
Abstract
Depolarization of the plasma membrane is a key mechanism of activation of contraction of vascular smooth muscle. This is commonly achieved in isolated, de-endothelialized vascular smooth muscle strips by increasing extracellular [K+] (replacing Na+ by K+) and leads to a rapid phasic contraction followed by a sustained tonic contraction. The initial phasic contractile response is due to opening of voltage-gated Ca2+ channels and entry of extracellular Ca2+, which binds to calmodulin, leading to activation of myosin light chain kinase, phosphorylation of the regulatory light chains of myosin II at Ser19 and cross-bridge cycling. The subsequent tonic contractile response involves, in addition to myosin light chain kinase activation, Ca2+-induced Ca2+ sensitization whereby Ca2+ entry activates the RhoA/Rho-associated kinase pathway leading to phosphorylation of MYPT1 (the myosin targeting subunit of myosin light chain phosphatase) and inhibition of the phosphatase. Investigations into the mechanism of activation of RhoA by Ca2+ have implicated a genistein-sensitive tyrosine kinase, and recent evidence indicates this to be the Ca2+-dependent tyrosine kinase, Pyk2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Depolarization-induced contraction of vascular smooth muscle
Depolarization of the plasma membrane evoked by neurotransmitter release is an important mechanism to elicit vascular smooth muscle contraction and thereby regulate blood flow to downstream tissues and organs. A typical contractile response to membrane depolarization is illustrated in Fig. 1. In this case, depolarization of de-endothelialized helical strips of the rat caudal artery was achieved by replacing extracellular NaCl by equimolar KCl, a common method to change the K+ equilibrium potential and clamp membrane potential above the resting level (Bolton 1979). In order to focus on events specific to the vascular smooth muscle cells, the problem of K+-induced activation of neurotransmitter release is avoided either by subjecting the preparation to several K+-induced contraction-relaxation cycles prior to evoking the test contraction, or by inclusion in the bathing solution of antagonists, most importantly α- and β-adrenoceptor antagonists (Mita and Walsh 1997; Ratz et al. 2005). K+-induced depolarization of the vascular smooth muscle sarcolemma opens predominantly voltage-gated L-type Ca2+ channels to allow Ca2+ to enter the cytosol down its electrochemical gradient, thereby increasing cytosolic free Ca2+ concentration ([Ca2+]i) from ~130 to ~700 nM (Williams and Fay 1986; Williams et al. 1987). Ca2+ binds to a specific pool of calmodulin (CaM) that is constitutively bound to the contractile machinery, inducing a conformational change in the Ca2+-binding protein, which interacts with the CaM-binding domain of smooth muscle myosin light chain kinase (MLCK) (Wilson et al. 2002). This interaction induces a conformational change in MLCK with removal of an autoinhibitory domain from the active site, thereby providing access to the substrate, the 20-kDa regulatory light chains (LC20) of myosin II. LC20 phosphorylation is facilitated by the proximity of kinase and substrate: MLCK, an elongated molecule, is anchored via three N-terminal actin-binding sites (with the sequence DFRxxL), and the active site, which is located near the C-terminus, interacts with and phosphorylates Ser19 of LC20, located in the neck region of the myosin motor (Stull et al. 1998; Mabuchi et al. 2010; Hong et al. 2011; Sutherland and Walsh 2012). LC20 phosphorylation abolishes asymmetric intramolecular interactions between the two myosin heads, thereby relieving inhibition of the MgATPase activity and enabling myosin cross-bridge interaction with actin and relative sliding of actin and myosin filaments driven by the energy derived from the hydrolysis of ATP (Wu et al. 1999; Wendt et al. 2001; Baumann et al. 2012; Taylor et al. 2014). At the level of the muscle strip, this is reflected by an increase in isometric force (as seen in Fig. 1) or shortening of the muscle under isotonic conditions.

Membrane depolarization-induced contraction of de-endothelialized rat caudal arterial smooth muscle. Caudal arteries were removed from male Sprague–Dawley rats (300–350 g) that had been anesthetized with halothane and euthanized according to protocols consistent with the standards of the Canadian Council on Animal Care and approved by the University of Calgary Animal Care and Use Committee. The arteries were cleaned of excess adventitia and adipose tissue in Ca2+-free HEPES-Tyrode’s solution (140.6 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 5.6 mM glucose, 10 mM HEPES, pH 7.4). Segments were placed over a 0.31-mm needle and moved back and forth 40 times to remove the endothelium, cut into helical strips (1.5 × 6 mm), mounted on a Grass isometric force transducer (model FT03C) connected to a PowerLab (ADInstruments) 8-channel recording device with a resting tension of 0.45 g, and incubated for 20 min in HEPES-Tyrode’s solution (137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5.6 mM glucose, 10 mM HEPES, pH 7.4) in a bath volume of 0.8 ml. The tissue was stimulated several times (for 1.5 min each) with HEPES-Tyrode’s solution containing 87 mM KCl (the last of these is indicated by the first bar labeled “K+”; the increase in KCl concentration was balanced by a decrease in NaCl concentration). The tissue relaxed upon return to HEPES-Tyrode’s solution and was then stimulated for 30 min with K+ prior to relaxation again in HEPES-Tyrode’s solution. The authors are grateful for the technical assistance of Ms Cindy Sutherland
Ca2+-induced Ca2+ sensitization in the tonic phase of depolarization-induced contraction
Examination of the prolonged time course of the contractile response to membrane depolarization indicates that, following the initial phasic contraction, force declines to a steady-state level that is sustained until removal of the depolarizing stimulus, whereupon force returns to resting levels (Fig. 1). Ca2+ entry and MLCK activity are required for both phasic and tonic components of depolarization-induced contraction since both phases are inhibited by removal of extracellular Ca2+ or pre-treatment with the L-type Ca2+ channel blocker, nicardipine, or the MLCK inhibitor, ML-9 (Mita et al. 2002). The time course of LC20 phosphorylation corresponds closely to that of force (Mita et al. 2002). The observation that protein kinase inhibitors, such as H-7 and HA-1077 (known at the time as protein kinase C (PKC) inhibitors), relaxed K+-induced contractions of vascular smooth muscle suggested the involvement of protein kinases other than MLCK in the depolarization-induced contractile response (Ratz 1990; Takizawa et al. 1993). Following the discovery of Rho-associated coiled-coil kinase (ROCK) (Leung et al. 1995; Uehata et al. 1997), it was recognized that these relaxant effects were due to inhibition of ROCK rather than PKC (Feng et al. 1999a; Uehata et al. 1997). Furthermore, studies with canine coronary arterial smooth muscle had previously indicated that membrane depolarization by K+ induced Ca2+ sensitization of contraction (Yanagisawa and Okada 1994). We demonstrated that pre-treatment of rat caudal arterial smooth muscle strips with inhibitors of ROCK, specifically Y-27632 and HA-1077, eliminated the tonic, but not the phasic contractile response to K+ (Mita et al. 2002) (Fig. 2a). This was confirmed in several subsequent studies with different vascular smooth muscle preparations (Sakamoto et al. 2003; Sakurada et al. 2003; Urban et al. 2003; Ratz et al. 2005; Seok et al. 2008). The loss of tonic contraction in the presence of ROCK inhibitors was explained by loss of the sustained increase in LC20 phosphorylation (Fig. 2b) with no effect on [Ca2+]i (Mita et al. 2002).

