JW, Somlyo AV, Goldman YE, Somlyo AP, Trentham DR (1987) Kinetics of smooth and skeletal muscle activation by laser pulse photolysis of caged inositol 1,4,5-trisphosphate. Nature 327:249–252
Wang X, Weisleder N, Collet C, Zhou J, Chu Y, Hirata Y, Zhao X, Pan Z, Brotto M, Cheng H, Ma J (2005) Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat Cell Biol 7:525–530
Wu F, Mi W, Hernández-Ochoa EO, Burns DK, Fu Y, Gray HF, Struyk AF, Schneider MF, Cannon SC (2012) A calcium channel mutant mouse model of hypokalemic periodic paralysis. J Clin Invest 122:4580–4591
Yamamoto M, Chen MZ, Wang YJ, Sun HQ, Wei Y, Martinez M, Yin HL (2006) Hypertonic stress increases phosphatidylinositol 4,5-bisphosphate levels by activating PIP5KIbeta. J Biol Chem 281:32630–32638
Zhou H, Yamaguchi N, Xu L, Wang Y, Sewry C, Jungbluth H, Zorzato F, Bertini E, Muntoni F, Meissner G, Treves S (2006) Characterization of recessive RYR1 mutations in core myopathies. Hum Mol Genet 15:2791–2803
Zhou H, Jungbluth H, Sewry CA, Feng L, Bertini E, Bushby K, Straub V, Roper H, Rose MR, Brockington M, Kinali M, Manzur A, Robb S, Appleton R, Messina S, D’Amico A, Quinlivan R, Swash M, Müller CR, Brown S, Treves S, Muntoni F (2007) Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 130:2024–2036
Zhou H, Lillis S, Loy RE, Ghassemi F, Rose MR, Norwood F, Mills K, Al-Sarraj S, Lane RJ, Feng L, Matthews E, Sewry CA, Abbs S, Buk S, Hanna M, Treves S, Dirksen RT, Meissner G, Muntoni F, Jungbluth H (2010) Multi-minicore disease and atypical periodic paralysis associated with novel mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord 20:166–173
Acknowledgments
This work was supported by grants from Centre National de la Recherche Scientifique (CNRS) and Université Lyon 1 to Centre de Génétique et de Physiologie Moléculaire et Cellulaire, by a grant from Association Française contre les Myopathies to V.J. (AFM # 18648), by the Hungarian National Science Fund (NN-107765) to L.C. and by the European Union, co-financed by the European Social Fund (TÁMOP-4.1.2.E-13/1/KONV-2013-0010).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Csernoch, L., Jacquemond, V. Phosphoinositides in Ca2+ signaling and excitation–contraction coupling in skeletal muscle: an old player and newcomers. J Muscle Res Cell Motil 36, 491–499 (2015). https://doi.org/10.1007/s10974-015-9422-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-015-9422-4