The effect of Rho-associated kinase inhibition on K+-induced contraction and LC20 phosphorylation in rat caudal arterial smooth muscle. After stable K+-induced contractions were achieved, muscle strips were incubated in HEPES-Tyrode’s solution at resting tension in the absence (filled circle) or presence (open circle) of Y-27632 (3 µM) for 20 min prior to K+ (60 mM) depolarization at time zero. a Force development is expressed as a percentage of the maximal force of the phasic contraction developed in response to K+ in the absence of ROCK inhibitor. Values indicate the mean ± SEM (n = 4). b Tissues treated as in a and harvested at the indicated times following K+ depolarization were immersed in 10 % trichloroacetic acid/10 mM dithiothreitol (DTT) in dry ice/acetone for 10 min. Frozen muscle strips were then washed with 10 mM DTT in acetone at 24 °C. The tissues were freeze-dried overnight and stored at −80 °C until LC20 extraction was carried out. Proteins were extracted from tissue strips in 30 µl of extraction buffer (6 M deionized urea, 20.1 mM Tris, 22.2 mM glycine, 10 mM DTT, 10 mM EGTA, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 0.6 M KI and 0.15 mM bromophenol blue) by constant rotation in a microcentrifuge tube for 90 min at 24 °C. After filtration and centrifugation, phosphorylated and unphosphorylated forms of LC20 were separated by urea/glycerol gel electrophoresis and transblotted to 0.2-µm nitrocellulose membrane in 10 mM sodium cyclohexylaminopropanesulfonic acid, pH 11, in a Mini Transblot Cell at 27 V and 5 °C for 16 h. LC20 was detected using a polyclonal antibody raised in rabbits against purified chicken gizzard LC20 (Mita and Walsh 1997; Weber et al. 1999), which recognizes phosphorylated and unphosphorylated LC20, with enhanced chemiluminescence detection. LC20 bands were quantified by densitometric scanning using a Pharmacia Image Master Desktop Scanning System. LC20 phosphorylation levels were calculated by dividing the chemiluminescence signal (absorbance × area of the band) of the phosphorylated LC20 peak by the total chemiluminescence signal of the phosphorylated and unphosphorylated LC20 peaks. The inset in b shows, with an expanded time-scale, the profiles of LC20 phosphorylation during the first 2 min following depolarization. Values indicate the mean ± SEM (n = 5 for the results in the absence of Y-27632 and n = 6 for the results in the presence of Y-27632). Statistically significant differences from control (no Y-27632) are indicated by asterisks: *p < 0.01, **p < 0.02, ***p < 0.05. This research was originally published in the Biochemical Journal. Mita et al. (2002) © the Biochemical Journal
It was already known from studies with contractile agonists that act via 7-transmembrane domain-containing G protein-coupled receptors (GPCRs), such as α1-adrenergic, thromboxane A2, serotonin, histamine and endothelin-1 receptors, that tonic contractile responses to such stimuli involve activation of the small GTPase, RhoA (Sakurada et al. 2001; Somlyo and Somlyo 2000). Replacement of RhoA-bound GDP by GTP results in activation of ROCK leading to inhibition of myosin light chain phosphatase (MLCP) via phosphorylation of the myosin targeting subunit of the phosphatase (MYPT1) at Thr697 and Thr855 (rat numbering) (Ichikawa et al. 1996; Feng et al. 1999b; Velasco et al. 2002; Murányi et al. 2005) or the PKC-potentiated inhibitory protein for protein phosphatase-1 of 17 kDa (CPI-17) at Thr38 (Koyama et al. 2000; Kitazawa et al. 2003). It should be noted, however, that expressed MLCP reconstituted in mammalian cells was inhibited by thiophosphorylation at Thr697 but not at Thr855 (Khasnis et al. 2014). Furthermore, using two mouse lines with Thr697 or Thr855 substituted with Ala, phosphorylation at Thr697 but not at Thr855 was found to mediate force maintenance via inhibition of MLCP activity and enhancement of LC20 phosphorylation in bladder smooth muscle in vivo (Chen et al. 2015). In addition, most contractile stimuli acting via GPCRs induce an increase in MYPT1 phosphorylation at Thr855 but not at Thr697 (e.g. Borysova et al. 2011; Kitazawa and Kitazawa 2012). The complete pathway of Ca2+ sensitization of contraction induced by such agonists is depicted in Fig. 3.

Agonist-induced Ca2+ sensitization of vascular smooth muscle contraction. A variety of contractile agonists act via 7-transmembrane domain-containing G protein-coupled receptors that are coupled to the G12/13 family of heterotrimeric G proteins. Ligand occupancy of these receptors activates G12/13, which in turn activates a RhoA guanine nucleotide exchange factor (RhoGEF). The activated RhoGEF converts inactive cytosolic RhoA, which is bound to a guanine nucleotide dissociation inhibitor (RhoGDI) and contains bound GDP, to active, membrane-bound RhoA-GTP. Dissociation of RhoA from RhoGDI exposes a geranylgeranyl moiety, which anchors activated RhoA to the plasma membrane. The principal target of RhoA-GTP is Rho-associated coiled-coil kinase (ROCK). Activated ROCK inhibits myosin light chain phosphatase (MLCP) activity by phosphorylating the myosin targeting subunit of the phosphatase (MYPT1) at Thr697 and Thr855 (rat numbering) and/or CPI-17 (protein kinase C-potentiated inhibitory protein for protein phosphatase-1 of 17 kDa) at Thr38. MLCP inhibition shifts the MLCK:MLCP activity ratio in favor of the kinase, resulting in an increase in LC20 phosphorylation at Ser19 and force
It is also clear from the data in Fig. 2a that the rate of relaxation after the peak of the contractile response to K+ is greater in the presence of a ROCK inhibitor than in the control (absence of inhibitor), consistent with ROCK inhibition preventing MYPT1 phosphorylation, thereby resulting in increased MLCP activity.
Depolarization-induced activation of the RhoA/ROCK pathway
These findings prompted us to investigate the molecular mechanisms underlying force maintenance during the tonic phase of K+-induced contraction. RhoA activation involves its translocation from the cytosol, where it exists in an inactive GDP-bound form in association with a guanine nucleotide dissociation inhibitor (RhoGDI), to the plasma membrane as free GTP-bound RhoA where it inserts into the membrane via its geranylgeranyl moiety (Gong et al. 1997; Somlyo and Somlyo 2000). Previous studies demonstrated that RhoA translocation is induced by K+ stimulation of rabbit renal artery, rabbit aorta and rat aorta (Sakurada et al. 2003; Urban et al. 2003; Seok et al. 2008) and the K+-induced activation of RhoA is inhibited by a CaM antagonist, suggesting the involvement of Ca2+ and CaM in RhoA activation (Sakurada et al. 2003). We demonstrated that K+ stimulation triggered the translocation of RhoA from the cytosolic to the particulate fraction and phosphorylation of MYPT1 at Thr697 and Thr855 on a time-scale that corresponded to the tonic rather than the phasic component of the contractile response (Mita et al. 2013). Pre-treatment with the ROCK inhibitor, Y-27632, prevented the K+-induced phosphorylation of MYPT1 at both Thr697 and Thr855, consistent with depolarization-induced RhoA translocation and activation leading to ROCK activation, phosphorylation of MYPT1 at the two ROCK sites, inhibition of MLCP activity, sustained LC20 phosphorylation and tonic contraction.
Involvement of a genistein-sensitive tyrosine kinase in the tonic contractile response to depolarization
But how is RhoA activated in response to membrane depolarization? It has long been known that tyrosine phosphorylation plays a role in smooth muscle contraction (Di Salvo et al. 1994; Hollenberg 1994; Laniyonu et al. 1994; Sasaki et al. 1998; Adegunloye et al. 2003). For example, tyrosine kinase inhibitors were shown to attenuate agonist-induced contraction and LC20 phosphorylation in intact smooth muscle (Di Salvo et al. 1993a) and to inhibit agonist-induced augmentation of Ca2+-induced contraction in permeabilized smooth muscle (Steusloff et al. 1995). Furthermore, receptor-mediated tyrosine kinase activation has been implicated in Ca2+ sensitization via activation of the RhoA/ROCK pathway in diverse smooth muscle tissues (Steusloff et al. 1995; Sakurada et al. 2001; Nakao et al. 2002; Seok et al. 2008). We considered the possibility, therefore, that a tyrosine kinase, acting upstream of RhoA, may be activated by depolarization. Pre-treatment of de-endothelialized rat caudal arterial smooth muscle strips with the non-selective tyrosine kinase inhibitor, genistein, but not the inactive analog, genistin, or the Src family tyrosine kinase inhibitor, PP2, abolished the tonic component of K+-induced contraction, while having a partial inhibitory effect on the phasic contractile response to depolarization (Mita et al. 2013). Addition of genistein once steady-state K+-induced force had been attained resulted in concentration-dependent relaxation (Mita et al. 2013). Furthermore, the K+-induced translocation of RhoA, phosphorylation of MYPT1 at Thr697 and Thr855, and sustained elevation of LC20 phosphorylation were also abolished by genistein at a concentration (10 µM) that had no effect on the K+-induced increase in [Ca2+]i (Mita et al. 2013).
The general tyrosine phosphatase inhibitor, sodium orthovanadate, induced a slow, sustained contraction of smooth muscle (Shimada et al. 1986; Di Salvo et al. 1993b; Masui and Wakabayashi 2000; Mori and Tsushima 2004), supporting a role for tyrosine kinase activity in tonic contraction, and this response was reversed by genistein treatment of rat caudal arterial smooth muscle that had been pre-contracted with vanadate in a concentration-dependent manner (Mita et al. 2013). The ROCK inhibitor, Y-27632, also reversed the vanadate-induced contraction. LC20 phosphorylation accompanied vanadate-induced contraction and was also sensitive to genistein and Y-27632 (Mita et al. 2013). These observations are consistent with ROCK being located downstream of the tyrosine kinase.
Identification of the tyrosine kinase as Pyk2
We considered the possibility, therefore, that sustained contraction induced by membrane depolarization may be mediated by Ca2+-dependent activation of a genistein-sensitive tyrosine kinase. A survey of the literature revealed the non-receptor proline-rich tyrosine kinase, Pyk2, also known as focal adhesion kinase 2 (FAK2), cell adhesion kinase β (CAKβ), related adhesion focal tyrosine kinase (RAFTK) and calcium-dependent protein tyrosine kinase (CADTK) (Lipinski and Loftus 2010), as the best candidate since it is regulated by Ca2+ (Yu et al. 1996; Avraham et al. 2000). It was also known to be expressed in vascular smooth muscle (Sabri et al. 1998). A useful tool to study Pyk2 function is sodium salicylate; well known as a cyclooxygenase inhibitor, salicylate is rather less potent, but quite selective, as an inhibitor of Pyk2 (Wang and Brecher 2001). Sodium salicylate was shown to induce vasodilation via inhibition of the RhoA/ROCK/MYPT1 pathway and decreased blood pressure in spontaneously hypertensive but not normotensive (Wistar Kyoto) rats (Ying et al. 2009a), i.e. salicylate has similar effects to ROCK inhibitors (Uehata et al. 1997). Furthermore, basal and angiotensin II-induced Pyk2 phosphorylation (activation) were greater in vascular smooth muscle cells from spontaneously hypertensive rats compared to Wistar Kyoto rats (Rocic et al. 2002). We demonstrated that salicylate reversed K+-induced rat caudal arterial smooth muscle contraction in a concentration-dependent manner, and pre-treatment of the tissue with salicylate eliminated the tonic component of depolarization-induced contraction without affecting the phasic contractile response (Fig. 4a) (Mills et al. 2015). Likewise, salicylate abolished the sustained elevation of LC20 phosphorylation without affecting the phasic K+-induced LC20 phosphorylation (Fig. 4b, c) (Mills et al. 2015). As observed for ROCK inhibition (Fig. 2a), salicylate increased the rate of relaxation following the peak of K+-induced force (Fig. 4a), consistent with enhanced MLCP activity.

The effect of sodium salicylate on K+-induced contraction and LC20 phosphorylation in rat caudal arterial smooth muscle. The time courses of K+-induced contraction (a) and LC20 phosphorylation (b, c) following pre-incubation for 30 min with vehicle (filled circle) or sodium salicylate (10 mM) (open circle). a Force is expressed as a percentage of the maximal force of the phasic contraction induced by K+ in the absence of salicylate. Values indicate the mean ± SEM (n = 8). b and c Tissues were quick-frozen at the indicated times following membrane depolarization for analysis of LC20 phosphorylation by Phos-tag SDS-PAGE, which separates phosphorylated and unphosphorylated forms of LC20 (Takeya et al. 2008). Phosphorylation stoichiometry was determined by densitometric scanning of the unphosphorylated and phosphorylated bands detected with anti-LC20. Values represent the mean ± SEM (n = 4–5). *p < 0.05 and **p < 0.01, significantly different from the level of LC20 phosphorylation in the absence of salicylate. c An expanded version of the first 2 min of the time courses depicted in (b). This research was originally published in the Journal of Biological Chemistry. Mills et al. (2015) © the American Society for Biochemistry and Molecular Biology
Although the mechanism of activation of Pyk2 is not known in detail, the FERM domain has been suggested to mediate Ca2+/CaM-dependent Pyk2 homodimerization (via binding of CaM to the α2-helix of the F2 subdomain) and transphosphorylation at Tyr402 to recruit Src, which binds via its SH2 domain and phosphorylates Pyk2 at Tyr579 and Tyr580 within the activation loop of the kinase domain to generate maximal Pyk2 activity (Kohno et al. 2008). Thus Pyk2 activation can be assessed in tissue by analysis of its autophosphorylation at Tyr402. Using phosphospecific antibodies to Pyk2-pTyr402, we observed that Pyk2 is activated in response to membrane depolarization with a time course that corresponds to the sustained contractile response (Mills et al. 2015).
The catalytic domain of Pyk2 is ~60 % identical in sequence to that of FAK (Sasaki et al. 1995); however, FAK activity is not Ca2+ dependent (Han et al. 2009). Nevertheless, it was important to determine if FAK plays a role in the depolarization-induced sustained contraction. A variety of FAK/Pyk2 inhibitors are available and can be used to distinguish between these two tyrosine kinases due to their relative potencies towards the two enzymes. We found a close correlation between the ability of these inhibitors to inhibit K+-induced sustained contraction and autophosphorylation (activation) of Pyk2 (Fig. 5a), but not FAK (Fig. 5b) (Mills et al. 2015). On this basis, we concluded that, although both Pyk2 and FAK are activated in response to membrane depolarization, it is Pyk2 and not FAK that is responsible for activation of the RhoA/ROCK pathway and the sustained contractile response to K+.

The relationship between inhibition of Pyk2 or FAK autophosphorylation and sustained K+-induced contraction for various FAK/Pyk2 inhibitors. Rat caudal arterial smooth muscle strips were pre-incubated for 30 min with PF-431396 (#1), PF-573228 (#2), PF-562271 (#3), NVP-TAE226 (#4) or sc-203950 (#5) (2 or 10 μM) or vehicle (#6) and then stimulated with K+ in the continued presence of inhibitor. Force was recorded continuously and values at 10 min after K+ addition were calculated relative to K+-induced contractions in the absence of inhibitor. Values for untreated tissue are also included (#7). Tissues were quick-frozen 10 min after the addition of K+ for analysis of Pyk2 autophosphorylation by western blotting with anti-Pyk2-pTyr402 and FAK autophosphorylation with anti-FAK-pTyr397. Actin was used as the loading control and anti-Pyk2 and anti-FAK were used to verify the total levels of the two kinases. The relationship between Pyk2 (a) and FAK (b) autophosphorylation and the magnitude of the tonic K+-induced contractile response are shown. Values represent the mean ± SEM (n = 4 (a) and n = 3 (b)). A close correlation (r2 = 0.87) was found between the ability of these inhibitors to inhibit Pyk2 autophosphorylation and K+-induced contraction. This was not true for FAK (r2 = 0.44). This research was originally published in the Journal of Biological Chemistry. Mills et al. (2015) © the American Society for Biochemistry and Molecular Biology
Western blotting with anti-phosphotyrosine revealed several phosphotyrosine-containing proteins in rat caudal arterial smooth muscle strips maintained at resting tension, the most prominent being a protein of ~135 kDa, which co-migrated with Pyk2 (identified by western blotting with several different antibodies) (Mills et al. 2015). Tyrosine phosphorylation of this protein was markedly enhanced by membrane depolarization and prevented by pre-treatment with the Pyk2/FAK inhibitor, PF-431396. FAK, on the other hand, was readily separated from Pyk2 by SDS-PAGE and was a minor tyrosine-phosphorylated protein at rest or following membrane depolarization. Vanadate also induced Pyk2 autophosphorylation at Tyr402 with a slow time course comparable to that of force development, and both vanadate-induced Pyk2 autophosphorylation and contraction were blocked by pre-treatment with PF-431396 (Mills et al. 2015). All these data support the conclusion that Pyk2 is the tyrosine kinase that mediates Ca2+-induced Ca2+ sensitization of vascular smooth muscle contraction.
Pyk2 is downstream of Ca2+ and upstream of RhoA
The Ca2+ ionophore, ionomycin, induces slow, sustained contraction of rat caudal arterial smooth muscle due to the influx of Ca2+. Ionomycin-induced contraction was attenuated by pre-treatment with genistein or Y-27632 (Mita et al. 2013), consistent with Ca2+ lying upstream of Pyk2 and ROCK activation. Ionomycin-induced contraction was accompanied by an increase in Pyk2 autophosphorylation at Tyr402, which was blocked by pre-treatment with PF-431396, consistent with Pyk2 being the key Ca2+-activated tyrosine kinase (Mills et al. 2015). In support of this conclusion, voltage-gated Ca2+ channel blockade with nifedipine inhibited both contraction and Pyk2 autophosphorylation. On the other hand, depolarization-induced Pyk2 activation was unaffected by pre-treatment with the MLCK inhibitor, ML-9, which, as expected, inhibited both peak and sustained contraction (Mills et al. 2015). Finally, K+-induced translocation of RhoA and phosphorylation of MYPT1 at Thr697 and Thr855 were prevented by pre-treatment with sodium salicylate, consistent with Pyk2 being located upstream of RhoA activation.
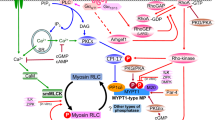
We conclude, therefore, that the tonic component of depolarization-induced contraction of vascular smooth muscle involves a Ca2+-induced Ca2+ sensitization mechanism whereby Ca2+ entry via voltage-gated Ca2+ channels activates Pyk2, leading to translocation of RhoA to the plasma membrane, activation of ROCK and inhibition of MLCP (Fig. 6). Inhibition of the phosphatase enables force maintenance at a level of activation of MLCK that is lower than during the phasic contractile response.

Proposed mechanism of Ca2+-induced Ca2+ sensitization of vascular smooth muscle contraction. Depolarization of the vascular smooth muscle cell plasma membrane induces Ca2+ entry via voltage-gated Ca2+ channels. The immediate response is activation of CaM-dependent MLCK, phosphorylation of LC20 and cross-bridge cycling, which accounts for the rapid, phasic contractile response. On a slower time-scale, Ca2+ activates the tyrosine kinase, Pyk2, which leads to the activation of RhoA and ROCK, and inhibition of MLCP activity. LC20 phosphorylation and force are thereby maintained at a lower level of activation of MLCK than is observed during the phasic contraction. CPI-17 is not included here as a ROCK substrate since KCl has been shown not to induce phosphorylation of CPI-17 at Thr38 (Kitazawa et al. 2000; Koyama et al. 2000; Kitazawa et al. 2003)
Challenges for the future
What is the mechanism of activation of Pyk2 by Ca2+? Evidence is required to determine whether Pyk2 is directly activated by Ca2+ or whether an intermediate, such as CaM (as has been suggested), is involved. A role for CaM is supported by the observation that the CaM antagonist, W-7, but not the inactive analog, W-5, suppressed K+-induced RhoA activation (Sakurada et al. 2003). If the Ca2+/CaM complex indeed activates Pyk2, this could be through direct interaction with the tyrosine kinase or may involve another CaM target protein, e.g. Ca2+/CaM-dependent kinase II (CaM kinase II), as an intermediary. In support of the latter possibility, the CaM kinase II inhibitor, KN-93, but not the inactive analog, KN-92, inhibited RhoA activation (Sakurada et al. 2003). However, while W-7 inhibited ionomycin-induced RhoA activation, KN-93 had no effect (Sakurada et al. 2003).
How does activated Pyk2 induce RhoA activation and translocation to the plasma membrane? Transfection experiments with primary cultured rat aortic vascular smooth muscle cells have suggested that the guanine nucleotide exchange activity of PDZ-RhoGEF (PDZ domain-containing Rho guanine nucleotide exchange factor) may be activated by tyrosine phosphorylation catalysed by Pyk2 (Ying et al. 2009b). This needs to be investigated in freshly isolated vascular smooth muscle tissue. Phosphoinositide 3-kinase class II α-isoform (PI3K-C2α) has been shown to be necessary for Ca2+-induced RhoA/ROCK-dependent inhibition of MLCP, LC20 phosphorylation and contraction of vascular smooth muscle tissue (rabbit thoracic aorta) and isolated rat aortic vascular smooth muscle cells (Wang et al. 2006; Yoshioka et al. 2007). PI3K is not activated directly by Ca2+ (Arcaro et al. 2000), but PI3K-C2α has been shown to be activated by tyrosine phosphorylation (Turner et al. 1998; Paulhe et al. 2002), raising the possibility that Pyk2 may lie upstream of PI3K-C2α, which generates phosphatidylinositol 3,4,5-trisphosphate, an activator of RhoA (Lu and Chen 1997; Missy et al. 1998). Ca2+-independent phospholipase A2 (iPLA2) has also been implicated in K+-induced Ca2+ sensitization of contraction of rabbit femoral and renal arterial rings (Ratz et al. 2009). However, pre-treatment of rat caudal arterial smooth muscle strips with the iPLA2 inhibitor bromoenol lactone (10 μM) had no effect on K+-induced contraction (Mita M, unpublished results), leading us to conclude that iPLA2 is not involved in depolarization-induced contraction in this tissue.
Is Pyk2 activation also coupled to dynamic remodeling of the actin cytoskeleton? Agonist stimulation of rat small mesenteric arteries induced Pyk2 phosphorylation at Tyr402 (and Tyr881), and, with a similar time course, paxillin phosphorylation at Tyr31 and Tyr118 (Ohanian et al. 2005). These events were accompanied by association of Pyk2 and paxillin with the Triton-insoluble fraction. Paxillin phosphorylation has been linked to actin polymerization in airway smooth muscle (Gunst and Zhang 2008), and actin polymerization in the sub-sarcolemmal domain is required for vascular smooth muscle contraction (reviewed by Walsh and Cole 2013). This mechanism has not been explored in the context of depolarization-induced vascular smooth muscle contraction.
Why does membrane depolarization induce less Ca2+ sensitization than agonists acting via GPCRs (Ratz et al. 2005)? Several possibilities can be considered, including: (i) GPCR activation, coupled to Gq/11 and G12/13 heterotrimeric G proteins, leads to the activation of PKC and ROCK, phosphorylation of CPI-17 and MYPT1, and inhibition of MLCP. We found that the PKC inhibitors, calphostin C and chelerythrine, had no effect on K+-induced contraction of rat caudal arterial smooth muscle (Mita et al. 2013) and that membrane depolarization did not induce CPI-17 phosphorylation (Mita M and Walsh MP, unpublished observation). GPCR activation, therefore, may elicit a higher degree of Ca2+ sensitization than membrane depolarization through enhanced inhibition of MLCP activity. (ii) Actin polymerization has been identified as another important mechanism involved in Ca2+ sensitization of vascular smooth muscle contraction, as mentioned above, and both ROCK and PKC have been implicated in agonist- and pressure-induced polymerization of cortical actin, leading to increased force development due to enhancement of the physical connections between the contractile machinery of the smooth muscle cell and the extracellular matrix (reviewed in Walsh and Cole 2013). It remains to be determined whether or not membrane depolarization induces actin polymerization in vascular smooth muscle, but if it does not, this could explain the greater Ca2+ sensitization induced by GPCR activation.
Finally, is Pyk2 a suitable therapeutic target for the treatment of cardiovascular diseases, such as hypertension, that are associated with hypercontractility? As indicated earlier, up-regulation of the RhoA/ROCK pathway has been implicated in the etiology of hypertension (Uehata et al. 1997; Kanda et al. 2006) and sodium salicylate lowers blood pressure in hypertensive rats at concentrations that inhibit Pyk2 (Ying et al. 2009a).
References
Adegunloye BJ, Su X, Camper EV, Moreland RS (2003) Sensitivity of rabbit aorta and mesenteric artery to norepinephrine: role of tyrosine kinases. Eur J Pharmacol 476:201–209
Arcaro A, Zvelebil MJ, Wallasch C, Ullrich A, Waterfield MD, Domin J (2000) Class II phosphoinositde 3-kinases are downstream targets of activated polypeptide growth factor receptors. Mol Cell Biol 20:3817–3830
Avraham H, Park SY, Schinkmann K, Avraham S (2000) RAFTK/Pyk2-mediated cellular signalling. Cell Signal 12:123–133
Baumann BAJ, Taylor DW, Huang Z, Tama F, Fagnant PM, Trybus KM, Taylor KA (2012) Phosphorylated smooth muscle heavy meromyosin shows an open conformation linked to activation. J Mol Biol 415:274–287
Bolton TB (1979) Mechanisms of action of transmitters and other substances on smooth muscle. Physiol Rev 59:606–718
Borysova L, Shabir S, Walsh MP, Burdyga T (2011) The importance of Rho-associated kinase-induced Ca2+ sensitization as a component of electromechanical and pharmacomechanical coupling in rat ureteric smooth muscle. Cell Calcium 50:393–405
Chen CP, Chen X, Qiao YN, Wang P, He WQ, Zhang CH, Zhao W, Gao YQ, Chen C, Tao T, Sun J, Wang Y, Gao N, Kamm KE, Stull JT, Zhu MS (2015) In vivo roles for myosin phosphatase targeting subunit-1 phosphorylation sites T694 and T852 in bladder smooth muscle contraction. J Physiol 593:681–700
Di Salvo J, Steusloff A, Semenchuk L, Satoh S, Kolquist K, Pfitzer G (1993a) Tyrosine kinase inhibitors suppress agonist-induced contraction in smooth muscle. Biochem Biophys Res Commun 190:968–974
Di Salvo J, Semenchuk LA, Lauer J (1993b) Vanadate-induced contraction of smooth muscle and enhanced protein tyrosine phosphorylation. Arch Biochem Biophys 304:386–391
Di Salvo J, Pfitzer G, Semenchuk LA (1994) Protein tyrosine phosphorylation, cellular Ca2+, and Ca2+ sensitivity for contraction of smooth muscle. Can J Physiol Pharmacol 72:1434–1439
Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, Okawa K, Iwamatsu A, Kaibuchi K, Hartshorne DJ, Nakano T (1999a) Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem 274:3744–3752
Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, Nakano T (1999b) Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem 274:37385–37390
Gong MC, Fujihara H, Somlyo AV, Somlyo AP (1997) Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J Biol Chem 272:10704–10709
Gunst SJ, Zhang W (2008) Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295:C576–C587
Han S, Mistry A, Chang JS, Cunningham D, Griffor M, Bonnette PC, Wang H, Chrunyk BA, Aspnes GE, Walker DP, Brosius AD, Buckbinder L (2009) Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J Biol Chem 284:13193–13201
Hollenberg MD (1994) Tyrosine kinase pathways and the regulation of smooth muscle contractility. Trends Pharmacol Sci 15:108–114
Hong F, Haldeman BD, Jackson D, Carter M, Baker JE, Cremo CR (2011) Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys 510:135–146
Ichikawa K, Ito M, Hartshorne DJ (1996) Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. J Biol Chem 271:4733–4740
Kanda T, Wakino S, Homma K, Yoshioka K, Tatematsu S, Hasegawa K, Takamatsu I, Sugano N, Hayashi K, Saruta T (2006) Rho kinase as a molecular target for insulin resistance and hypertension. FASEB J 20:169–171
Khasnis M, Nakatomi A, Gumpper K, Eto M (2014) Reconstituted human myosin light chain phosphatase reveals distinct roles of two inhibitory phosphorylation sites of the regulatory subunit, MYPT1. Biochemistry 53:2701–2709
Kitazawa T, Kitazawa K (2012) Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J Physiol 590:5401–5423
Kitazawa T, Eto M, Woodsome TP, Brautigan DL (2000) Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem 275:9897–9900
Kitazawa T, Eto M, Woodsome TP, Khalequzzaman M (2003) Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca2+ sensitization in rabbit smooth muscle. J Physiol 546:879–889
Kohno T, Matsuda E, Sasaki H, Sasaki T (2008) Protein tyrosine kinase CAKβ/PYK2 is activated by binding Ca2+/calmodulin to FERM F2 α2 helix and thus forming its dimer. Biochem J 410:513–523
Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ, Nakano T (2000) Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett 475:197–200
Laniyonu AA, Saifeddine M, Yang SG, Hollenberg MD (1994) Tyrosine kinase inhibitors and the contractile action of G-protein-linked vascular agonists. Can J Physiol Pharmacol 72:1075–1085
Leung T, Manser E, Tan L, Lim L (1995) A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem 270:29051–29054
Lipinski CA, Loftus JC (2010) The Pyk2 FERM domain: a novel therapeutic target. Expert Opin Ther Targets 14:95–108
Lu PJ, Chen CS (1997) Selective recognition of phosphatidylinositol 3,4,5-trisphosphate by a synthetic peptide. J Biol Chem 272:466–472
Mabuchi Y, Mabuchi K, Stafford WF, Grabarek Z (2010) Modular structure of smooth muscle myosin light chain kinase: hydrodynamic modeling and functional implications. Biochemistry 49:2903–2917
Masui H, Wakabayashi I (2000) Tyrosine phosphorylation increases Ca2+ sensitivity of vascular smooth muscle contraction. Life Sci 68:363–372
Mills RD, Mita M, Nakagawa J, Shoji M, Sutherland C, Walsh MP (2015) A role for the tyrosine kinase Pyk2 in depolarization-induced contraction of vascular smooth muscle. J Biol Chem 290:8677–8692
Missy K, Van Poucke V, Raynal P, Viala C, Mauco G, Plantavid G, Chap H, Payrastre B (1998) Lipid products of phosphoinositide 3-kinase interact with Rac1 GTPase and stimulate GDP dissociation. J Biol Chem 273:30279–30286
Mita M, Walsh MP (1997) α1-Adrenoceptor-mediated phosphorylation of myosin in rat-tail arterial smooth muscle. Biochem J 327:669–674
Mita M, Yanagihara H, Hishinuma S, Saito M, Walsh MP (2002) Membrane depolarization-induced contraction of rat caudal arterial smooth muscle involves Rho-associated kinase. Biochem J 364:431–440
Mita M, Tanaka H, Yanagihara H, Nakagawa J, Hishinuma S, Sutherland C, Walsh MP, Shoji M (2013) Membrane depolarization-induced RhoA/Rho-associated kinase activation and sustained contraction of rat caudal arterial smooth muscle involves genistein-sensitive tyrosine phosphorylation. J Smooth Muscle Res 49:26–45
Mori M, Tsushima H (2004) Vanadate activates RhoA translocation in association with contracting effects in ileal longitudinal smooth muscle of guinea pig. J Pharmacol Sci 95:443–451
Murányi A, Derkach D, Erdődi F, Kiss A, Ito M, Hartshorne DJ (2005) Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett 579:6611–6615
Nakao F, Kobayashi S, Mogami K, Mizukami Y, Shirao S, Miwa S, Todoroki-Ikeda N, Ito M, Matsuzaki M (2002) Involvement of Src family protein tyrosine kinases in Ca2+ sensitization of coronary artery contraction mediated by a sphingosylphosphorylcholine-Rho-kinase pathway. Circ Res 91:953–960
Ohanian V, Gatfield K, Ohanian J (2005) Role of the actin cytoskeleton in G-protein-coupled receptor activation of PYK2 and paxillin in vascular smooth muscle. Hypertension 46:93–99
Paulhe F, Perret B, Chap H, Iberg N, Morand O, Racaud-Sultan C (2002) Phosphoinositide 3-kinase C2α is activated upon smooth muscle cell migration and regulated by α v β 3 integrin engagement. Biochem Biophys Res Commun 297:261–266
Ratz PH (1990) Effect of the kinase inhibitor, H-7, on stress, crossbridge phosphorylation, muscle shortening and inositol phosphate production in rabbit arteries. J Pharmacol Exp Ther 252:253–259
Ratz PH, Berg KM, Urban NH, Miner AS (2005) Regulation of smooth muscle calcium sensitivity: KCl as a calcium-sensitizing stimulus. Am J Physiol Cell Physiol 288:C769–C783
Ratz PH, Miner AS, Barbour SE (2009) Calcium-independent phospholipase A2 participates in KCl-induced calcium sensitization of vascular smooth muscle. Cell Calcium 46:65–72
Rocic P, Griffin TM, McRae CN, Lucchesi PA (2002) Altered Pyk2 phosphorylation by ANG II in hypertensive vascular smooth muscle. Am J Physiol Heart Circ Physiol 282:H457–H465
Sabri A, Govindarajan G, Griffin TM, Byron KL, Samarel AM, Lucchesi PA (1998) Calcium and protein kinase C-dependent activation of the tyrosine kinase PYK2 by angiotensin II in vascular smooth muscle. Circ Res 83:841–851
Sakamoto K, Hori M, Izumi M, Oka T, Kohama K, Ozaki H, Karaki H (2003) Inhibition of high K+-induced contraction by the ROCKs inhibitor Y-27632 in vascular smooth muscle: possible involvement of ROCKs in a signal transduction pathway. J Pharmacol Sci 92:56–69
Sakurada S, Okamoto H, Takuwa N, Sugimoto N, Takuwa Y (2001) Rho activation in excitatory agonist-stimulated vascular smooth muscle. Am J Physiol Cell Physiol 281:C571–C578
Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y (2003) Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res 93:548–556
Sasaki H, Kazuko N, Ishino M, Tobioka H, Kotani K, Sasaki T (1995) Cloning and characterization of cell adhesion kinase β, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem 270:21206–21219
Sasaki M, Hattori Y, Tomita F, Moriishi K, Kanno M, Kohya T, Oguma K, Kitabatake A (1998) Tyrosine phosphorylation as a convergent pathway of heterotrimeric G protein- and rho protein-mediated Ca2+ sensitization of smooth muscle of rabbit mesenteric artery. Br J Pharmacol 125:1651–1660
Seok YM, Baek I, Kim YH, Jeong YS, Lee IJ, Shin DH, Hwang YH, Kim IK (2008) Isoflavone attenuates vascular contraction through inhibition of the RhoA/Rho-kinase signaling pathway. J Pharmacol Exp Ther 326:991–998
Shimada T, Shimamura K, Sunano S (1986) Effects of sodium vanadate on various types of vascular smooth muscles. Blood Vessels 23:113–124
Somlyo AP, Somlyo AV (2000) Signal transduction by G-proteins, Rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol 522:177–185
Steusloff A, Paul E, Semenchuk LA, Di Salvo J, Pfitzer G (1995) Modulation of Ca2+ sensitivity in smooth muscle by genistein and protein tyrosine phosphorylation. Arch Biochem Biophys 320:236–242
Stull JT, Lin PJ, Krueger JK, Trewhella J, Zhi G (1998) Myosin light chain kinase: functional domains and structural motifs. Acta Physiol Scand 164:471–482
Sutherland C, Walsh MP (2012) Myosin regulatory light chain diphosphorylation slows relaxation of arterial smooth muscle. J Biol Chem 287:24064–24076
Takeya K, Loutzenhiser K, Shiraishi M, Loutzenhiser RD, Walsh MP (2008) A highly sensitive technique to measure myosin regulatory light chain phosphorylation: the first quantification in renal arterioles. Am J Physiol Renal Physiol 294:F1487–F1492
Takizawa S, Hori M, Ozaki H, Karaki H (1993) Effects of isoquinoline derivatives, HA 1077 and H-7, on cytosolic Ca2+ level and contraction in vascular smooth muscle. Eur J Pharmacol 250:431–437
Taylor KA, Feig M, Brooks CL III, Fagnant PM, Lowey S, Trybus KM (2014) Role of the essential light chain in the activation of smooth muscle myosin by regulatory light chain phosphorylation. J Struct Biol 185:375–382
Turner SJ, Domin J, Waterfield MD, Ward SG, Westwick J (1998) The CC chemokine monocyte chemotactic peptide-1 activates both the class I p85/p110 phosphatidylinositol 3-kinase and the class II PI3K-C2α. J Biol Chem 273:25987–25995
Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S (1997) Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389:990–994
Urban NH, Berg KM, Ratz PH (2003) K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol 285:C1377–C1385
Velasco G, Armstrong C, Morrice N, Frame S, Cohen P (2002) Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1 M at Thr850 induces its dissociation from myosin. FEBS Lett 527:101–104
Walsh MP, Cole WC (2013) The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab 33:1–12
Wang Z, Brecher P (2001) Salicylate inhibits phosphorylation of the nonreceptor tyrosine kinases, proline-rich tyrosine kinase-2 and c-Src. Hypertension 37:148–153
Wang Y, Yoshioka K, Azam MA, Takuwa N, Sakurada S, Kayaba Y, Sugimoto N, Inoki I, Kimura T, Kuwaki T, Takuwa Y (2006) Class II phosphoinositide 3-kinase α-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem J 394:581–592
Weber LP, Van Lierop JE, Walsh MP (1999) Ca2+-independent phosphorylation of myosin in rat caudal artery and chicken gizzard myofilaments. J Physiol 516:805–824
Wendt T, Taylor D, Trybus KM, Taylor K (2001) Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc Natl Acad Sci USA 98:4361–4366
Williams DA, Fay FS (1986) Calcium transients and resting levels in isolated smooth muscle cells as monitored with quin 2. Am J Physiol Cell Physiol 250:C779–C791
Williams DA, Becker PL, Fay FS (1987) Regional changes in calcium underlying contraction of single smooth muscle cells. Science 235:1644–1648
Wilson DP, Sutherland C, Walsh MP (2002) Ca2+ activation of smooth muscle contraction: evidence for the involvement of calmodulin that is bound to the Triton-insoluble fraction even in the absence of Ca2+. J Biol Chem 277:2186–2192
Wu X, Clack BA, Zhi G, Stull JT, Cremo CR (1999) Phosphorylation-dependent structural changes in the regulatory light chain domain of smooth muscle heavy meromyosin. J Biol Chem 274:20328–20335
Yanagisawa T, Okada Y (1994) KCl depolarization increases Ca2+ sensitivity of contractile elements in coronary arterial smooth muscle. Am J Physiol Heart Circ Physiol 267:H614–H621
Ying Z, Giachini FRC, Tostes RC, Webb RC (2009a) Salicylates dilate blood vessels through inhibiting PYK2-mediated RhoA/Rho-kinase activation. Cardiovasc Res 83:155–162
Ying Z, Giachini FRC, Tostes RC, Webb RC (2009b) PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol 29:1657–1663
Yoshioka K, Sugimoto N, Takuwa N, Takuwa Y (2007) Essential role for class II phosphoinositide 3-kinase α-isoform in Ca2+-induced, Rho- and Rho kinase-dependent regulation of myosin phosphatase and contraction in isolated vascular smooth muscle cells. Mol Pharmacol 71:912–920
Yu H, Li X, Marchetto GS, Dy R, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ, Graves LM, Earp HS (1996) Activation of a novel calcium-dependent protein-tyrosine kinase. J Biol Chem 271:29993–29998
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (MOP-111262) to M.P. Walsh and by grants from the High-Tech Research Center Project (S0801043) and the Meiyaku Open Research Project, the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan to M. Mita. M.P. Walsh was recipient of a Canada Research Chair (Tier 1) in Vascular Smooth Muscle Research and an Alberta Innovates—Health Solutions Scientist Award. R.D. Mills was recipient of a Kertland Family Postdoctoral Fellowship in Vascular Biology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mills, R.D., Mita, M. & Walsh, M.P. A role for the Ca2+-dependent tyrosine kinase Pyk2 in tonic depolarization-induced vascular smooth muscle contraction. J Muscle Res Cell Motil 36, 479–489 (2015). https://doi.org/10.1007/s10974-015-9416-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-015-9416-2